
Molecular Landscape of Tourette’s Disorder

 ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Input Omics Datasets and Candidate Genes
2.2. Tissue and Cell Type Specificity
2.2.1. Genomic Data
2.2.2. Brain Transcriptomic Data
2.3. Functional Enrichment Analyses
2.4. Shared Genetic Etiology Analyses with Levels of Blood and Cerebrospinal Fluid Metabolites
2.4.1. Polygenic Risk Score (PRS)-Based Analyses
2.4.2. SNP Effect Concordance Analysis (SECA)
2.5. Molecular Landscape of TD
2.5.1. Description of the TD Landscape
Presynaptic and Postsynaptic Neurons
- Extracellular matrix (ECM)
- Cell membrane
- Cytoskeleton
- Cytoplasm
- Organelles
- Nucleus
Microglial Cells and Astrocytes
- Microglial cells
- Astrocytes
3. Discussion
4. Materials and Methods
4.1. Literature Search and Selection of Omics Datasets
4.2. Analyses of TD GWAS summary Statistics
4.2.1. Genome-Wide Gene-Based Analysis in MAGMA
4.2.2. SNP Functional Annotation and Gene Mapping in FUMA
- SNP functional annotation
- Gene mapping
4.2.3. Transcriptome-Wide Association Study
4.2.4. Shared Genetic Etiology Analyses with Levels of Blood and Cerebrospinal Fluid Metabolites
- Polygenic risk score (PRS)-based analyses
- SNP effect concordance analyses (SECA)
4.3. Integration, Annotation, and Prioritization of Omics Studies Results
4.3.1. Integration of Omics Studies Results
4.3.2. Gene-Level Annotation of Omics Studies Results
4.3.3. Prioritization of Omics Studies Results
4.4. Tissue and Cell Type Specificity Analyses
4.5. Functional Enrichment Analyses
4.6. Molecular Landscape of TD
4.7. Selection of Putative Drug Targets from the Built Molecular Landscape of TD
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robertson, M.M.; Eapen, V.; Singer, H.S.; Martino, D.; Scharf, J.M.; Paschou, P.; Roessner, V.; Woods, D.W.; Hariz, M.; Mathews, C.A.; et al. Gilles de la Tourette syndrome. Nat. Rev. Dis. Prim. 2017, 3, 16097. [Google Scholar] [CrossRef] [PubMed]
- Martino, D.; Ganos, C.; Pringsheim, T.M. Chapter Fifty-Three-Tourette Syndrome and Chronic Tic Disorders: The Clinical Spectrum Beyond Tics. In International Review of Neurobiology; Chaudhuri, K.R., Titova, N., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 134, pp. 1461–1490. [Google Scholar]
- Mataix-Cols, D.; Isomura, K.; Pérez-Vigil, A.; Chang, Z.; Rück, C.; Larsson, K.J.; Leckman, J.F.; Serlachius, E.; Larsson, H.; Lichtenstein, P. Familial Risks of Tourette Syndrome and Chronic Tic Disorders. A Population-Based Cohort Study. JAMA Psychiatry 2015, 72, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Tischfield, J.A.; King, R.A.; Heiman, G.A. Functional Evaluations of Genes Disrupted in Patients with Tourette’s Disorder. Front. Psychiatry 2016, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Tsetsos, F.; Yu, D.; Sul, J.H.; Huang, A.Y.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; Greenberg, E.; Muller-Vahl, K.R.; et al. Synaptic processes and immune-related pathways implicated in Tourette syndrome. Transl. Psychiatry 2021, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.A.; Scharf, J.M.; Miller, L.L.; Macdonald-Wallis, C.; Lawlor, D.A.; Ben-Shlomo, Y. Association between pre- and perinatal exposures and Tourette syndrome or chronic tic disorder in the ALSPAC cohort. Br. J. Psychiatry 2014, 204, 40–45. [Google Scholar] [CrossRef]
- Tagwerker Gloor, F.; Walitza, S. Tic Disorders and Tourette Syndrome: Current Concepts of Etiology and Treatment in Children and Adolescents. Neuropediatrics 2016, 47, 84–96. [Google Scholar] [CrossRef]
- Harris, K.; Singer, H.S. Tic disorders: Neural circuits, neurochemistry, and neuroimmunology. J. Child Neurol. 2006, 21, 678–689. [Google Scholar] [CrossRef]
- Andrén, P.; Jakubovski, E.; Murphy, T.L.; Woitecki, K.; Tarnok, Z.; Zimmerman-Brenner, S.; van de Griendt, J.; Debes, N.M.; Viefhaus, P.; Robinson, S.; et al. European clinical guidelines for Tourette syndrome and other tic disorders-version 2.0. Part II: Psychological interventions. Eur. Child Adolesc. Psychiatry 2021, 31, 403–423. [Google Scholar] [CrossRef]
- Müller-Vahl, K.R.; Szejko, N.; Verdellen, C.; Roessner, V.; Hoekstra, P.J.; Hartmann, A.; Cath, D.C. European clinical guidelines for Tourette syndrome and other tic disorders: Summary statement. Eur. Child Adolesc. Psychiatry 2022, 31, 377–382. [Google Scholar] [CrossRef]
- Pringsheim, T.; Okun, M.S.; Müller-Vahl, K.; Martino, D.; Jankovic, J.; Cavanna, A.E.; Woods, D.W.; Robinson, M.; Jarvie, E.; Roessner, V.; et al. Practice guideline recommendations summary: Treatment of tics in people with Tourette syndrome and chronic tic disorders. Neurology 2019, 92, 896–906. [Google Scholar] [CrossRef]
- Roessner, V.; Eichele, H.; Stern, J.S.; Skov, L.; Rizzo, R.; Debes, N.M.; Nagy, P.; Cavanna, A.E.; Termine, C.; Ganos, C.; et al. European clinical guidelines for Tourette syndrome and other tic disorders-version 2.0. Part III: Pharmacological treatment. Eur. Child Adolesc. Psychiatry 2022, 31, 425–441. [Google Scholar] [CrossRef]
- King, E.A.; Davis, J.W.; Degner, J.F. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019, 15, e1008489. [Google Scholar] [CrossRef]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef]
- Johnson, E.C.; Border, R.; Melroy-Greif, W.E.; de Leeuw, C.A.; Ehringer, M.A.; Keller, M.C. No Evidence That Schizophrenia Candidate Genes Are More Associated with Schizophrenia Than Noncandidate Genes. Biol. Psychiatry 2017, 82, 702–708. [Google Scholar] [CrossRef]
- Farrell, M.S.; Werge, T.; Sklar, P.; Owen, M.J.; Ophoff, R.A.; O’Donovan, M.C.; Corvin, A.; Cichon, S.; Sullivan, P.F. Evaluating historical candidate genes for schizophrenia. Mol. Psychiatry 2015, 20, 555–562. [Google Scholar] [CrossRef]
- Poelmans, G.; Pauls, D.L.; Buitelaar, J.K.; Franke, B. Integrated genome-wide association study findings: Identification of a neurodevelopmental network for attention deficit hyperactivity disorder. Am. J. Psychiatry 2011, 168, 365–377. [Google Scholar] [CrossRef]
- Poelmans, G.; Franke, B.; Pauls, D.L.; Glennon, J.C.; Buitelaar, J.K. AKAPs integrate genetic findings for autism spectrum disorders. Transl. Psychiatry 2013, 3, e270. [Google Scholar] [CrossRef]
- Van de Vondervoort, I.; Poelmans, G.; Aschrafi, A.; Pauls, D.L.; Buitelaar, J.K.; Glennon, J.C.; Franke, B. An integrated molecular landscape implicates the regulation of dendritic spine formation through insulin-related signalling in obsessive-compulsive disorder. J. Psychiatry Neurosci. 2016, 41, 280–285. [Google Scholar] [CrossRef]
- Klemann, C.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Park. Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Ramautar, R.; Berger, R.; van der Greef, J.; Hankemeier, T. Human metabolomics: Strategies to understand biology. Curr. Opin. Chem. Biol. 2013, 17, 841–846. [Google Scholar] [CrossRef]
- Hagenbeek, F.A.; Pool, R.; van Dongen, J.; Draisma, H.H.M.; Jan Hottenga, J.; Willemsen, G.; Abdellaoui, A.; Fedko, I.O.; den Braber, A.; Visser, P.J.; et al. Heritability estimates for 361 blood metabolites across 40 genome-wide association studies. Nat. Commun. 2020, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; David, D.E.; Johnson, E.W.; Konecki, D.; Burmester, J.K.; Ledbetter, D.H.; Weber, J.L. Breakpoint sequences of an 1;8 translocation in a family with Gilles de la Tourette syndrome. Eur. J. Hum. Genet. 2000, 8, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Petek, E.; Windpassinger, C.; Vincent, J.B.; Cheung, J.; Boright, A.P.; Scherer, S.W.; Kroisel, P.M.; Wagner, K. Disruption of a novel gene (IMMP2L) by a breakpoint in 7q31 associated with Tourette syndrome. Am. J. Hum. Genet. 2001, 68, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Mathews, C.A.; Joosse, M.; Eussen, B.H.; Heutink, P.; Oostra, B.A. Tourette Syndrome Association International Consortium for, G. CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics 2003, 82, 1–9. [Google Scholar] [CrossRef]
- Abelson, J.F.; Kwan, K.Y.; O’Roak, B.J.; Baek, D.Y.; Stillman, A.A.; Morgan, T.M.; Mathews, C.A.; Pauls, D.L.; Rasin, M.R.; Gunel, M.; et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science 2005, 310, 317–320. [Google Scholar] [CrossRef]
- Breedveld, G.J.; Fabbrini, G.; Oostra, B.A.; Berardelli, A.; Bonifati, V. Tourette disorder spectrum maps to chromosome 14q31.1 in an Italian kindred. Neurogenetics 2010, 11, 417–423. [Google Scholar] [CrossRef]
- Patel, C.; Cooper-Charles, L.; McMullan, D.J.; Walker, J.M.; Davison, V.; Morton, J. Translocation breakpoint at 7q31 associated with tics: Further evidence for IMMP2L as a candidate gene for Tourette syndrome. Eur. J. Hum. Genet. 2011, 19, 634–639. [Google Scholar] [CrossRef]
- Hooper, S.D.; Johansson, A.C.; Tellgren-Roth, C.; Stattin, E.L.; Dahl, N.; Cavelier, L.; Feuk, L. Genome-wide sequencing for the identification of rearrangements associated with Tourette syndrome and obsessive-compulsive disorder. BMC Med. Genet. 2012, 13, 123. [Google Scholar] [CrossRef]
- Bertelsen, B.; Melchior, L.; Jensen, L.R.; Groth, C.; Nazaryan, L.; Debes, N.M.; Skov, L.; Xie, G.; Sun, W.; Brondum-Nielsen, K.; et al. A t(3;9)(q25.1;q34.3) translocation leading to OLFM1 fusion transcripts in Gilles de la Tourette syndrome, OCD and ADHD. Psychiatry Res. 2015, 225, 268–275. [Google Scholar] [CrossRef]
- Ercan-Sencicek, A.G.; Stillman, A.A.; Ghosh, A.K.; Bilguvar, K.; O’Roak, B.J.; Mason, C.E.; Abbott, T.; Gupta, A.; King, R.A.; Pauls, D.L.; et al. L-histidine decarboxylase and Tourette’s syndrome. N. Engl. J. Med. 2010, 362, 1901–1908. [Google Scholar] [CrossRef]
- Sundaram, S.K.; Huq, A.M.; Sun, Z.; Yu, W.; Bennett, L.; Wilson, B.J.; Behen, M.E.; Chugani, H.T. Exome sequencing of a pedigree with Tourette syndrome or chronic tic disorder. Ann. Neurol. 2011, 69, 901–904. [Google Scholar] [CrossRef]
- Eriguchi, Y.; Kuwabara, H.; Inai, A.; Kawakubo, Y.; Nishimura, F.; Kakiuchi, C.; Tochigi, M.; Ohashi, J.; Aoki, N.; Kato, K.; et al. Identification of candidate genes involved in the etiology of sporadic Tourette syndrome by exome sequencing. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 712–723. [Google Scholar] [CrossRef]
- Willsey, A.J.; Fernandez, T.V.; Yu, D.; King, R.A.; Dietrich, A.; Xing, J.; Sanders, S.J.; Mandell, J.D.; Huang, A.Y.; Richer, P.; et al. De Novo Coding Variants Are Strongly Associated with Tourette Disorder. Neuron 2017, 94, 486–499. [Google Scholar] [CrossRef]
- Sun, N.; Nasello, C.; Deng, L.; Wang, N.; Zhang, Y.; Xu, Z.; Song, Z.; Kwan, K.; King, R.A.; Pang, Z.P.; et al. The PNKD gene is associated with Tourette Disorder or Tic disorder in a multiplex family. Mol. Psychiatry 2018, 23, 1487–1495. [Google Scholar] [CrossRef]
- Wang, S.; Mandell, J.D.; Kumar, Y.; Sun, N.; Morris, M.T.; Arbelaez, J.; Nasello, C.; Dong, S.; Duhn, C.; Zhao, X.; et al. De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis. Cell Rep. 2018, 24, 3441–3454. [Google Scholar] [CrossRef]
- Carias, K.V.; Wevrick, R. Clinical and genetic analysis of children with a dual diagnosis of Tourette syndrome and autism spectrum disorder. J. Psychiatr. Res. 2019, 111, 145–153. [Google Scholar] [CrossRef]
- Depienne, C.; Ciura, S.; Trouillard, O.; Bouteiller, D.; Leitao, E.; Nava, C.; Keren, B.; Marie, Y.; Guegan, J.; Forlani, S.; et al. Association of Rare Genetic Variants in Opioid Receptors with Tourette Syndrome. Tremor Other Hyperkinet. Mov. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Vadgama, N.; Pittman, A.; Simpson, M.; Nirmalananthan, N.; Murray, R.; Yoshikawa, T.; De Rijk, P.; Rees, E.; Kirov, G.; Hughes, D.; et al. De novo single-nucleotide and copy number variation in discordant monozygotic twins reveals disease-related genes. Eur. J. Hum. Genet. 2019, 27, 1121–1133. [Google Scholar] [CrossRef]
- Liu, S.; Tian, M.; He, F.; Li, J.; Xie, H.; Liu, W.; Zhang, Y.; Zhang, R.; Yi, M.; Che, F.; et al. Mutations in ASH1L confer susceptibility to Tourette syndrome. Mol. Psychiatry 2020, 25, 476–490. [Google Scholar] [CrossRef]
- Yuan, A.; Wang, Z.; Xu, W.; Ding, Q.; Zhao, Y.; Han, J.; Sun, J. A Rare Novel CLCN2 Variation and Risk of Gilles de la Tourette Syndrome: Whole-Exome Sequencing in a Multiplex Family and a Follow-Up Study in a Chinese Population. Front. Psychiatry 2020, 11, 543911. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, S.; Hao, J.; Zhu, P.; Zhang, X.; Wu, M. A Whole-Exome Sequencing Study of Tourette Disorder in a Chinese Population. DNA Cell Biol. 2020, 39, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Zhang, Y.; Abdulkadir, M.; Deng, L.; Fernandez, T.V.; Garcia-Delgar, B.; Hagstrom, J.; Hoekstra, P.J.; King, R.A.; Koesterich, J.; et al. Whole-exome sequencing identifies genes associated with Tourette’s disorder in multiplex families. Mol. Psychiatry 2021, 26, 6937–6951. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, M.; Szatkiewicz, J.; Mudgal, P.; Yu, D.; Psychiatric Genomics Consortium TS/OCD Working Group; Nordsletten, A.E.; Mataix-Cols, D.; Mathews, C.A.; Scharf, J.M.; Mattheisen, M.; et al. Elevated common variant genetic risk for tourette syndrome in a densely-affected pedigree. Mol. Psychiatry 2021, 26, 7522–7529. [Google Scholar] [CrossRef] [PubMed]
- Lawson-Yuen, A.; Saldivar, J.S.; Sommer, S.; Picker, J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur. J. Hum. Genet. 2008, 16, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.K.; Huq, A.M.; Wilson, B.J.; Chugani, H.T. Tourette syndrome is associated with recurrent exonic copy number variants. Neurology 2010, 74, 1583–1590. [Google Scholar] [CrossRef]
- Fernandez, T.V.; Sanders, S.J.; Yurkiewicz, I.R.; Ercan-Sencicek, A.G.; Kim, Y.S.; Fishman, D.O.; Raubeson, M.J.; Song, Y.; Yasuno, K.; Ho, W.S.; et al. Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol. Psychiatry 2012, 71, 392–402. [Google Scholar] [CrossRef]
- Melchior, L.; Bertelsen, B.; Debes, N.M.; Groth, C.; Skov, L.; Mikkelsen, J.D.; Brøndum-Nielsen, K.; Tümer, Z. Microduplication of 15q13.3 and Xq21.31 in a family with Tourette syndrome and comorbidities. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013, 162, 825–831. [Google Scholar] [CrossRef]
- Nag, A.; Bochukova, E.G.; Kremeyer, B.; Campbell, D.D.; Muller, H.; Valencia-Duarte, A.V.; Cardona, J.; Rivas, I.C.; Mesa, S.C.; Cuartas, M.; et al. CNV analysis in Tourette syndrome implicates large genomic rearrangements in COL8A1 and NRXN1. PLoS ONE 2013, 8, e59061. [Google Scholar] [CrossRef]
- McGrath, L.M.; Yu, D.; Marshall, C.; Davis, L.K.; Thiruvahindrapuram, B.; Li, B.; Cappi, C.; Gerber, G.; Wolf, A.; Schroeder, F.A.; et al. Copy number variation in obsessive-compulsive disorder and tourette syndrome: A cross-disorder study. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53, 910–919. [Google Scholar] [CrossRef]
- Prontera, P.; Napolioni, V.; Ottaviani, V.; Rogaia, D.; Fusco, C.; Augello, B.; Serino, D.; Parisi, V.; Bernardini, L.; Merla, G.; et al. DPP6 gene disruption in a family with Gilles de la Tourette syndrome. Neurogenetics 2014, 15, 237–242. [Google Scholar] [CrossRef]
- Huang, A.Y.; Yu, D.; Davis, L.K.; Sul, J.H.; Tsetsos, F.; Ramensky, V.; Zelaya, I.; Ramos, E.M.; Osiecki, L.; Chen, J.A.; et al. Rare Copy Number Variants in NRXN1 and CNTN6 Increase Risk for Tourette Syndrome. Neuron 2017, 94, 1101–1111. [Google Scholar] [CrossRef]
- Maccarini, S.; Cipani, A.; Bertini, V.; Skripac, J.; Salvi, A.; Borsani, G.; Marchina, E. Inherited duplication of the pseudoautosomal region Xq28 in a subject with Gilles de la Tourette syndrome and intellectual disability: A case report. Mol. Cytogenet. 2020, 13, 23. [Google Scholar] [CrossRef]
- Yu, D.; Sul, J.H.; Tsetsos, F.; Nawaz, M.S.; Huang, A.Y.; Zelaya, I.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; et al. Interrogating the Genetic Determinants of Tourette’s Syndrome and Other Tic Disorders Through Genome-Wide Association Studies. Am. J. Psychiatry 2019, 176, 217–227. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 2019, 179, 1469–1482. [Google Scholar] [CrossRef]
- Reay, W.R.; Cairns, M.J. Pairwise common variant meta-analyses of schizophrenia with other psychiatric disorders reveals shared and distinct gene and gene-set associations. Transl. Psychiatry 2020, 10, 134. [Google Scholar] [CrossRef]
- Peyrot, W.J.; Price, A.L. Identifying loci with different allele frequencies among cases of eight psychiatric disorders using CC-GWAS. Nat. Genet. 2021, 53, 445–454. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, H.; Lee, P.H.; Tsetsos, F.; Davis, L.K.; Yu, D.; Lee, S.H.; Dalsgaard, S.; Haavik, J.; Barta, C.; et al. Investigating Shared Genetic Basis Across Tourette Syndrome and Comorbid Neurodevelopmental Disorders Along the Impulsivity-Compulsivity Spectrum. Biol. Psychiatry 2021, 90, 317–327. [Google Scholar] [CrossRef]
- Tsetsos, F.; Topaloudi, A.; Jain, P.; Yang, Z.; Yu, D.; Kolovos, P.; Tumer, Z.; Rizzo, R.; Hartmann, A.; Depienne, C.; et al. Genome-wide Association Study identifies two novel loci for Gilles de la Tourette Syndrome. medRxiv 2021. [Google Scholar] [CrossRef]
- Zilhão, N.R.; Padmanabhuni, S.S.; Pagliaroli, L.; Barta, C.; Smit, D.J.; Cath, D.; Nivard, M.G.; Baselmans, B.M.; van Dongen, J.; Paschou, P.; et al. Epigenome-Wide Association Study of Tic Disorders. Twin Res. Hum. Genet. 2015, 18, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Hildonen, M.; Levy, A.M.; Hansen, C.S.; Bybjerg-Grauholm, J.; Skytthe, A.; Debes, N.M.; Tan, Q.; Tümer, Z. EWAS of Monozygotic Twins Implicate a Role of mTOR Pathway in Pathogenesis of Tic Spectrum Disorder. Genes 2021, 12, 1510. [Google Scholar] [CrossRef] [PubMed]
- Lennington, J.B.; Coppola, G.; Kataoka-Sasaki, Y.; Fernandez, T.V.; Palejev, D.; Li, Y.; Huttner, A.; Pletikos, M.; Sestan, N.; Leckman, J.F.; et al. Transcriptome Analysis of the Human Striatum in Tourette Syndrome. Biol. Psychiatry 2016, 79, 372–382. [Google Scholar] [CrossRef]
- Lit, L.; Gilbert, D.L.; Walker, W.; Sharp, F.R. A subgroup of Tourette’s patients overexpress specific natural killer cell genes in blood: A preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Lit, L.; Enstrom, A.; Sharp, F.R.; Gilbert, D.L. Age-related gene expression in Tourette syndrome. J. Psychiatr. Res. 2009, 43, 319–330. [Google Scholar] [CrossRef]
- Tian, Y.; Gunther, J.R.; Liao, I.H.; Liu, D.; Ander, B.P.; Stamova, B.S.; Lit, L.; Jickling, G.C.; Xu, H.; Zhan, X.; et al. GABA- and acetylcholine-related gene expression in blood correlate with tic severity and microarray evidence for alternative splicing in Tourette syndrome: A pilot study. Brain Res. 2011, 1381, 228–236. [Google Scholar] [CrossRef]
- Tian, Y.; Liao, I.H.; Zhan, X.; Gunther, J.R.; Ander, B.P.; Liu, D.; Lit, L.; Jickling, G.C.; Corbett, B.A.; Bos-Veneman, N.G.; et al. Exon expression and alternatively spliced genes in Tourette Syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 72–78. [Google Scholar] [CrossRef]
- Xi, L.; Zhou, F.; Sha, H.; Zhu, W.; Hu, X.; Ruan, J.; Huang, Y.; Zhang, Y.; Long, H. Potential Plasma Metabolic Biomarkers of Tourette Syndrome Discovery Based on Integrated Non-Targeted and Targeted Metabolomics Screening. Res. Sq. 2020, 1–21. [Google Scholar] [CrossRef]
- Murgia, F.; Gagliano, A.; Tanca, M.G.; Or-Geva, N.; Hendren, A.; Carucci, S.; Pintor, M.; Cera, F.; Cossu, F.; Sotgiu, S.; et al. Metabolomic Characterization of Pediatric Acute-Onset Neuropsychiatric Syndrome (PANS). Front. Neurosci. 2021, 15, 597. [Google Scholar] [CrossRef]
- Piras, C.; Pintus, R.; Pruna, D.; Dessì, A.; Atzori, L.; Fanos, V. Pediatric Acute-onset Neuropsychiatric Syndrome and Mycoplasma pneumoniae Infection: A Case Report Analysis with a Metabolomics Approach. Curr. Pediatr. Rev. 2020, 16, 183–193. [Google Scholar] [CrossRef]
- Xi, W.; Gao, X.; Zhao, H.; Luo, X.; Li, J.; Tan, X.; Wang, L.; Zhao, J.B.; Wang, J.; Yang, G.; et al. Depicting the composition of gut microbiota in children with tic disorders: An exploratory study. J. Child Psychol Psychiatry 2021, 62, 1246–1254. [Google Scholar] [CrossRef]
- Quagliariello, A.; Del Chierico, F.; Russo, A.; Reddel, S.; Conte, G.; Lopetuso, L.R.; Ianiro, G.; Dallapiccola, B.; Cardona, F.; Gasbarrini, A.; et al. Gut Microbiota Profiling and Gut–Brain Crosstalk in Children Affected by Pediatric Acute-Onset Neuropsychiatric Syndrome and Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections. Front. Microbiol. 2018, 9, 675. [Google Scholar] [CrossRef]
- Xu, X.; Nehorai, A.; Dougherty, J. Cell Type Specific Analysis of Human Brain Transcriptome Data to Predict Alterations in Cellular Composition. Syst. Biomed. 2013, 1, 151–160. [Google Scholar] [CrossRef]
- Vanderah, T.W.; Gould, D.J. Organization of the Brainstem. In Nolte’s The Human Brain, 8th ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 258–284. [Google Scholar]
- Ahola-Olli, A.V.; Würtz, P.; Havulinna, A.S.; Aalto, K.; Pitkänen, N.; Lehtimäki, T.; Kähönen, M.; Lyytikäinen, L.P.; Raitoharju, E.; Seppälä, I.; et al. Genome-wide Association Study Identifies 27 Loci Influencing Concentrations of Circulating Cytokines and Growth Factors. Am. J. Hum. Genet. 2017, 100, 40–50. [Google Scholar] [CrossRef]
- Draisma, H.H.M.; Pool, R.; Kobl, M.; Jansen, R.; Petersen, A.-K.; Vaarhorst, A.A.M.; Yet, I.; Haller, T.; Demirkan, A.; Esko, T.; et al. Genome-wide association study identifies novel genetic variants contributing to variation in blood metabolite levels. Nat. Commun. 2015, 6, 7208. [Google Scholar] [CrossRef]
- Kettunen, J.; Demirkan, A.; Würtz, P.; Draisma, H.H.; Haller, T.; Rawal, R.; Vaarhorst, A.; Kangas, A.J.; Lyytikäinen, L.P.; Pirinen, M.; et al. Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat. Commun. 2016, 7, 11122. [Google Scholar] [CrossRef]
- Panyard, D.J.; Kim, K.M.; Darst, B.F.; Deming, Y.K.; Zhong, X.; Wu, Y.; Kang, H.; Carlsson, C.M.; Johnson, S.C.; Asthana, S.; et al. Cerebrospinal fluid metabolomics identifies 19 brain-related phenotype associations. Commun. Biol. 2021, 4, 63. [Google Scholar] [CrossRef]
- Rhee, E.P.; Ho, J.E.; Chen, M.H.; Shen, D.; Cheng, S.; Larson, M.G.; Ghorbani, A.; Shi, X.; Helenius, I.T.; O’Donnell, C.J.; et al. A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab. 2013, 18, 130–143. [Google Scholar] [CrossRef]
- Aramideh, J.A.; Vidal-Itriago, A.; Morsch, M.; Graeber, M.M.B. Cytokine Signalling at the Microglial Penta-Partite Synapse. Int. J. Mol. Sci. 2021, 22, 13186. [Google Scholar] [CrossRef]
- Ferrer-Ferrer, M.; Dityatev, A. Shaping Synapses by the Neural Extracellular Matrix. Front. Neuroanat. 2018, 12, 40. [Google Scholar] [CrossRef]
- Krishnaswamy, V.R.; Benbenishty, A.; Blinder, P.; Sagi, I. Demystifying the extracellular matrix and its proteolytic remodeling in the brain: Structural and functional insights. Cell. Mol. Life Sci. 2019, 76, 3229–3248. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Colangelo, A.M.; Virtuoso, A.; Alberghina, L.; Papa, M. Neurons, Glia, Extracellular Matrix and Neurovascular Unit: A Systems Biology Approach to the Complexity of Synaptic Plasticity in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1539. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, K.A.; Cheyette, B.N. Neurodevelopmental Perspectives on Wnt Signaling in Psychiatry. Mol. Neuropsychiatry 2017, 2, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Almén, M.S.; Nordström, K.J.; Fredriksson, R.; Schiöth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Gerber, K.J.; Squires, K.E.; Hepler, J.R. Roles for Regulator of G Protein Signaling Proteins in Synaptic Signaling and Plasticity. Mol. Pharm. 2016, 89, 273–286. [Google Scholar] [CrossRef]
- Liu, J.; Yang, L.; Li, H.; Cai, Y.; Feng, J.; Hu, Z. Conditional ablation of protein tyrosine phosphatase receptor U in midbrain dopaminergic neurons results in reduced neuronal size. J. Chem. Neuroanat. 2022, 124, 102135. [Google Scholar] [CrossRef]
- Daniels, M.P. The role of agrin in synaptic development, plasticity and signaling in the central nervous system. Neurochem. Int. 2012, 61, 848–853. [Google Scholar] [CrossRef]
- Hilgenberg, L.G.; Su, H.; Gu, H.; O’Dowd, D.K.; Smith, M.A. Alpha3Na+/K+-ATPase is a neuronal receptor for agrin. Cell 2006, 125, 359–369. [Google Scholar] [CrossRef]
- Xie, X.; Mahmood, S.R.; Gjorgjieva, T.; Percipalle, P. Emerging roles of cytoskeletal proteins in regulating gene expression and genome organization during differentiation. Nucleus 2020, 11, 53–65. [Google Scholar] [CrossRef]
- Herrmann, H.; Strelkov, S.V.; Burkhard, P.; Aebi, U. Intermediate filaments: Primary determinants of cell architecture and plasticity. J. Clin. Investig. 2009, 119, 1772–1783. [Google Scholar] [CrossRef]
- Parato, J.; Bartolini, F. The microtubule cytoskeleton at the synapse. Neurosci. Lett. 2021, 753, 135850. [Google Scholar] [CrossRef]
- Rapoport, S.I.; Primiani, C.T.; Chen, C.T.; Ahn, K.; Ryan, V.H. Coordinated Expression of Phosphoinositide Metabolic Genes during Development and Aging of Human Dorsolateral Prefrontal Cortex. PLoS ONE 2015, 10, e0132675. [Google Scholar] [CrossRef]
- Posor, Y.; Jang, W.; Haucke, V. Phosphoinositides as membrane organizers. Nat. Rev. Mol. Cell Biol. 2022, 23, 797–816. [Google Scholar] [CrossRef]
- Parthasarathy, L.K.; Ratnam, L.; Seelan, S.; Tobias, C.; Casanova, M.F.; Parthasarathy, R.N. Mammalian Inositol 3-phosphate Synthase: Its Role in the Biosynthesis of Brain Inositol and its Clinical Use as a Psychoactive Agent. In Biology of Inositols and Phosphoinositides: Subcellular Biochemistry; Majumder, A.L., Biswas, B.B., Eds.; Springer: Boston, MA, USA, 2006; pp. 293–314. [Google Scholar]
- Gupta, M.K.; Randhawa, P.K.; Masternak, M.M. Role of BAG5 in Protein Quality Control: Double-Edged Sword? Front. Aging 2022, 3, 844168. [Google Scholar] [CrossRef]
- Prashad, S.; Gopal, P.P. RNA-binding proteins in neurological development and disease. RNA Biol. 2021, 18, 972–987. [Google Scholar] [CrossRef]
- Aibara, S.; Singh, V.; Modelska, A.; Amunts, A. Structural basis of mitochondrial translation. eLife 2020, 9, e58362. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Nguyen, N.D.; Wang, D. Multiview learning for understanding functional multiomics. PLoS Comput. Biol. 2020, 16, e1007677. [Google Scholar] [CrossRef]
- Carulli, D.; Laabs, T.; Geller, H.M.; Fawcett, J.W. Chondroitin sulfate proteoglycans in neural development and regeneration. Curr. Opin. Neurobiol. 2005, 15, 116–120. [Google Scholar] [CrossRef]
- Ebersole, B.; Petko, J.; Woll, M.; Murakami, S.; Sokolina, K.; Wong, V.; Stagljar, I.; Lüscher, B.; Levenson, R. Effect of C-Terminal S-Palmitoylation on D2 Dopamine Receptor Trafficking and Stability. PLoS ONE 2015, 10, e0140661. [Google Scholar] [CrossRef]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol 2017, 18, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Francette, A.M.; Tripplehorn, S.A.; Arndt, K.M. The Paf1 Complex: A Keystone of Nuclear Regulation Operating at the Interface of Transcription and Chromatin. J. Mol. Biol. 2021, 433, 166979. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, B.N.; Guhathakurta, S.; Akhtar, A. The non-specific lethal (NSL) complex at the crossroads of transcriptional control and cellular homeostasis. EMBO Rep. 2019, 20, e47630. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Barres, B.A. Glia—More than just brain glue. Nature 2009, 457, 675–677. [Google Scholar] [CrossRef]
- Eroglu, C.; Barres, B.A. Regulation of synaptic connectivity by glia. Nature 2010, 468, 223–231. [Google Scholar] [CrossRef]
- Mauch, D.H.; Nägler, K.; Schumacher, S.; Göritz, C.; Müller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef]
- Dai, Y.B.; Tan, X.J.; Wu, W.F.; Warner, M.; Gustafsson, J. Liver X receptor β protects dopaminergic neurons in a mouse model of Parkinson disease. Proc. Natl. Acad. Sci. USA 2012, 109, 13112–13117. [Google Scholar] [CrossRef]
- Breschi, A.; Muñoz-Aguirre, M.; Wucher, V.; Davis, C.A.; Garrido-Martín, D.; Djebali, S.; Gillis, J.; Pervouchine, D.D.; Vlasova, A.; Dobin, A.; et al. A limited set of transcriptional programs define major cell types. Genome Res. 2020, 30, 1047–1059. [Google Scholar] [CrossRef]
- Caligiore, D.; Mannella, F.; Arbib, M.A.; Baldassarre, G. Dysfunctions of the basal ganglia-cerebellar-thalamo-cortical system produce motor tics in Tourette syndrome. PLoS Comput. Biol. 2017, 13, e1005395. [Google Scholar] [CrossRef]
- Wan, X.; Zhang, S.; Wang, W.; Su, X.; Li, J.; Yang, X.; Tan, Q.; Yue, Q.; Gong, Q. Gray matter abnormalities in Tourette Syndrome: A meta-analysis of voxel-based morphometry studies. Transl. Psychiatry 2021, 11, 287. [Google Scholar] [CrossRef]
- Kreitzer, A.C. Physiology and pharmacology of striatal neurons. Annu. Rev. Neurosci. 2009, 32, 127–147. [Google Scholar] [CrossRef]
- Smith, A.C.W.; Jonkman, S.; Difeliceantonio, A.G.; O’Connor, R.M.; Ghoshal, S.; Romano, M.F.; Everitt, B.J.; Kenny, P.J. Opposing roles for striatonigral and striatopallidal neurons in dorsolateral striatum in consolidating new instrumental actions. Nat. Commun. 2021, 12, 5121. [Google Scholar] [CrossRef]
- Sun, Z.; Wu, M.; Ren, W. Striatal D2: Where habits and newly learned actions meet. Learn. Behav. 2022, 50, 267–268. [Google Scholar] [CrossRef]
- Leckman, J.F.; Riddle, M.A. Tourette’s Syndrome: When Habit-Forming Systems Form Habits of Their Own? Neuron 2000, 28, 349–354. [Google Scholar] [CrossRef]
- Maia, T.V.; Conceicao, V.A. The Roles of Phasic and Tonic Dopamine in Tic Learning and Expression. Biol. Psychiatry 2017, 82, 401–412. [Google Scholar] [CrossRef]
- Delorme, C.; Salvador, A.; Valabrègue, R.; Roze, E.; Palminteri, S.; Vidailhet, M.; de Wit, S.; Robbins, T.; Hartmann, A.; Worbe, Y. Enhanced habit formation in Gilles de la Tourette syndrome. Brain 2015, 139, 605–615. [Google Scholar] [CrossRef]
- Shephard, E.; Groom, M.J.; Jackson, G.M. Implicit sequence learning in young people with Tourette syndrome with and without co-occurring attention-deficit/hyperactivity disorder. J. Neuropsychol. 2019, 13, 529–549. [Google Scholar] [CrossRef]
- Beste, C.; Münchau, A. Tics and Tourette syndrome—Surplus of actions rather than disorder? Mov. Disord. 2018, 33, 238–242. [Google Scholar] [CrossRef]
- Fründt, O.; Woods, D.; Ganos, C. Behavioral therapy for Tourette syndrome and chronic tic disorders. Neurol. Clin. Pract. 2017, 7, 148. [Google Scholar] [CrossRef]
- Petruo, V.; Bodmer, B.; Bluschke, A.; Münchau, A.; Roessner, V.; Beste, C. Comprehensive Behavioral Intervention for Tics reduces perception-action binding during inhibitory control in Gilles de la Tourette syndrome. Sci. Rep. 2020, 10, 1174. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.M.; Flores, C. Mesocorticolimbic Dopamine Pathways Across Adolescence: Diversity in Development. Front. Neural Circuits 2021, 15, 735625. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, O.J.; McGuirt, A.F.; Mosharov, E.V.; Pigulevskiy, I.; Hobson, B.D.; Choi, S.; Frier, M.D.; Santini, E.; Borgkvist, A.; Sulzer, D. Dopamine Triggers the Maturation of Striatal Spiny Projection Neuron Excitability during a Critical Period. Neuron 2018, 99, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Haycock, J.W.; Becker, L.; Ang, L.; Furukawa, Y.; Hornykiewicz, O.; Kish, S.J. Marked disparity between age-related changes in dopamine and other presynaptic dopaminergic markers in human striatum. J. Neurochem. 2003, 87, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Minzer, K.; Lee, O.; Hong, J.J.; Singer, H.S. Increased prefrontal D2 protein in Tourette syndrome: A postmortem analysis of frontal cortex and striatum. J. Neurol. Sci. 2004, 219, 55–61. [Google Scholar] [CrossRef]
- Yoon, D.Y.; Gause, C.D.; Leckman, J.F.; Singer, H.S. Frontal dopaminergic abnormality in Tourette syndrome: A postmortem analysis. J. Neurol. Sci. 2007, 255, 50–56. [Google Scholar] [CrossRef]
- Wolf, S.S.; Jones, D.W.; Knable, M.B.; Gorey, J.G.; Lee, K.S.; Hyde, T.M.; Coppola, R.; Weinberger, D.R. Tourette syndrome: Prediction of phenotypic variation in monozygotic twins by caudate nucleus D2 receptor binding. Science 1996, 273, 1225–1227. [Google Scholar] [CrossRef]
- Singer, H.S. Treatment of tics and tourette syndrome. Curr. Treat. Options Neurol. 2010, 12, 539–561. [Google Scholar] [CrossRef]
- Kataoka, Y.; Kalanithi, P.S.; Grantz, H.; Schwartz, M.L.; Saper, C.; Leckman, J.F.; Vaccarino, F.M. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J. Comp. Neurol. 2010, 518, 277–291. [Google Scholar] [CrossRef]
- Favier, M.; Janickova, H.; Justo, D.; Kljakic, O.; Runtz, L.; Natsheh, J.Y.; Pascoal, T.A.; Germann, J.; Gallino, D.; Kang, J.I.; et al. Cholinergic dysfunction in the dorsal striatum promotes habit formation and maladaptive eating. J. Clin. Investig. 2020, 130, 6616–6630. [Google Scholar] [CrossRef]
- Aoki, S.; Liu, A.W.; Akamine, Y.; Zucca, A.; Zucca, S.; Wickens, J.R. Cholinergic interneurons in the rat striatum modulate substitution of habits. Eur. J. Neurosci. 2018, 47, 1194–1205. [Google Scholar] [CrossRef]
- Gritton, H.J.; Howe, W.M.; Romano, M.F.; DiFeliceantonio, A.G.; Kramer, M.A.; Saligrama, V.; Bucklin, M.E.; Zemel, D.; Han, X. Unique contributions of parvalbumin and cholinergic interneurons in organizing striatal networks during movement. Nat. Neurosci. 2019, 22, 586–597. [Google Scholar] [CrossRef]
- Quik, M.; Boyd, J.T.; Bordia, T.; Perez, X. Potential Therapeutic Application for Nicotinic Receptor Drugs in Movement Disorders. Nicotine Tob. Res. 2019, 21, 357–369. [Google Scholar] [CrossRef]
- Schramm, M.; Selinger, Z. Message transmission: Receptor controlled adenylate cyclase system. Science 1984, 225, 1350–1356. [Google Scholar] [CrossRef]
- Taskén, K.; Skålhegg, B.S.; Taskén, K.A.; Solberg, R.; Knutsen, H.K.; Levy, F.O.; Sandberg, M.; Orstavik, S.; Larsen, T.; Johansen, A.K.; et al. Structure, function, and regulation of human cAMP-dependent protein kinases. Adv. Second Messenger Phosphoprot. Res. 1997, 31, 191–204. [Google Scholar] [CrossRef]
- Singer, H.S.; Hahn, I.H.; Krowiak, E.; Nelson, E.; Moran, T. Tourette’s syndrome: A neurochemical analysis of postmortem cortical brain tissue. Ann. Neurol. 1990, 27, 443–446. [Google Scholar] [CrossRef]
- Singer, H.S.; Hahn, I.H.; Moran, T.H. Abnormal dopamine uptake sites in postmortem striatum from patients with Tourette’s syndrome. Ann. Neurol. 1991, 30, 558–562. [Google Scholar] [CrossRef]
- Korff, S.; Stein, D.J.; Harvey, B.H. Cortico-striatal cyclic AMP-phosphodiesterase-4 signalling and stereotypy in the deer mouse: Attenuation after chronic fluoxetine treatment. Pharmacol. Biochem. Behav. 2009, 92, 514–520. [Google Scholar] [CrossRef]
- Andersen, S.L. Changes in the second messenger cyclic AMP during development may underlie motoric symptoms in attention deficit/hyperactivity disorder (ADHD). Behav. Brain Res. 2002, 130, 197–201. [Google Scholar] [CrossRef]
- Vendel, E.; de Lange, E.C. Functions of the CB1 and CB 2 receptors in neuroprotection at the level of the blood-brain barrier. Neuromol. Med. 2014, 16, 620–642. [Google Scholar] [CrossRef]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Szejko, N.; Fichna, J.P.; Safranow, K.; Dziuba, T.; Żekanowski, C.; Janik, P. Association of a Variant of CNR1 Gene Encoding Cannabinoid Receptor 1 With Gilles de la Tourette Syndrome. Front. Genet. 2020, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Gadzicki, D.; Müller-Vahl, K.R.; Heller, D.; Ossege, S.; Nöthen, M.M.; Hebebrand, J.; Stuhrmann, M. Tourette syndrome is not caused by mutations in the central cannabinoid receptor (CNR1) gene. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004, 127, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Müller-Vahl, K.R.; Bindila, L.; Lutz, B.; Musshoff, F.; Skripuletz, T.; Baumgaertel, C.; Sühs, K.W. Cerebrospinal fluid endocannabinoid levels in Gilles de la Tourette syndrome. Neuropsychopharmacology 2020, 45, 1323–1329. [Google Scholar] [CrossRef]
- Watson, S.; Chambers, D.; Hobbs, C.; Doherty, P.; Graham, A. The endocannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol. Cell. Neurosci. 2008, 38, 89–97. [Google Scholar] [CrossRef]
- Martella, A.; Sepe, R.M.; Silvestri, C.; Zang, J.; Fasano, G.; Carnevali, O.; De Girolamo, P.; Neuhauss, S.C.; Sordino, P.; Di Marzo, V. Important role of endocannabinoid signaling in the development of functional vision and locomotion in zebrafish. FASEB J. 2016, 30, 4275–4288. [Google Scholar] [CrossRef]
- Gianessi, C.A.; Groman, S.M.; Taylor, J.R. The effects of fatty acid amide hydrolase inhibition and monoacylglycerol lipase inhibition on habit formation in mice. Eur. J. Neurosci. 2022, 55, 922–938. [Google Scholar] [CrossRef]
- Hilário, M.R.; Clouse, E.; Yin, H.H.; Costa, R.M. Endocannabinoid signaling is critical for habit formation. Front. Integr. Neurosci. 2007, 1, 6. [Google Scholar] [CrossRef]
- Wade, M.R.; Tzavara, E.T.; Nomikos, G.G. Cannabinoids reduce cAMP levels in the striatum of freely moving rats: An in vivo microdialysis study. Brain Res. 2004, 1005, 117–123. [Google Scholar] [CrossRef]
- Pacheco, M.; Childers, S.R.; Arnold, R.; Casiano, F.; Ward, S.J. Aminoalkylindoles: Actions on specific G-protein-linked receptors. J. Pharmacol. Exp. Ther. 1991, 257, 170–183. [Google Scholar]
- Glass, M.; Felder, C.C. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997, 17, 5327–5333. [Google Scholar] [CrossRef]
- Sandyk, R.; Awerbuch, G. Marijuana and Tourette’s syndrome. J. Clin. Psychopharmacol. 1988, 8, 444–445. [Google Scholar] [CrossRef]
- Artukoglu, B.B.; Bloch, M.H. The Potential of Cannabinoid-Based Treatments in Tourette Syndrome. CNS Drugs 2019, 33, 417–430. [Google Scholar] [CrossRef]
- Szejko, N.; Saramak, K.; Lombroso, A.; Müller-Vahl, K. Cannabis-based medicine in treatment of patients with Gilles de la Tourette syndrome. Neurol. Neurochir. Pol. 2022, 56, 28–38. [Google Scholar] [CrossRef]
- Fride, E. The endocannabinoid-CB receptor system: Importance for development and in pediatric disease. Neuroendocrinol. Lett 2004, 25, 24–30. [Google Scholar]
- Hasan, A.; Rothenberger, A.; Münchau, A.; Wobrock, T.; Falkai, P.; Roessner, V. Oral delta 9-tetrahydrocannabinol improved refractory Gilles de la Tourette syndrome in an adolescent by increasing intracortical inhibition: A case report. J. Clin. Psychopharmacol. 2010, 30, 190–192. [Google Scholar] [CrossRef]
- Szejko, N.; Jakubovski, E.; Fremer, C.; Kunert, K.; Mueller-Vahl, K. Delta-9-tetrahydrocannabinol for the treatment of a child with Tourette syndrome—Case report. Eur. J. Med. Case Rep. 2018, 2, 39–41. [Google Scholar] [CrossRef]
- Szejko, N.; Jakubovski, E.; Fremer, C.; Müller-Vahl, K.R. Vaporized Cannabis Is Effective and Well-Tolerated in an Adolescent with Tourette Syndrome. Med. Cannabis Cannabinoids 2019, 2, 60–64. [Google Scholar] [CrossRef]
- Wu, S.W.; Gilbert, D.L. Altered neurophysiologic response to intermittent theta burst stimulation in Tourette syndrome. Brain Stimul. 2012, 5, 315–319. [Google Scholar] [CrossRef]
- Suppa, A.; Belvisi, D.; Bologna, M.; Marsili, L.; Berardelli, I.; Moretti, G.; Pasquini, M.; Fabbrini, G.; Berardelli, A. Abnormal cortical and brain stem plasticity in Gilles de la Tourette syndrome. Mov. Disord. 2011, 26, 1703–1710. [Google Scholar] [CrossRef]
- Brandt, V.C.; Niessen, E.; Ganos, C.; Kahl, U.; Bäumer, T.; Münchau, A. Altered synaptic plasticity in Tourette’s syndrome and its relationship to motor skill learning. PLoS ONE 2014, 9, e98417. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, D.E.; Wilcken, B.; Dudman, N.P.; Tyrrell, P.A. Homocystinuria—The effects of betaine in the treatment of patients not responsive to pyridoxine. N. Engl. J. Med. 1983, 309, 448–453. [Google Scholar] [CrossRef] [PubMed]
- McKeever, M.P.; Weir, D.G.; Molloy, A.; Scott, J.M. Betaine-homocysteine methyltransferase: Organ distribution in man, pig and rat and subcellular distribution in the rat. Clin. Sci. 1991, 81, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Gómez Padilla, P.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.C.; Yang, F.; Capecci, L.M.; Gu, Z.; Schafer, A.I.; Durante, W.; Yang, X.F.; Wang, H. Regulation of homocysteine metabolism and methylation in human and mouse tissues. FASEB J. 2010, 24, 2804–2817. [Google Scholar] [CrossRef]
- Wang, L.; Alachkar, A.; Sanathara, N.; Belluzzi, J.D.; Wang, Z.; Civelli, O. A Methionine-Induced Animal Model of Schizophrenia: Face and Predictive Validity. Int. J. Neuropsychopharmacol. 2015, 18, pyv054. [Google Scholar] [CrossRef]
- Taylor, M. Dietary modification of amphetamine stereotyped behaviour: The action of tryptophan, methionine, and lysine. Psychopharmacology 1979, 61, 81–83. [Google Scholar] [CrossRef]
- Muller, U.J.; Frick, B.; Winkler, C.; Fuchs, D.; Wenning, G.K.; Poewe, W.; Mueller, J. Homocysteine and serum markers of immune activation in primary dystonia. Mov. Disord. 2005, 20, 1663–1667. [Google Scholar] [CrossRef]
- Müller, T.; Woitalla, D.; Hunsdiek, A.; Kuhn, W. Elevated plasma levels of homocysteine in dystonia. Acta Neurol. Scand. 2000, 101, 388–390. [Google Scholar] [CrossRef]
- Ueland, P.M.; Ulvik, A.; Rios-Avila, L.; Midttun, Ø.; Gregory, J.F. Direct and Functional Biomarkers of Vitamin B6 Status. Annu. Rev. Nutr. 2015, 35, 33–70. [Google Scholar] [CrossRef]
- Garcia-Lopez, R.; Perea-Milla, E.; Garcia, C.R.; Rivas-Ruiz, F.; Romero-Gonzalez, J.; Moreno, J.L.; Faus, V.; Aguas Gdel, C.; Diaz, J.C. New therapeutic approach to Tourette Syndrome in children based on a randomized placebo-controlled double-blind phase IV study of the effectiveness and safety of magnesium and vitamin B6. Trials 2009, 10, 16. [Google Scholar] [CrossRef]
- García-López, R.; Romero-González, J.; Perea-Milla, E.; Ruiz-García, C.; Rivas-Ruiz, F.; de Las Mulas Béjar, M. An open study evaluating the efficacy and security of magnesium and vitamin B(6) as a treatment of Tourette syndrome in children. Med. Clin. 2008, 131, 689–691. [Google Scholar] [CrossRef]
- Mauri, D.N.; Ebner, R.; Montgomery, R.I.; Kochel, K.D.; Cheung, T.C.; Yu, G.L.; Ruben, S.; Murphy, M.; Eisenberg, R.J.; Cohen, G.H.; et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 1998, 8, 21–30. [Google Scholar] [CrossRef]
- Garcia-Delgar, B.; Morer, A.; Luber, M.J.; Coffey, B.J. Obsessive-Compulsive Disorder, Tics, and Autoinflammatory Diseases: Beyond PANDAS. J. Child Adolesc. Psychopharmacol. 2016, 26, 847–850. [Google Scholar] [CrossRef]
- Kratz, A.; Campos-Neto, A.; Hanson, M.S.; Ruddle, N.H. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J. Exp. Med. 1996, 183, 1461–1472. [Google Scholar] [CrossRef]
- Gommerman, J.L.; Browning, J.L. Lymphotoxin/LIGHT, lymphoid microenvironments and autoimmune disease. Nat. Rev. Immunol. 2003, 3, 642–655. [Google Scholar] [CrossRef]
- Ying, X.; Chan, K.; Shenoy, P.; Hill, M.; Ruddle, N.H. Lymphotoxin plays a crucial role in the development and function of nasal-associated lymphoid tissue through regulation of chemokines and peripheral node addressin. Am. J. Pathol. 2005, 166, 135–146. [Google Scholar] [CrossRef]
- Park, H.S.; Francis, K.P.; Yu, J.; Cleary, P.P. Membranous cells in nasal-associated lymphoid tissue: A portal of entry for the respiratory mucosal pathogen group A streptococcus. J. Immunol. 2003, 171, 2532–2537. [Google Scholar] [CrossRef]
- Dileepan, T.; Smith, E.D.; Knowland, D.; Hsu, M.; Platt, M.; Bittner-Eddy, P.; Cohen, B.; Southern, P.; Latimer, E.; Harley, E.; et al. Group A Streptococcus intranasal infection promotes CNS infiltration by streptococcal-specific Th17 cells. J. Clin. Investig. 2016, 126, 303–317. [Google Scholar] [CrossRef]
- Hutanu, A.; Reddy, L.N.; Mathew, J.; Avanthika, C.; Jhaveri, S.; Tummala, N. Pediatric Autoimmune Neuropsychiatric Disorders Associated with Group A Streptococci: Etiopathology and Diagnostic Challenges. Cureus 2022, 14, e27729. [Google Scholar] [CrossRef]
- Kim, S.W.; Grant, J.E.; Kim, S.I.; Swanson, T.A.; Bernstein, G.A.; Jaszcz, W.B.; Williams, K.A.; Schlievert, P.M. A possible association of recurrent streptococcal infections and acute onset of obsessive-compulsive disorder. J. Neuropsychiatry Clin. Neurosci. 2004, 16, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, A.R.; Salgado-Pabón, W.; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, D.; Kwok, L.Y.; Lütjen, S.; Soltek, S.; Hoffmann, S.; Körner, H.; Deckert, M. Both lymphotoxin-alpha and TNF are crucial for control of Toxoplasma gondii in the central nervous system. J. Immunol. 2003, 170, 6172–6182. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.; Matz, J.; Weidinger, E.; Wagner, J.; Wildenauer, A.; Obermeier, M.; Riedel, M.; Müller, N. Association between intracellular infectious agents and Tourette’s syndrome. Eur. Arch. Psychiatry Clin. Neurosci. 2010, 260, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Akaltun, İ.; Kara, T.; Sertan Kara, S.; Ayaydın, H. Seroprevalance Anti-Toxoplasma gondii antibodies in children and adolescents with tourette syndrome/chronic motor or vocal tic disorder: A case-control study. Psychiatry Res. 2018, 263, 154–157. [Google Scholar] [CrossRef]
- Croze, M.L.; Soulage, C.O. Potential role and therapeutic interests of myo-inositol in metabolic diseases. Biochimie 2013, 95, 1811–1827. [Google Scholar] [CrossRef]
- Fisher, S.K.; Novak, J.E.; Agranoff, B.W. Inositol and higher inositol phosphates in neural tissues: Homeostasis, metabolism and functional significance. J. Neurochem. 2002, 82, 736–754. [Google Scholar] [CrossRef]
- Devito, T.J.; Drost, D.J.; Pavlosky, W.; Neufeld, R.W.J.; Rajakumar, N.; McKinlay, B.D.; Williamson, P.C.; Nicolson, R.O.B. Brain Magnetic Resonance Spectroscopy in Tourette’s Disorder. J. Am. Acad. Child Adolesc. Psychiatry 2005, 44, 1301–1308. [Google Scholar] [CrossRef]
- Cryns, K.; Shamir, A.; Van Acker, N.; Levi, I.; Daneels, G.; Goris, I.; Bouwknecht, J.A.; Andries, L.; Kass, S.; Agam, G.; et al. IMPA1 is essential for embryonic development and lithiu.um-like pilocarpine sensitivity. Neuropsychopharmacology 2008, 33, 674–684. [Google Scholar] [CrossRef]
- Lauritzen, L.; Hansen, H.S.; Jørgensen, M.H.; Michaelsen, K.F. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog. Lipid Res. 2001, 40, 1–94. [Google Scholar] [CrossRef]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef]
- Nishizaki, T.; Nomura, T.; Matsuoka, T.; Tsujishita, Y. Arachidonic acid as a messenger for the expression of long-term potentiation. Biochem. Biophys. Res. Commun. 1999, 254, 446–449. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, L.; Ding, J.; Huang, J.; Shao, A.; Tang, B. The Role of Formyl Peptide Receptors in Neurological Diseases via Regulating Inflammation. Front. Cell. Neurosci. 2021, 15, 753832. [Google Scholar] [CrossRef]
- Pamplona, F.A.; Ferreira, J.; Menezes de Lima, O., Jr.; Duarte, F.S.; Bento, A.F.; Forner, S.; Villarinho, J.G.; Bellocchio, L.; Wotjak, C.T.; Lerner, R.; et al. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 21134–21139. [Google Scholar] [CrossRef]
- Lands, W.E.; Libelt, B.; Morris, A.; Kramer, N.C.; Prewitt, T.E.; Bowen, P.; Schmeisser, D.; Davidson, M.H.; Burns, J.H. Maintenance of lower proportions of (n-6) eicosanoid precursors in phospholipids of human plasma in response to added dietary (n-3) fatty acids. Biochim. Biophys. Acta 1992, 1180, 147–162. [Google Scholar] [CrossRef]
- Rudkowska, I.; Paradis, A.M.; Thifault, E.; Julien, P.; Tchernof, A.; Couture, P.; Lemieux, S.; Barbier, O.; Vohl, M.C. Transcriptomic and metabolomic signatures of an n-3 polyunsaturated fatty acids supplementation in a normolipidemic/normocholesterolemic Caucasian population. J. Nutr. Biochem. 2013, 24, 54–61. [Google Scholar] [CrossRef]
- Gabbay, V.; Babb, J.S.; Klein, R.G.; Panzer, A.M.; Katz, Y.; Alonso, C.M.; Petkova, E.; Wang, J.; Coffey, B.J. A double-blind, placebo-controlled trial of ω-3 fatty acids in Tourette’s disorder. Pediatrics 2012, 129, e1493–e1500. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, H.; Jing, M.; Hu, X.; Wang, J.; Hua, Y. Gut Microbiome Composition Abnormalities Determined Using High-Throughput Sequencing in Children with Tic Disorder. Front. Pediatr. 2022, 10, 831944. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.-J.; Xu, Q.; Yan, S.-S.; Han, B.-X.; Zhang, H.; Wei, X.-T.; Feng, G.-J.; Zhao, M.; Pei, Y.-F.; Zhang, L. Gut Microbiota and Psychiatric Disorders: A Two-Sample Mendelian Randomization Study. Front. Microbiol. 2021, 12, 737197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shi, Y.; Luo, X.; Peng, L.; Yang, Y.; Zou, L. The Effect of Fecal Microbiota Transplantation on a Child with Tourette Syndrome. Case Rep. Med. 2017, 2017, 6165239. [Google Scholar] [CrossRef] [PubMed]
- Vijay, N.; Morris, M.E. Role of monocarboxylate transporters in drug delivery to the brain. Curr. Pharm. Des. 2014, 20, 1487–1498. [Google Scholar] [CrossRef]
- Huuskonen, J.; Suuronen, T.; Nuutinen, T.; Kyrylenko, S.; Salminen, A. Regulation of microglial inflammatory response by sodium butyrate and short-chain fatty acids. Br. J. Pharm. 2004, 141, 874–880. [Google Scholar] [CrossRef]
- Candido, E.P.M.; Reeves, R.; Davie, J.R. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 1978, 14, 105–113. [Google Scholar] [CrossRef]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef]
- Wang, A.; Si, H.; Liu, D.; Jiang, H. Butyrate activates the cAMP-protein kinase A-cAMP response element-binding protein signaling pathway in Caco-2 cells. J. Nutr. 2012, 142, 1–6. [Google Scholar] [CrossRef]
- Vijay, A.; Kouraki, A.; Gohir, S.; Turnbull, J.; Kelly, A.; Chapman, V.; Barrett, D.A.; Bulsiewicz, W.J.; Valdes, A.M. The anti-inflammatory effect of bacterial short chain fatty acids is partially mediated by endocannabinoids. Gut Microbes 2021, 13, 1997559. [Google Scholar] [CrossRef]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F.; et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl. Psychiatry 2018, 8, 42. [Google Scholar] [CrossRef]
- Kratsman, N.; Getselter, D.; Elliott, E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 2016, 102, 136–145. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J. Experimental therapeutics in transgenic mouse models of Huntington’s disease. Nat. Rev. Neurosci. 2004, 5, 373–384. [Google Scholar] [CrossRef]
- Naia, L.; Cunha-Oliveira, T.; Rodrigues, J.; Rosenstock, T.R.; Oliveira, A.; Ribeiro, M.; Carmo, C.; Oliveira-Sousa, S.I.; Duarte, A.I.; Hayden, M.R.; et al. Histone Deacetylase Inhibitors Protect Against Pyruvate Dehydrogenase Dysfunction in Huntington’s Disease. J. Neurosci. 2017, 37, 2776–2794. [Google Scholar] [CrossRef]
- St Laurent, R.; O’Brien, L.M.; Ahmad, S.T. Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson’s disease. Neuroscience 2013, 246, 382–390. [Google Scholar] [CrossRef]
- Langley, B.; Gensert, J.M.; Beal, M.F.; Ratan, R.R. Remodeling chromatin and stress resistance in the central nervous system: Histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 41–50. [Google Scholar] [CrossRef]
- Sharma, S.; Taliyan, R.; Singh, S. Beneficial effects of sodium butyrate in 6-OHDA induced neurotoxicity and behavioral abnormalities: Modulation of histone deacetylase activity. Behav. Brain Res. 2015, 291, 306–314. [Google Scholar] [CrossRef]
- Fischer, A.; Sananbenesi, F.; Wang, X.; Dobbin, M.; Tsai, L.H. Recovery of learning and memory is associated with chromatin remodelling. Nature 2007, 447, 178–182. [Google Scholar] [CrossRef]
- Watson, H.; Mitra, S.; Croden, F.C.; Taylor, M.; Wood, H.M.; Perry, S.L.; Spencer, J.A.; Quirke, P.; Toogood, G.J.; Lawton, C.L.; et al. A randomised trial of the effect of omega-3 polyunsaturated fatty acid supplements on the human intestinal microbiota. Gut 2018, 67, 1974–1983. [Google Scholar] [CrossRef]
- Li, H.; Dong, J.; Cai, M.; Xu, Z.; Cheng, X.D.; Qin, J.J. Protein degradation technology: A strategic paradigm shift in drug discovery. J. Hematol. Oncol. 2021, 14, 138. [Google Scholar] [CrossRef]
- Jarome, T.J.; Helmstetter, F.J. The ubiquitin-proteasome system as a critical regulator of synaptic plasticity and long-term memory formation. Neurobiol. Learn. Mem. 2013, 105, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Whartenby, K.A.; Small, D.; Calabresi, P.A. FLT3 inhibitors for the treatment of autoimmune disease. Expert Opin. Investig. Drugs 2008, 17, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Vuokila, V.; Catoire, H.; Akçimen, F.; Ross, J.P.; Bourassa, C.V.; Dion, P.A.; Meijer, I.A.; Rouleau, G.A. Transcriptome-wide association study reveals increased neuronal FLT3 expression is associated with Tourette’s syndrome. Commun. Biol. 2022, 5, 289. [Google Scholar] [CrossRef] [PubMed]
- Mataix-Cols, D.; Frans, E.; Pérez-Vigil, A.; Kuja-Halkola, R.; Gromark, C.; Isomura, K.; Fernández de la Cruz, L.; Serlachius, E.; Leckman, J.F.; Crowley, J.J.; et al. A total-population multigenerational family clustering study of autoimmune diseases in obsessive-compulsive disorder and Tourette’s/chronic tic disorders. Mol. Psychiatry 2018, 23, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Fernández de la Cruz, L.; Mataix-Cols, D. General health and mortality in Tourette syndrome and chronic tic disorder: A mini-review. Neurosci. Biobehav. Rev. 2020, 119, 514–520. [Google Scholar] [CrossRef]
- Tylee, D.S.; Sun, J.; Hess, J.L.; Tahir, M.A.; Sharma, E.; Malik, R.; Worrall, B.B.; Levine, A.J.; Martinson, J.J.; Nejentsev, S.; et al. Genetic correlations among psychiatric and immune-related phenotypes based on genome-wide association data. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 641–657. [Google Scholar] [CrossRef]
- Tang, X.; Drotar, J.; Li, K.; Clairmont, C.D.; Brumm, A.S.; Sullins, A.J.; Wu, H.; Liu, X.S.; Wang, J.; Gray, N.S.; et al. Pharmacological enhancement of <i>KCC2</i> gene expression exerts therapeutic effects on human Rett syndrome neurons and <i>Mecp2</i> mutant mice. Sci. Transl. Med. 2019, 11, eaau0164. [Google Scholar] [CrossRef]
- Rivat, C.; Sar, C.; Mechaly, I.; Leyris, J.P.; Diouloufet, L.; Sonrier, C.; Philipson, Y.; Lucas, O.; Mallié, S.; Jouvenel, A.; et al. Inhibition of neuronal FLT3 receptor tyrosine kinase alleviates peripheral neuropathic pain in mice. Nat. Commun. 2018, 9, 1042. [Google Scholar] [CrossRef]
- Bzdega, T.; Crowe, S.L.; Ramadan, E.R.; Sciarretta, K.H.; Olszewski, R.T.; Ojeifo, O.A.; Rafalski, V.A.; Wroblewska, B.; Neale, J.H. The cloning and characterization of a second brain enzyme with NAAG peptidase activity. J. Neurochem. 2004, 89, 627–635. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Pearl, P.L. (Eds.) General Principles and a Phenomenology-Based Approach to Movement Disorders and Inherited Metabolic Disorders. In Movement Disorders and Inherited Metabolic Disorders: Recognition, Understanding, Improving Outcomes; Cambridge University Press: Cambridge, UK, 2020; pp. 1–170. [Google Scholar]
- Morland, C.; Nordengen, K. N-Acetyl-Aspartyl-Glutamate in Brain Health and Disease. Int. J. Mol. Sci. 2022, 23, 1268. [Google Scholar] [CrossRef]
- Kanaan, A.S.; Gerasch, S.; García-García, I.; Lampe, L.; Pampel, A.; Anwander, A.; Near, J.; Möller, H.E.; Müller-Vahl, K. Pathological glutamatergic neurotransmission in Gilles de la Tourette syndrome. Brain 2017, 140, 218–234. [Google Scholar] [CrossRef]
- Naaijen, J.; Forde, N.J.; Lythgoe, D.J.; Akkermans, S.E.; Openneer, T.J.; Dietrich, A.; Zwiers, M.P.; Hoekstra, P.J.; Buitelaar, J.K. Fronto-striatal glutamate in children with Tourette’s disorder and attention-deficit/hyperactivity disorder. Neuroimage Clin. 2017, 13, 16–23. [Google Scholar] [CrossRef]
- Mahone, E.M.; Puts, N.A.; Edden, R.A.E.; Ryan, M.; Singer, H.S. GABA and glutamate in children with Tourette syndrome: A (1)H MR spectroscopy study at 7T. Psychiatry Res. Neuroimaging 2018, 273, 46–53. [Google Scholar] [CrossRef]
- Kang, Y.; Tiziani, S.; Park, G.; Kaul, M.; Paternostro, G. Cellular protection using Flt3 and PI3Kα inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat. Commun. 2014, 5, 3672. [Google Scholar] [CrossRef]
- Zhong, C.; Zhao, X.; Van, K.C.; Bzdega, T.; Smyth, A.; Zhou, J.; Kozikowski, A.P.; Jiang, J.; O’Connor, W.T.; Berman, R.F.; et al. NAAG peptidase inhibitor increases dialysate NAAG and reduces glutamate, aspartate and GABA levels in the dorsal hippocampus following fluid percussion injury in the rat. J. Neurochem. 2006, 97, 1015–1025. [Google Scholar] [CrossRef]
- Olszewski, R.T.; Bukhari, N.; Zhou, J.; Kozikowski, A.P.; Wroblewski, J.T.; Shamimi-Noori, S.; Wroblewska, B.; Bzdega, T.; Vicini, S.; Barton, F.B.; et al. NAAG peptidase inhibition reduces locomotor activity and some stereotypes in the PCP model of schizophrenia via group II mGluR. J. Neurochem. 2004, 89, 876–885. [Google Scholar] [CrossRef]
- Olszewski, R.T.; Wegorzewska, M.M.; Monteiro, A.C.; Krolikowski, K.A.; Zhou, J.; Kozikowski, A.P.; Long, K.; Mastropaolo, J.; Deutsch, S.I.; Neale, J.H. Phencyclidine and dizocilpine induced behaviors reduced by N-acetylaspartylglutamate peptidase inhibition via metabotropic glutamate receptors. Biol. Psychiatry 2008, 63, 86–91. [Google Scholar] [CrossRef]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef]
- Limatola, C.; Ransohoff, R.M. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front. Cell. Neurosci. 2014, 8, 229. [Google Scholar] [CrossRef]
- Zhan, Y.; Paolicelli, R.C.; Sforazzini, F.; Weinhard, L.; Bolasco, G.; Pagani, F.; Vyssotski, A.L.; Bifone, A.; Gozzi, A.; Ragozzino, D.; et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 2014, 17, 400–406. [Google Scholar] [CrossRef]
- Gunner, G.; Cheadle, L.; Johnson, K.M.; Ayata, P.; Badimon, A.; Mondo, E.; Nagy, M.A.; Liu, L.; Bemiller, S.M.; Kim, K.W.; et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019, 22, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Garton, K.J.; Gough, P.J.; Blobel, C.P.; Murphy, G.; Greaves, D.R.; Dempsey, P.J.; Raines, E.W. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J. Biol. Chem. 2001, 276, 37993–38001. [Google Scholar] [CrossRef] [PubMed]
- Hundhausen, C.; Misztela, D.; Berkhout, T.A.; Broadway, N.; Saftig, P.; Reiss, K.; Hartmann, D.; Fahrenholz, F.; Postina, R.; Matthews, V.; et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 2003, 102, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Su, Z.; Wang, Z.; Li, Z.; Shang, Z.; Du, H.; Liu, G.; Qi, D.; Yang, Z.; Xu, Z.; et al. Transcriptional profiling reveals the transcription factor networks regulating the survival of striatal neurons. Cell Death Dis. 2021, 12, 262. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, G.K.; Murphy, K.J. Neuron-glia crosstalk in health and disease: Fractalkine and CX3CR1 take centre stage. Open Biol. 2013, 3, 130181. [Google Scholar] [CrossRef]
- Shan, S.; Hong-Min, T.; Yi, F.; Jun-Peng, G.; Yue, F.; Yan-Hong, T.; Yun-Ke, Y.; Wen-Wei, L.; Xiang-Yu, W.; Jun, M.; et al. New evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson’s disease. Neurobiol. Aging 2011, 32, 443–458. [Google Scholar] [CrossRef]
- Morganti, J.M.; Nash, K.R.; Grimmig, B.A.; Ranjit, S.; Small, B.; Bickford, P.C.; Gemma, C. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J. Neurosci. 2012, 32, 14592–14601. [Google Scholar] [CrossRef]
- Horiuchi, M.; Smith, L.; Maezawa, I.; Jin, L.W. CX(3)CR1 ablation ameliorates motor and respiratory dysfunctions and improves survival of a Rett syndrome mouse model. Brain Behav. Immun. 2017, 60, 106–116. [Google Scholar] [CrossRef]
- Wang, J.B.; Johnson, P.S.; Persico, A.M.; Hawkins, A.L.; Griffin, C.A.; Uhl, G.R. Human mu opiate receptor. cDNA and genomic clones, pharmacologic characterization and chromosomal assignment. FEBS Lett. 1994, 338, 217–222. [Google Scholar] [CrossRef]
- Law, P.Y.; Wong, Y.H.; Loh, H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 389–430. [Google Scholar] [CrossRef]
- Haber, S.N.; Kowall, N.W.; Vonsattel, J.P.; Bird, E.D.; Richardson, E.P., Jr. Gilles de la Tourette’s syndrome. A postmortem neuropathological and immunohistochemical study. J. Neurol. Sci. 1986, 75, 225–241. [Google Scholar] [CrossRef]
- Leckman, J.F.; Riddle, M.A.; Berrettini, W.H.; Anderson, G.M.; Hardin, M.; Chappell, P.; Bissette, G.; Nemeroff, C.B.; Goodman, W.K.; Cohen, D.J. Elevated CSF dynorphin A [1–8] in Tourette’s syndrome. Life Sci. 1988, 43, 2015–2023. [Google Scholar] [CrossRef]
- Sandyk, R. The effects of naloxone in Tourette’s syndrome. Ann. Neurol. 1985, 18, 367–368. [Google Scholar] [CrossRef]
- Sandyk, R. Naloxone withdrawal exacerbates Tourette syndrome. J. Clin. Psychopharmacol. 1986, 6, 58–59. [Google Scholar] [CrossRef]
- Sandyk, R. Naloxone abolishes obsessive-compulsive behavior in Tourette’s syndrome. Int. J. Neurosci. 1987, 35, 93–94. [Google Scholar] [CrossRef]
- Kurlan, R.; Majumdar, L.; Deeley, C.; Mudholkar, G.S.; Plumb, S.; Como, P.G. A controlled trial of propoxyphene and naltrexone in patients with Tourette’s syndrome. Ann. Neurol. 1991, 30, 19–23. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Liu, M.R.; Woods, J.H.; Medzihradsky, F. Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J. Pharmacol. Exp. Ther. 1994, 271, 1630–1637. [Google Scholar]
- Chappell, P.B.; Leckman, J.F.; Riddle, M.A.; Anderson, G.M.; Listwack, S.J.; Ort, S.I.; Hardin, M.T.; Scahill, L.D.; Cohen, D.J. Neuroendocrine and behavioral effects of naloxone in Tourette syndrome. Adv. Neurol. 1992, 58, 253–262. [Google Scholar]
- Van Wattum, P.J.; Chappell, P.B.; Zelterman, D.; Scahill, L.D.; Leckman, J.F. Patterns of response to acute naloxone infusion in Tourette’s syndrome. Mov. Disord. 2000, 15, 1252–1254. [Google Scholar] [CrossRef]
- Meuldijk, R.; Colon, E.J. Methadone treatment of Tourette’s disorder. Am. J. Psychiatry 1992, 149, 139–140. [Google Scholar] [CrossRef]
- Sarajlija, M.; Raketic, D.; Nesic, N. Heroin Addiction in Serbian Patients with Tourette Syndrome. J. Psychiatr. Pract. 2018, 24, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Sandyk, R.; Bamford, C.R. Opioid modulation of gonadotrophin release in Tourette’s syndrome. Int. J. Neurosci. 1988, 39, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Grossman, A.; Moult, P.J.; Cunnah, D.; Besser, M. Different opioid mechanisms are involved in the modulation of ACTH and gonadotrophin release in man. Neuroendocrinology 1986, 42, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Karagiannidis, I.; Dehning, S.; Sandor, P.; Tarnok, Z.; Rizzo, R.; Wolanczyk, T.; Madruga-Garrido, M.; Hebebrand, J.; Nöthen, M.M.; Lehmkuhl, G.; et al. Support of the histaminergic hypothesis in Tourette syndrome: Association of the histamine decarboxylase gene in a large sample of families. J. Med. Genet. 2013, 50, 760–764. [Google Scholar] [CrossRef]
- Alexander, J.; Potamianou, H.; Xing, J.; Deng, L.; Karagiannidis, I.; Tsetsos, F.; Drineas, P.; Tarnok, Z.; Rizzo, R.; Wolanczyk, T.; et al. Targeted Re-Sequencing Approach of Candidate Genes Implicates Rare Potentially Functional Variants in Tourette Syndrome Etiology. Front. Neurosci. 2016, 10, 428. [Google Scholar] [CrossRef]
- Cheng, Y.-H.; Zheng, Y.; He, F.; Yang, J.-H.; Li, W.-B.; Wang, M.-L.; Cui, D.-Y.; Chen, Y. Detection of Autoantibodies and Increased Concentrations of Interleukins in Plasma from Patients with Tourette’s Syndrome. J. Mol. Neurosci. 2012, 48, 219–224. [Google Scholar] [CrossRef]
- Yeon, S.-M.; Lee, J.H.; Kang, D.; Bae, H.; Lee, K.Y.; Jin, S.; Kim, J.R.; Jung, Y.W.; Park, T.W. A cytokine study of pediatric Tourette’s disorder without obsessive compulsive disorder. Psychiatry Res. 2017, 247, 90–96. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Naganuma, F.; Iida, T.; Nakamura, T.; Harada, R.; Mohsen, A.S.; Kasajima, A.; Sasano, H.; Yanai, K. Molecular mechanism of histamine clearance by primary human astrocytes. Glia 2013, 61, 905–916. [Google Scholar] [CrossRef]
- Baldan, L.C.; Williams, K.A.; Gallezot, J.D.; Pogorelov, V.; Rapanelli, M.; Crowley, M.; Anderson, G.M.; Loring, E.; Gorczyca, R.; Billingslea, E.; et al. Histidine decarboxylase deficiency causes tourette syndrome: Parallel findings in humans and mice. Neuron 2014, 81, 77–90. [Google Scholar] [CrossRef]
- Dai, H.; Zhang, Z.; Zhu, Y.; Shen, Y.; Hu, W.; Huang, Y.; Luo, J.; Timmerman, H.; Leurs, R.; Chen, Z. Histamine protects against NMDA-induced necrosis in cultured cortical neurons through H2 receptor/cyclic AMP/protein kinase A and H3 receptor/GABA release pathways. J. Neurochem. 2006, 96, 1390–1400. [Google Scholar] [CrossRef]
- Liao, R.-J.; Jiang, L.; Wang, R.-R.; Zhao, H.-W.; Chen, Y.; Li, Y.; Wang, L.; Jie, L.-Y.; Zhou, Y.-D.; Zhang, X.-N.; et al. Histidine provides long-term neuroprotection after cerebral ischemia through promoting astrocyte migration. Sci. Rep. 2015, 5, 15356. [Google Scholar] [CrossRef]
- Ito, C.; Onodera, K.; Watanabe, T.; Sato, M. Effects of histamine agents on methamphetamine-induced stereotyped behavior and behavioral sensitization in rats. Psychopharmacology 1997, 130, 362–367. [Google Scholar] [CrossRef]
- Joshi, V.V.; Balsara, J.J.; Jadhav, J.H.; Chandorkar, A.G. Effect of L-histidine and chlorcyclizine on apomorphine-induced climbing behaviour and methamphetamine stereotypy in mice. Eur. J. Pharmacol. 1981, 69, 499–502. [Google Scholar] [CrossRef]
- Itoh, Y.; Nishibori, M.; Oishi, R.; Saeki, K. Neuronal histamine inhibits methamphetamine-induced locomotor hyperactivity in mice. Neurosci. Lett. 1984, 48, 305–309. [Google Scholar] [CrossRef]
- Clapham, J.; Kilpatrick, G.J. Thioperamide, the selective histamine H3 receptor antagonist, attenuates stimulant-induced locomotor activity in the mouse. Eur J. Pharm. 1994, 259, 107–114. [Google Scholar] [CrossRef]
- Kitanaka, J.; Kitanaka, N.; Tatsuta, T.; Morita, Y.; Takemura, M. Blockade of brain histamine metabolism alters methamphetamine-induced expression pattern of stereotypy in mice via histamine H1 receptors. Neuroscience 2007, 147, 765–777. [Google Scholar] [CrossRef]
- Kitanaka, J.; Kitanaka, N.; Hall, F.S.; Uhl, G.R.; Tatsuta, T.; Morita, Y.; Tanaka, K.; Nishiyama, N.; Takemura, M. Histamine H3 receptor agonists decrease hypothalamic histamine levels and increase stereotypical biting in mice challenged with methamphetamine. Neurochem. Res. 2011, 36, 1824–1833. [Google Scholar] [CrossRef]
- Kitanaka, N.; Hall, F.S.; Kobori, S.; Kushihara, S.; Oyama, H.; Sasaoka, Y.; Takechi, M.; Tanaka, K.I.; Tomita, K.; Igarashi, K.; et al. Metoprine, a histamine N-methyltransferase inhibitor, attenuates methamphetamine-induced hyperlocomotion via activation of histaminergic neurotransmission in mice. Pharmacol. Biochem. Behav. 2021, 209, 173257. [Google Scholar] [CrossRef]
- Moro, J.; Tomé, D.; Schmidely, P.; Demersay, T.C.; Azzout-Marniche, D. Histidine: A Systematic Review on Metabolism and Physiological Effects in Human and Different Animal Species. Nutrients 2020, 12, 1414. [Google Scholar] [CrossRef]
- Yamakami, J.; Sakurai, E.; Sakurada, T.; Maeda, K.; Hikichi, N. Stereoselective blood-brain barrier transport of histidine in rats. Brain Res. 1998, 812, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, L.; Tilling, K.; Davey Smith, G. Genetic epidemiology and Mendelian randomization for informing disease therapeutics: Conceptual and methodological challenges. PLoS Genet. 2017, 13, e1006944. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, C.; Araújo, D.; Faulk, L.; Gou, Y.; Hamelers, A.; Huang, Z.; Ide-Smith, M.; Levchenko, M.; Marinos, N.; Nambiar, R.; et al. Europe PMC in 2020. Nucleic Acids Res. 2021, 49, D1507–D1514. [Google Scholar] [CrossRef] [PubMed]
- Morer, A.; Lázaro, L.; Sabater, L.; Massana, J.; Castro, J.; Graus, F. Antineuronal antibodies in a group of children with obsessive-compulsive disorder and Tourette syndrome. J. Psychiatr. Res. 2008, 42, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef]
- De Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Wang, X.; Tucker, N.R.; Rizki, G.; Mills, R.; Krijger, P.H.; de Wit, E.; Subramanian, V.; Bartell, E.; Nguyen, X.X.; Ye, J.; et al. Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. Elife 2016, 5, e10557. [Google Scholar] [CrossRef]
- Hammond, R.K.; Pahl, M.C.; Su, C.; Cousminer, D.L.; Leonard, M.E.; Lu, S.; Doege, C.A.; Wagley, Y.; Hodge, K.M.; Lasconi, C.; et al. Biological constraints on GWAS SNPs at suggestive significance thresholds reveal additional BMI loci. Elife 2021, 10, e62206. [Google Scholar] [CrossRef]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Kerimov, N.; Hayhurst, J.D.; Peikova, K.; Manning, J.R.; Walter, P.; Kolberg, L.; Samovica, M.; Sakthivel, M.P.; Kuzmin, I.; Trevanion, S.J.; et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 2021, 53, 1290–1299. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Straub, R.E.; Shin, J.H.; Tao, R.; Gao, Y.; Collado-Torres, L.; Kam-Thong, T.; Xi, H.S.; Quan, J.; Chen, Q.; et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat. Neurosci. 2018, 21, 1117–1125. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Foskolou, S.; Kilpinen, H.; Rodrigues, J.; Alasoo, K.; Knights, A.J.; Patel, M.; Goncalves, A.; Ferreira, R.; Benn, C.L.; et al. Molecular and functional variation in iPSC-derived sensory neurons. Nat. Genet. 2018, 50, 54–61. [Google Scholar] [CrossRef]
- Wang, D.; Liu, S.; Warrell, J.; Won, H.; Shi, X.; Navarro, F.C.P.; Clarke, D.; Gu, M.; Emani, P.; Yang, Y.T.; et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018, 362, eaat8464. [Google Scholar] [CrossRef]
- Ng, B.; White, C.C.; Klein, H.U.; Sieberts, S.K.; McCabe, C.; Patrick, E.; Xu, J.; Yu, L.; Gaiteri, C.; Bennett, D.A.; et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 2017, 20, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.S.; Kavanagh, D.H.; Perumal, T.M.; Ruderfer, D.M.; Oh, E.C.; Topol, A.; Shah, H.R.; et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.D.; Hu, M.; Jung, I.; Xu, Z.; Qiu, Y.; Tan, C.L.; Li, Y.; Lin, S.; Lin, Y.; Barr, C.L.; et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep. 2016, 17, 2042–2059. [Google Scholar] [CrossRef] [PubMed]
- Giusti-Rodríguez, P.; Lu, L.; Yang, Y.; Crowley, C.A.; Liu, X.; Juric, I.; Martin, J.S.; Abnousi, A.; Allred, S.C.; Ancalade, N.; et al. Using three-dimensional regulatory chromatin interactions from adult and fetal cortex to interpret genetic results for psychiatric disorders and cognitive traits. bioRxiv 2019. [Google Scholar] [CrossRef]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.; Jansen, R.; de Geus, E.J.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef]
- Barbeira, A.N.; Dickinson, S.P.; Bonazzola, R.; Zheng, J.; Wheeler, H.E.; Torres, J.M.; Torstenson, E.S.; Shah, K.P.; Garcia, T.; Edwards, T.L.; et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018, 9, 1825. [Google Scholar] [CrossRef]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef]
- Gusev, A.; Lawrenson, K.; Lin, X.; Lyra, P.C., Jr.; Kar, S.; Vavra, K.C.; Segato, F.; Fonseca, M.A.S.; Lee, J.M.; Pejovic, T.; et al. A transcriptome-wide association study of high-grade serous epithelial ovarian cancer identifies new susceptibility genes and splice variants. Nat. Genet. 2019, 51, 815–823. [Google Scholar] [CrossRef]
- Wainberg, M.; Sinnott-Armstrong, N.; Mancuso, N.; Barbeira, A.N.; Knowles, D.A.; Golan, D.; Ermel, R.; Ruusalepp, A.; Quertermous, T.; Hao, K.; et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 2019, 51, 592–599. [Google Scholar] [CrossRef]
- Euesden, J.; Lewis, C.M.; O’Reilly, P.F. PRSice: Polygenic Risk Score software. Bioinformatics 2015, 31, 1466–1468. [Google Scholar] [CrossRef]
- Ng, E.; Lind, P.M.; Lindgren, C.; Ingelsson, E.; Mahajan, A.; Morris, A.; Lind, L. Genome-wide association study of toxic metals and trace elements reveals novel associations. Hum. Mol. Genet. 2015, 24, 4739–4745. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Nyholt, D.R. SECA: SNP effect concordance analysis using genome-wide association summary results. Bioinformatics 2014, 30, 2086–2088. [Google Scholar] [CrossRef]
- Oh, S.; Abdelnabi, J.; Al-Dulaimi, R.; Aggarwal, A.; Ramos, M.; Davis, S.; Riester, M.; Waldron, L. HGNChelper: Identification and correction of invalid gene symbols for human and mouse. F1000Research 2020, 9, 1493. [Google Scholar] [CrossRef]
- Waldron, L.; Riester, M. HGNChelper: Identify and Correct Invalid HGNC Human Gene Symbols and MGI Mouse Gene Symbols; R Rackage Version 0.8.1; R Core Team: Vienna, Austria, 2019. [Google Scholar]
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human; R Package Version 3.12.0; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Tweedie, S.; Braschi, B.; Gray, K.; Jones, T.E.M.; Seal, R.L.; Yates, B.; Bruford, E.A. Genenames.org: The HGNC and VGNC resources in 2021. Nucleic Acids Res. 2021, 49, D939–D946. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Bartha, I.; di Iulio, J.; Venter, J.C.; Telenti, A. Human gene essentiality. Nat. Rev. Genet. 2018, 19, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Rackham, O.J.; Shihab, H.A.; Johnson, M.R.; Petretto, E. EvoTol: A protein-sequence based evolutionary intolerance framework for disease-gene prioritization. Nucleic Acids Res. 2015, 43, e33. [Google Scholar] [CrossRef] [PubMed]
- Samocha, K.E.; Robinson, E.B.; Sanders, S.J.; Stevens, C.; Sabo, A.; McGrath, L.M.; Kosmicki, J.A.; Rehnström, K.; Mallick, S.; Kirby, A.; et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014, 46, 944–950. [Google Scholar] [CrossRef]
- Fadista, J.; Oskolkov, N.; Hansson, O.; Groop, L. LoFtool: A gene intolerance score based on loss-of-function variants in 60,706 individuals. Bioinformatics 2017, 33, 471–474. [Google Scholar] [CrossRef]
- Bartha, I.; Rausell, A.; McLaren, P.J.; Mohammadi, P.; Tardaguila, M.; Chaturvedi, N.; Fellay, J.; Telenti, A. The Characteristics of Heterozygous Protein Truncating Variants in the Human Genome. PLoS Comput. Biol. 2015, 11, e1004647. [Google Scholar] [CrossRef]
- Cassa, C.A.; Weghorn, D.; Balick, D.J.; Jordan, D.M.; Nusinow, D.; Samocha, K.E.; O’Donnell-Luria, A.; MacArthur, D.G.; Daly, M.J.; Beier, D.R.; et al. Estimating the selective effects of heterozygous protein-truncating variants from human exome data. Nat. Genet. 2017, 49, 806–810. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Blomen, V.A.; Májek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, eaag1166. [Google Scholar] [CrossRef]
- Wells, A.; Kopp, N.; Xu, X.; O’Brien, D.R.; Yang, W.; Nehorai, A.; Adair-Kirk, T.L.; Kopan, R.; Dougherty, J.D. The anatomical distribution of genetic associations. Nucleic Acids Res. 2015, 43, 10804–10820. [Google Scholar] [CrossRef]
- Dougherty, J.D.; Schmidt, E.F.; Nakajima, M.; Heintz, N. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res. 2010, 38, 4218–4230. [Google Scholar] [CrossRef]
- Xu, X.; Wells, A.B.; O’Brien, D.R.; Nehorai, A.; Dougherty, J.D. Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J. Neurosci. 2014, 34, 1420–1431. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Miller, J.A.; Ding, S.L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef]
- Klemann, C.; Visser, J.E.; Van Den Bosch, L.; Martens, G.J.M.; Poelmans, G. Integrated molecular landscape of amyotrophic lateral sclerosis provides insights into disease etiology. Brain Pathol. 2018, 28, 203–211. [Google Scholar] [CrossRef]
- Türei, D.; Korcsmáros, T.; Saez-Rodriguez, J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966–967. [Google Scholar] [CrossRef]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Yu, H. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 2012, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Kotlyar, M.; Pastrello, C.; Ahmed, Z.; Chee, J.; Varyova, Z.; Jurisica, I. IID 2021: Towards context-specific protein interaction analyses by increased coverage, enhanced annotation and enrichment analysis. Nucleic Acids Res. 2022, 50, D640–D647. [Google Scholar] [CrossRef] [PubMed]
- Veres, D.V.; Gyurkó, D.M.; Thaler, B.; Szalay, K.Z.; Fazekas, D.; Korcsmáros, T.; Csermely, P. ComPPI: A cellular compartment-specific database for protein-protein interaction network analysis. Nucleic Acids Res. 2015, 43, D485–D493. [Google Scholar] [CrossRef]
- Rachlin, J.; Cohen, D.D.; Cantor, C.; Kasif, S. Biological context networks: A mosaic view of the interactome. Mol. Syst. Biol. 2006, 2, 66. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Hurle, M.R.; Agarwal, P. Systematic interrogation of diverse Omic data reveals interpretable, robust, and generalizable transcriptomic features of clinically successful therapeutic targets. PLoS Comput. Biol. 2018, 14, e1006142. [Google Scholar] [CrossRef]
- Ryaboshapkina, M.; Hammar, M. Tissue-specific genes as an underutilized resource in drug discovery. Sci. Rep. 2019, 9, 7233. [Google Scholar] [CrossRef]
- Plenge, R.M.; Scolnick, E.M.; Altshuler, D. Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 2013, 12, 581–594. [Google Scholar] [CrossRef]
- Glassberg, E.C.; Gao, Z.; Harpak, A.; Lan, X.; Pritchard, J.K. Evidence for Weak Selective Constraint on Human Gene Expression. Genetics 2019, 211, 757–772. [Google Scholar] [CrossRef]


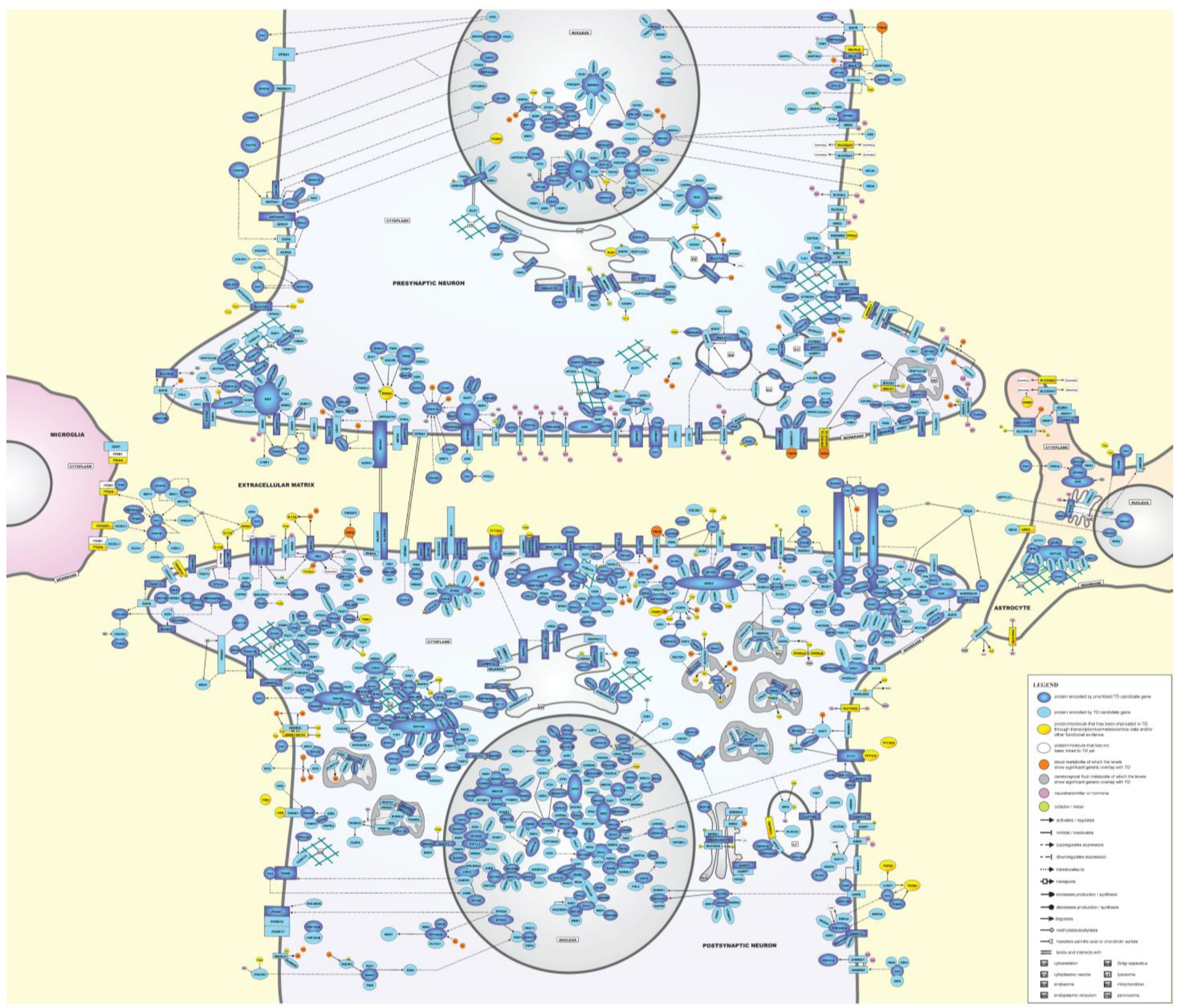{kind=link}
| Canonical Pathway | FDR p-Value | Ratio | Dataset Genes in the Pathway |
|---|---|---|---|
| cAMP-mediated signaling | 1.95 × 10−2 | 22/235 | ADCY2, AKAP9, CAMK1D, CRHR1, DRD2, DUSP6, FFAR3, FPR1, FPR2, GRK4, HRH2, MAPK3, MPPE1, OPRD1, OPRK1, OPRM1, PALM2AKAP2, PDE4A, PDE6B, PDE9A, PRKAR2A, RGS12 |
| Endocannabinoid Neuronal Synapse Pathway | 2.19 × 10−2 | 16/149 | ADCY2, CACNA1D, CACNA1I, CACNA1S, CACNA2D3, DAGLA, DNAH1, DNAH10, DNAH3, GNB1L, GRIN2A, MAPK3, PLCH1, PRKAG1, PRKAR2A, RIMS1 |
| Molecular and Cellular Functions | Physiological System Development and Function | ||||
|---|---|---|---|---|---|
| Functional Annotation | FDR p-Value | Genes | Functional Annotation | FDR p-Value | Genes |
| Cell movement of neurons | 5.73 × 10−9 | 40 | Cognition | 8.33 × 10−12 | 72 |
| Development of neurons * | 2.00 × 10−8 | 92 | Learning | 4.23 × 10−9 | 62 |
| Neuritogenesis * | 2.03 × 10−8 | 48 | Morphology of nervous system | 9.54 × 10−9 | 107 |
| Migration of neurons * | 2.11 × 10−8 | 38 | Development of head | 1.32 × 10−8 | 108 |
| Development of neural cells * | 2.90 × 10−8 | 95 | Morphogenesis of nervous tissue | 1.95 × 10−8 | 76 |
| Neurotransmission * | 7.57 × 10−8 | 54 | Development of body axis | 2.66 × 10−8 | 112 |
| Organization of cytoplasm | 2.01 × 10−7 | 148 | Morphology of brain | 2.94 × 10−8 | 66 |
| Organization of cytoskeleton | 4.55 × 10−7 | 136 | Morphology of central nervous system | 4.82 × 10−8 | 70 |
| Microtubule dynamics | 6.08 × 10−7 | 121 | Organismal death | 5.02 × 10−8 | 220 |
| Formation of cellular protrusions | 9.93 × 10−7 | 38 | Development of central nervous system | 1.29 × 10−6 | 73 |
| Migration of neural cells | 2.02 × 10−6 | 40 | Formation of brain | 1.54 × 10−6 | 60 |
| Quantity of neurotransmitter | 5.87 × 10−6 | 27 | Abnormal morphology of brain | 1.38 × 10−5 | 50 |
| Transport of molecule | 6.29 × 10−6 | 141 | Spatial learning | 1.44 × 10−5 | 28 |
| Synaptic transmission * | 2.07 × 10−5 | 41 | Emotional behavior | 2.01 × 10−5 | 38 |
| Cell movement of brain cells | 3.39 × 10−5 | 18 | Conditioning | 2.17 × 10−5 | 30 |
| Axonogenesis * | 3.68 × 10−5 | 37 | Abnormal morphology of nervous system | 2.23 × 10−5 | 81 |
| Branching of cells | 4.59 × 10−5 | 75 | Morphology of head | 2.26 × 10−5 | 101 |
| Action potential of cells * | 4.61 × 10−5 | 23 | Morphology of nervous tissue | 2.44 × 10−5 | 72 |
| Proliferation of neural cells * | 5.92 × 10−5 | 70 | Abnormal morphology of central nervous system | 2.46 × 10−5 | 53 |
| Length of cells | 6.46 × 10−5 | 31 | Prepulse inhibition | 2.68 × 10−5 | 17 |
| Morphology of neurons * | 7.13 × 10−5 | 38 | Quantity of neurons | 3.93 × 10−5 | 51 |
| Sprouting | 1.06 × 10−4 | 99 | Vocalization | 4.81 × 10−5 | 12 |
| Proliferation of neuronal cells * | 1.11 × 10−4 | 56 | Vertical rearing | 9.39 × 10−5 | 16 |
| Length of neurons * | 1.83 × 10−4 | 47 | Movement of rodents | 1.35 × 10−4 | 26 |
| Shape change of neurites | 2.85 × 10−4 | 20 | Social exploration | 2.34 × 10−4 | 12 |
| Abnormal quantity of neurotransmitter | 3.28 × 10−4 | 9 | Social behavior | 2.61 × 10−4 | 18 |
| Branching of neurons * | 3.36 × 10−4 | 18 | Exploratory behavior | 2.65 × 10−4 | 16 |
| Length of neurites * | 3.52 × 10−4 | 16 | Abnormal morphology of body cavity | 3.43 × 10−4 | 126 |
| Migration of brain cells | 3.80 × 10−4 | 15 | Nest-building behavior | 4.37 × 10−4 | 8 |
| Branching of neurites * | 4.15 × 10−4 | 65 | Abnormal morphology of head | 4.81 × 10−4 | 86 |
| Organization of cells | 5.56 × 10−4 | 22 | Locomotion | 5.03 × 10−4 | 39 |
| Quantity of monoamines | 5.90 × 10−4 | 24 | Morphology of body cavity | 6.05 × 10−4 | 139 |
| Quantity of catecholamine | 5.95 × 10−4 | 20 | Development of body trunk | 6.47 × 10−4 | 104 |
| Action potential of neurons * | 7.23 × 10−4 | 19 | Quantity of cells | 7.26 × 10−4 | 157 |
| Uptake of dopamine | 8.48 × 10−4 | 7 | Self-abusive behavior | 7.51 × 10−4 | 4 |
| Passive avoidance learning | 9.72 × 10−4 | 9 | |||
| Metabolite | Superpathway | Pathway | PT | p-Value | R2 | N SNPs | Concordance p-Value | Concordance with TD | GWAS | N GWAS | Biofluid |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Betaine a | Amino acid | Glycine, Serine, and Threonine Metabolism | 0.5 | 4.05 × 10−7 | 1.34% | 160,355 | 9.99 × 10−4 | + | Rhee | 1802 | P |
| Indoxyl sulfate a | Amino acid | Tryptophan Metabolism | 0.5 | 7.08 × 10−6 | 1.29% | 160,350 | 9.99 × 10−4 | - | Rhee | 1455 | P |
| Homocitrulline | Amino acid | Urea cycle; Arginine and Proline Metabolism | 0.5 | 4.41 × 10−6 | 0.50% | 135,356 | 9.99 × 10−4 | - | Shin | 3950 | P, S |
| Valine a | Amino acid | Valine, leucine, and isoleucine metabolism | 0.4 | 4.44 × 10−6 | 0.30% | 130,625 | 9.99 × 10−4 | + | Draaisma | 6538 | S |
| Pyridoxate a | Cofactors and vitamins | Vitamin B6 Metabolism | 0.05 | 4.91 × 10−6 | 1.34% | 25,642 | 9.99 × 10−4 | - | Rhee | 1453 | P |
| Tumor necrosis factor-beta | Cytokine | NA | 0.3 | 3.75 × 10−6 | 1.28% | 148,461 | 9.99 × 10−4 | + | Ahola-Olli | 1559 | P, S |
| Phosphatidylcholine diacyl c38:4 | Lipid | Glycerophospholipids | 0.4 | 1.54 × 10−8 | 0.41% | 130,941 | 9.99 × 10−4 | + | Draaisma | 7474 | S |
| Phosphatidylcholine 32:1 a | Lipid | Glycerophospholipids | 0.5 | 2.94 × 10−7 | 1.38% | 160,355 | 9.99 × 10−4 | - | Rhee | 1797 | P |
| Phosphatidylcholine 38:5 a | Lipid | Glycerophospholipids | 0.5 | 3.19 × 10−7 | 1.37% | 160,355 | 9.99 × 10−4 | + | Rhee | 1797 | P |
| 1-arachidonoylglycerophosphoethanolamine * | Lipid | Glycerophospholipids | 0.4 | 3.47 × 10−7 | 0.33% | 115,933 | 9.99 × 10−4 | + | Shin | 7350 | P, S |
| 1-stearoylglycerophosphoethanolamine | Lipid | Glycerophospholipids | 0.05 | 1.50 × 10−6 | 0.31% | 22,467 | 9.99 × 10−4 | + | Shin | 6929 | P, S |
| Phosphatidylcholine diacyl c38:6 | Lipid | Glycerophospholipids | 0.5 | 6.30 × 10−6 | 0.25% | 153,336 | 9.99 × 10−4 | + | Draaisma | 7475 | S |
| 1-arachidonoylglycerophosphocholine * | Lipid | Glycerophospholipids | 0.3 | 6.35 × 10−6 | 0.27% | 93,688 | 9.99 × 10−4 | + | Shin | 7063 | P, S |
| Phosphatidylcholine diacyl c36:4 | Lipid | Glycerophospholipids | 0.4 | 6.58 × 10−6 | 0.25% | 130,937 | 9.99 × 10−4 | + | Draaisma | 7476 | S |
| Myo-inositol | Lipid | Inositol Metabolism | 0.5 | 8.25 × 10−10 | 0.49% | 135,380 | 9.99 × 10−4 | - | Shin | 7354 | P, S |
| Total lipids in very small VLDL | Lipid | Lipid ratios | 0.4 | 1.73 × 10−6 | 0.11% | 390,060 | 9.99 × 10−4 | + | Shin | 2859 | P, S |
| Concentration of very small VLDL particles | Lipid | Lipid ratios | 0.2 | 3.27 × 10−6 | 0.11% | 223,305 | 9.99 × 10−4 | + | Kettunen | 19,273 | P, S |
| Phospholipids in very small VLDL | Lipid | Lipid ratios | 0.4 | 3.37 × 10−6 | 0.11% | 390,017 | 9.99 × 10−4 | + | Kettunen | 19,273 | P, S |
| Sum SM | Lipid | Lipid ratios | 0.3 | 4.21 × 10−6 | 1.10% | 109,794 | 9.99 × 10−4 | + | Rhee | 1797 | P |
| Ratio total PC: total LPC | Lipid | Lipid ratios | 0.3 | 5.96 × 10−6 | 1.06% | 109,794 | 9.99 × 10−4 | - | Rhee | 1797 | P |
| Stearate (18:0) | Lipid | Long Chain Fatty Acid | 0.2 | 3.46 × 10−8 | 0.39% | 68,520 | 9.99 × 10−4 | + | Shin | 7355 | P, S |
| X-12442—5,8-tetradecadienoate | Lipid | Long Chain Fatty Acid | 0.1 | 8.00 × 10−7 | 0.31% | 39,528 | 9.99 × 10−4 | + | Shin | 7334 | P, S |
| Palmitate (16:0) | Lipid | Long Chain Fatty Acid | 0.3 | 2.73 × 10−6 | 0.28% | 93,694 | 9.99 × 10−4 | + | Shin | 7352 | P, S |
| Laurate (12:0) | Lipid | Medium Chain Fatty Acid | 0.1 | 3.77 × 10−7 | 0.33% | 39,530 | 9.99 × 10−4 | + | Shin | 7346 | P, S |
| Linoleate (18:2n6) a | Lipid | Polyunsaturated Fatty Acid (n3 and n6) | 0.2 | 4.67 × 10−8 | 0.39% | 68,518 | 9.99 × 10−4 | + | Shin | 7333 | P, S |
| Arachidonate (20:4n6) | Lipid | Polyunsaturated Fatty Acid (n3 and n6) | 0.5 | 1.09 × 10−7 | 0.36% | 135,383 | 9.99 × 10−4 | + | Shin | 7367 | P, S |
| Linolenate [alpha or gamma; (18:3n3 or 6)] | Lipid | Polyunsaturated Fatty Acid (n3 and n6) | 0.2 | 1.38 × 10−6 | 0.30% | 68,524 | 9.99 × 10−4 | + | Shin | 7338 | P, S |
| Dihomo-linoleate (20:2n6) | Lipid | Polyunsaturated Fatty Acid (n3 and n6) | 0.3 | 6.47 × 10−6 | 0.26% | 93,696 | 9.99 × 10−4 | + | Shin | 7353 | P, S |
| Sphingomyelin 18:1 a | Lipid | Sphingolipid Metabolism | 0.4 | 3.87 × 10−10 | 2.08% | 136,596 | 9.99 × 10−4 | + | Rhee | 1797 | P |
| Sphingomyelin 18:0 a | Lipid | Sphingolipid Metabolism | 0.3 | 4.64 × 10−7 | 1.33% | 109,794 | 9.99 × 10−4 | + | Rhee | 1797 | P |
| Triacylglycerol 50:2 | Lipid | Triacylglycerol | 0.5 | 2.52 × 10−6 | 1.15% | 160,355 | 9.99 × 10−4 | - | Rhee | 1797 | P |
| Triacylglycerol 48:0 | Lipid | Triacylglycerol | 0.5 | 5.66 × 10−6 | 1.07% | 160,355 | 9.99 × 10−4 | - | Rhee | 1797 | P |
| Triacylglycerol 48:1 | Lipid | Triacylglycerol | 0.5 | 7.18 × 10−6 | 1.04% | 160,355 | 9.99 × 10−4 | - | Rhee | 1797 | P |
| X-11381 | Unknown | Unknown | 0.1 | 1.06 × 10−6 | 0.31% | 39,527 | 9.99 × 10−4 | - | Shin | 7308 | P, S |
| X-04494 | Unknown | Unknown | 0.4 | 1.21 × 10−6 | 0.47% | 115,916 | 9.99 × 10−4 | - | Shin | 4689 | P, S |
| X-12116 | Unknown | Unknown | 0.5 | 2.21 × 10−6 | 0.73% | 135,350 | 9.99 × 10−4 | - | Shin | 2859 | P, S |
| X-09706 | Unknown | Unknown | 0.5 | 3.32 × 10−6 | 0.28% | 135,378 | 9.99 × 10−4 | - | Shin | 7256 | P, S |
| N-acetyl-aspartyl-glutamate (NAAG) | Amino acid | Glutamate Metabolism | 0.5 | 1.80 × 10−8 | 9.90% | 277,618 | 9.99 × 10−4 | - | Panyard | 291 | CSF |
| Butyrate (4:0) | Lipid | Short Chain Fatty Acid | 0.5 | 2.29 × 10−6 | 6.97% | 277,618 | 9.99 × 10−4 | - | Panyard | 291 | CSF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Widomska, J.; De Witte, W.; Buitelaar, J.K.; Glennon, J.C.; Poelmans, G. Molecular Landscape of Tourette’s Disorder. Int. J. Mol. Sci. 2023, 24, 1428. https://doi.org/10.3390/ijms24021428
Widomska J, De Witte W, Buitelaar JK, Glennon JC, Poelmans G. Molecular Landscape of Tourette’s Disorder. International Journal of Molecular Sciences. 2023; 24(2):1428. https://doi.org/10.3390/ijms24021428
Chicago/Turabian StyleWidomska, Joanna, Ward De Witte, Jan K. Buitelaar, Jeffrey C. Glennon, and Geert Poelmans. 2023. "Molecular Landscape of Tourette’s Disorder" International Journal of Molecular Sciences 24, no. 2: 1428. https://doi.org/10.3390/ijms24021428
APA StyleWidomska, J., De Witte, W., Buitelaar, J. K., Glennon, J. C., & Poelmans, G. (2023). Molecular Landscape of Tourette’s Disorder. International Journal of Molecular Sciences, 24(2), 1428. https://doi.org/10.3390/ijms24021428