
mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases


Abstract
:1. Introduction
2. Mitochondrial Proteostasis
2.1. Mitochondrial Biogenesis
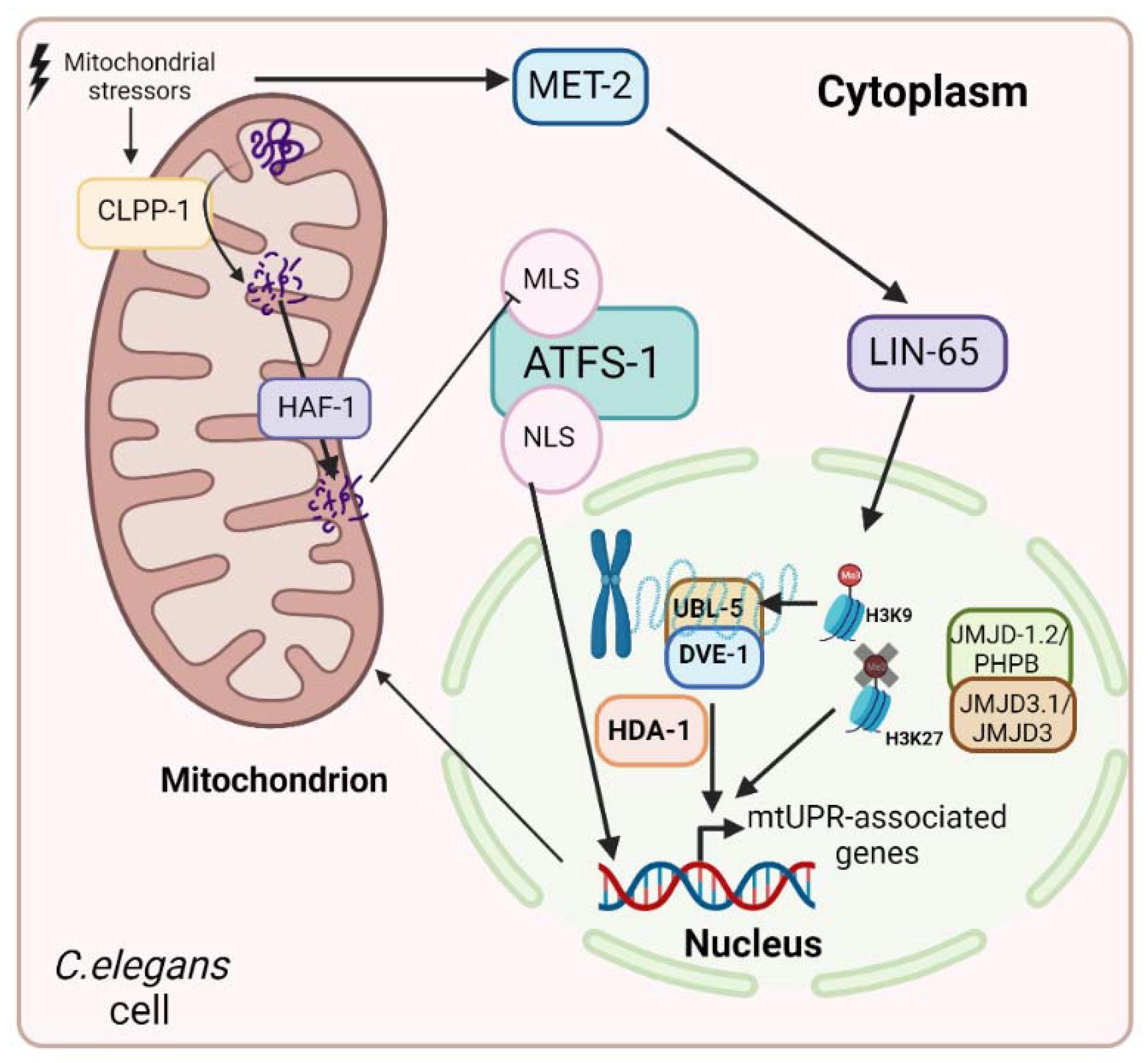
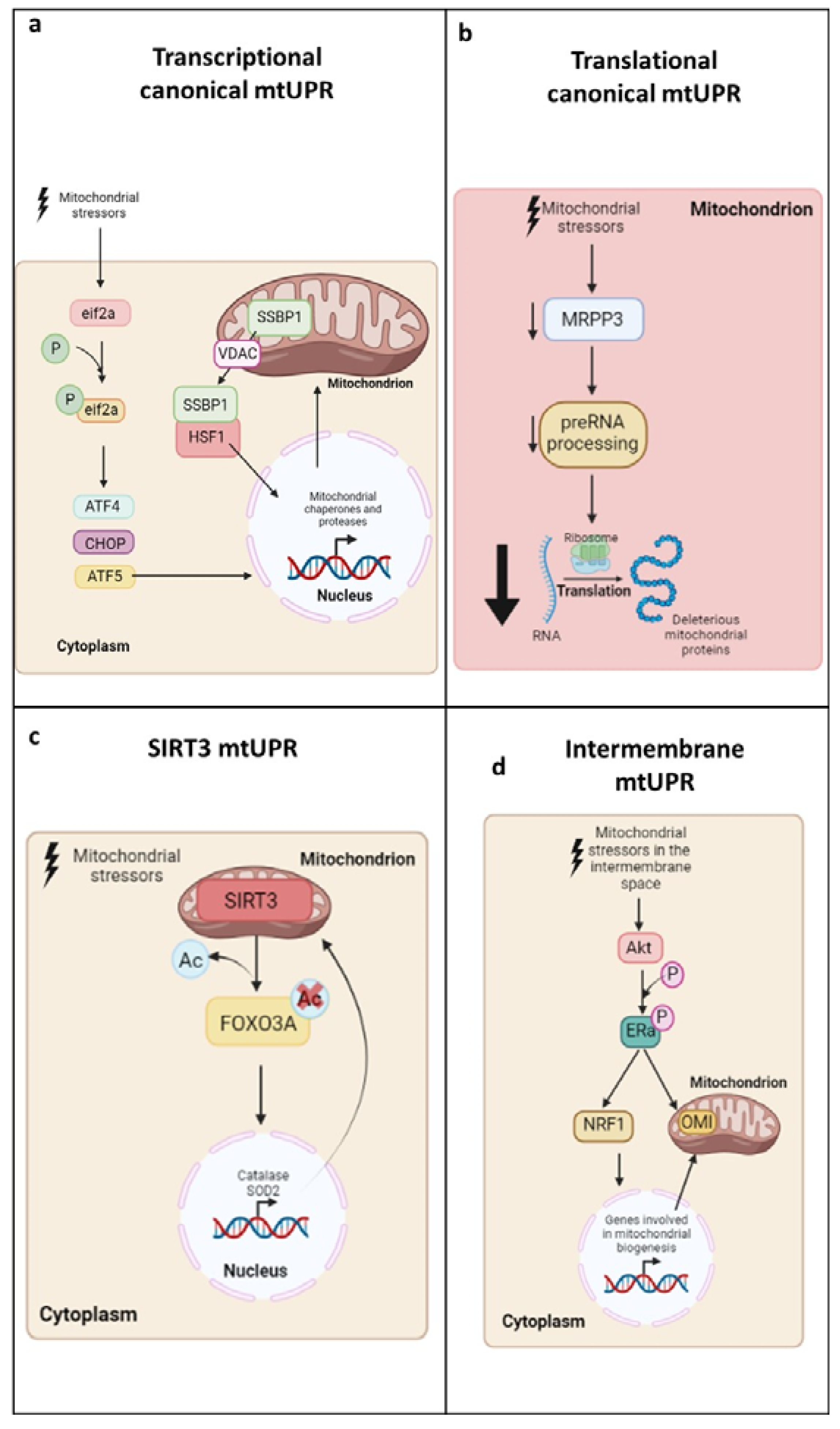
2.2. Mitochondrial Unfolded Protein Response (mtUPR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activators of mtUPR | Examples | Model Organism | mtUPR Biological Markers | References |
|---|---|---|---|---|
| Malfunctioning or inhibition of mitochondrial chaperones and proteases | Drosophila Melanogaster | Hsp60 Hsp70 ClPP | [72] | |
| Gamitrinib | Cellular models | Akt LonP1 Hsp60 Hsp70 | [73,74] | |
| Overexpression or depletion of ornithine transcarbamylase | Cellular models | ClPP LonP1 CHOP Hsp70 | [75] | |
| C. elegans | ATFS-1 Hsp60 | [75] | ||
| Overexpression of endonuclease G | Cellular models | Akt OMI | [76] | |
| Paraquat | C. elegans | ATFS-1 Hsp60 SOD2 CLPP-1 LonP1 | [78,79] | |
| Cellular models | ClPP LonP1 ATF5 Hsp60 | [77] | ||
| Toxins produced by pathogenic microorganisms | Cyanide | C. elegans | ATFS-1 Hsp60 | [80] |
| Depletion of mitochondrial import systems | Cellular models | eif2α ATF5 | [81] | |
| Inhibitors of mitochondrial translation | Tetracyclines (doxycycline and minocycline, methacycline), retapamulin, and chloramphenicol | Cellular models | LonP1 ATF5 Hsp60 Hsp70 ClPP LonP1 CHOP eif2α P-eif2α ATF4 SIRT3 | [45,82,83,84,85,87] |
| Inhibitors of the mtETC | Cancer cells | SIRT1 Hsp60 | [88,89] | |
| C. elegans | Hsp60 Hsp70 UBL-5 DVE-1 | [71] | ||
| Depletion of mtDNA or the downregulation of ribosomal protein expression | Ethidium bromide | C. elegans | CLPP-1 LonP1 Hsp60 | [79] |
| Mus musculus | Hsp60 Hsp70 UBL-5 | [42] | ||
| Itaconate | C. elegans | ATFS-1 Hsp60 UBL-5 CLPP-1 | [90] | |
| Activation of sirtuins | Nicotinamide | Cellular models | SIRT3 OMI FOXO3a SOD2 | [66] |
| C. elegans | SIRT3 FOXO3a SIRT1 SOD2 | [68] | ||
| Mus musculus | SIRT3 SOD2 SIRT1 FOXO3a Catalase Hsp60 Hsp70 CHOP | [67,92,94,99] | ||
| Pterostilbene | Cellular models | eif2α P- eif2α ATF4 ATF5 CHOP Hsp60 Hsp70 SIRT3 Nrf2 | [93] | |
| ε-viniferin | Cellular models and Drosophila melanogaster | SIRT3 SOD2 | [95] | |
| Caloric restriction | Mus musculus | Hsp60 ClPP | [95] | |
| Inhibitors of HMG-CoA reductase | Fluvastatin and rosuvastatin | C. elegans | ATFS-1 DVE-1 Hsp60 Hsp70 | [96] |
| D2 dopamine receptor antagonists | Chlorprothixene | C. elegans | ATFS-1 Hsp60 | [86] |
| Antirheumatic agents | Auranofin | C. elegans | ATFS-1 Hsp60 | [86] |
| Antioxidants | Auraptene | Cellular models | ATF4 ClPP CHOP | [97] |
| Choline | Rat | SIRT3 Hsp60 LonP1 | [98] |
3. mtUPR in Primary and Secondary Mitochondrial Diseases
3.1. mtUPR and Aging
3.2. mtUPR and Neurodegenerative Diseases
3.3. mtUPR and Cardiovascular Diseases
3.4. mtUPR and Primary Mitochondrial Disease
3.5. mtUPR and Metabolic Diseases
3.6. mtUPR and Cancer
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schatz, G. Mitochondria: Beyond Oxidative Phosphorylation. BBA Mol. Basis Dis. 1995, 1271, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Vandecasteele, G.; Gyö, G.; Szabadkai, G.; Rizzuto, R. Critical Review Mitochondrial Calcium Homeostasis: Mechanisms and Molecules. IUBMB Life 2001, 52, 213–219. [Google Scholar] [CrossRef]
- Li, Q.; Gao, Z.; Chen, Y.; Guan, M.X. The Role of Mitochondria in Osteogenic, Adipogenic and Chondrogenic Differentiation of Mesenchymal Stem Cells. Protein Cell 2017, 8, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Loscalzo, J. Redox Regulation of Mitochondrial Function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [Green Version]
- Barupala, D.P.; Dzul, S.P.; Riggs-Gelasco, P.J.; Stemmler, T.L. Synthesis, Delivery and Regulation of Eukaryotic Heme and Fe-S Cluster Cofactors. Arch. Biochem. Biophys. 2016, 592, 60–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.; Parikh, S.M. Mitochondrial Metabolism in Acute Kidney Injury. In Seminars in Nephrology; W.B. Saunders: Philadelphia, PA, USA, 2020; pp. 101–113. [Google Scholar] [CrossRef]
- al Ageeli, E. Alterations of Mitochondria and Related Metabolic Pathways in Leukemia: A Narrative Review. Saudi J. Med. Med. Sci. 2020, 8, 3. [Google Scholar] [CrossRef]
- Guda, P.; Guda, C.; Subramaniam, S. Reconstruction of Pathways Associated with Amino Acid Metabolism in Human Mitochondria. Genom. Proteom. Bioinform. 2007, 5, 166–176. [Google Scholar] [CrossRef]
- Karnkowska, A.; Vacek, V.; Zubáčová, Z.; Treitli, S.C.; Petrželková, R.; Eme, L.; Novák, L.; Žárský, V.; Barlow, L.D.; Herman, E.K.; et al. A Eukaryote without a Mitochondrial Organelle. Curr. Biol. 2016, 26, 1274–1284. [Google Scholar] [CrossRef]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial Protein Import: From Proteomics to Functional Mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Contreras, M.; Sweetwyne, M.T.; Kohrn, B.F.; Tsantilas, K.A.; Hipp, M.J.; Schmidt, E.K.; Fredrickson, J.; Whitson, J.A.; Campbell, M.D.; Rabinovitch, P.S.; et al. A Replication-Linked Mutational Gradient Drives Somatic Mutation Accumulation and Influences Germline Polymorphisms and Genome Composition in Mitochondrial DNA. Nucleic Acids Res. 2021, 49, 11103–11118. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G. Somatic Mitochondrial DNA Mutations in Mammalian Aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.I.; Alexeyev, M.F. Oxidative Stress Induces Degradation of Mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Bai, F. The Association of Tau with Mitochondrial Dysfunction in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef] [Green Version]
- Kasapoǧlu, I.; Seli, E. Mitochondrial Dysfunction and Ovarian Aging. Endocrinology 2020, 161, bqaa001. [Google Scholar] [CrossRef] [Green Version]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial Dysfunction in Obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef]
- Kujoth, C.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Medicine: Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of Mitochondria Derived Vesicle Formation Demonstrates Selective Enrichment of Oxidized Cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef] [Green Version]
- Popov, L.D. Mitochondrial-Derived Vesicles: Recent Insights. J. Cell. Mol. Med. 2022, 26, 3323–3328. [Google Scholar] [CrossRef]
- Song, J.; Herrmann, J.M.; Becker, T. Quality Control of the Mitochondrial Proteome. Nat. Rev. Mol. Cell Biol. 2020, 22, 54–70. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial Biogenesis in Neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (Ros): A Complex Relationship Regulated by the Camp/Pka Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Scarpulla, R.C. NRF-I: A Trans-Activator of Nuclear-Encoded Respiratory Genes in Animal Cells. Genes Dev. 1990, 4, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional Integration of Mitochondrial Biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial Biogenesis: A Therapeutic Target for Neurodevelopmental Disorders and Neurodegenerative Diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Kesimer, M.; Tolun, G.; Zheng, X.; Xu, Q.; Lu, J.; Sheehan, J.K.; Griffith, J.D.; Li, X. The NAD +-Dependent Protein Deacetylase Activity of SIRT1 Is Regulated by Its Oligomeric Status. Sci. Rep. 2012, 2, 640. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Cai, Z. SIRT3 Regulates Mitochondrial Biogenesis in Aging-Related Diseases. J. Biomed. Res. 2022, 36, 1. [Google Scholar] [CrossRef]
- Liu, H.; Li, S.; Liu, X.; Chen, Y.; Deng, H. SIRT3 Overexpression Inhibits Growth of Kidney Tumor Cells and Enhances Mitochondrial Biogenesis. J. Proteome Res. 2018, 17, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Igura, M.; Obita, T.; Ose, T.; Kojima, R.; Maenaka, K.; Endo, T.; Kohda, D. Tom20 Recognizes Mitochondrial Presequences through Dynamic Equilibrium among Multiple Bound States. EMBO J. 2007, 26, 4777–4787. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.; Mahlke, K.; Pfanner, N. Role of an Energized Inner Membrane in Mitochondrial Protein Import: ΔΨ Drives the Movement of Presequences. J. Biol. Chem. 1991, 266, 18051–18057. [Google Scholar] [CrossRef] [PubMed]
- Voisine, C.; Craig, E.A.; Zufall, N.; von Ahsen, O.; Pfanner, N.; Voos, W. The Protein Import Motor of Mitochondria: Unfolding and Trapping of Preproteins Are Distinct and Separable Functions of Matrix Hsp70. Cell 1999, 97, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.Y.; Hartl, F.; Martin, J.; Pollock, R.A.; Kalousek, F.; Neuper, W.; Hallberg, E.M.; Hallberg, R.L. Mitochondrial Heat-Shock Protein Hsp60 Is Essential for Assembly of Proteins Im-Ported into Yeast Mitochondria. Nature 1989, 337, 620–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttinger, L.; Oeljeklaus, S.; Guiard, B.; Rospert, S.; Warscheid, B.; Becker, T. Mitochondrial Heat Shock Protein (Hsp) 70 and Hsp10 Cooperate in the Formation of Hsp60 Complexes. J. Biol. Chem. 2015, 290, 11611–11622. [Google Scholar] [CrossRef] [Green Version]
- Quiles, J.M.; Gustafsson, Å.B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Naresh, N.U.; Haynes, C.M. Signaling and Regulation of the Mitochondrial Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033944. [Google Scholar] [CrossRef]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Høj, P.B.; Hoogenraad, N.J. Selective Induction of Mitochondrial Chaperones in Response to Loss of the Mitochondrial Genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear Protein Imbalance as a Conserved Longevity Mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, H.S.; Chang, J.Y.; Shong, M. The Mitochondrial Unfolded Protein Response and Mitohormesis: A Perspective on Metabolic Diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.T.; Lim, Y.; McCall, M.N.; Huang, K.T.; Haynes, C.M.; Nehrke, K.; Brookes, P.S. Cardioprotection by the Mitochondrial Unfolded Protein Response Requires ATF5. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H472–H478. [Google Scholar] [CrossRef]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A Mitochondrial Specific Stress Response in Mammalian Cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Shpilka, T.; Du, Y.G.; Yang, Q.; Melber, A.; Uma Naresh, N.; Lavelle, J.; Kim, S.; Liu, P.; Weidberg, H.; Li, R.; et al. UPRmt Scales Mitochondrial Network Expansion with Protein Synthesis via Mitochondrial Import in Caenorhabditis Elegans. Nat. Commun. 2021, 12, 479. [Google Scholar] [CrossRef]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the Mitochondrial Unfolded Protein Response. Biochim. Biophys. ActaMol. Cell Res. 2013, 1833, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.M.; Hao, Q.; Liu, S.Y.; Chang, Y.Z.; Duan, X.L.; Tan, K. The Molecular Mechanisms of Mitochondrial Unfolded Protein Response. Prog. Biochem. Biophys. 2017, 44, 477–485. [Google Scholar]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPRmt. Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, L.W.; Peng, Q.; Dong, M.; Gao, K.; Li, Y.; Li, Y.; Li, C.Y.; Liu, Y. Histone Deacetylase HDA-1 Modulates Mitochondrial Stress Response and Longevity. Nat. Commun. 2020, 11, 4639. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-Omics Analysis Identifies ATF4 as a Key Regulator of the Mitochondrial Stress Response in Mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP Induces Activating Transcription Factor 5 (ATF5) to Trigger Apoptosis in Response to Perturbations in Protein Homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of Genes Activated by the Mitochondrial Unfolded Protein Response (MtUPR) and Cognate Promoter Elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef]
- Fusakio, M.E.; Willy, J.A.; Wang, Y.; Mirek, E.T.; Baghdadi, R.J.T.A.; Adams, C.M.; Anthony, T.G.; Wek, R.C. Transcription Factor ATF4 Directs Basal and Stress-Induced Gene Expression in the Unfolded Protein Response and Cholesterol Metabolism in the Liver. Mol. Biol. Cell. 2016, 27, 1536–1551. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP Induces Death by Promoting Protein Synthesis and Oxidation in the Stressed Endoplasmic Reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef]
- Katiyar, A.; Fujimoto, M.; Tan, K.; Kurashima, A.; Srivastava, P.; Okada, M.; Takii, R.; Nakai, A. HSF1 Is Required for Induction of Mitochondrial Chaperones during the Mitochondrial Unfolded Protein Response. FEBS Open Bio 2020, 10, 1135–1148. [Google Scholar] [CrossRef]
- Tan, K.; Fujimoto, M.; Takii, R.; Takaki, E.; Hayashida, N.; Nakai, A. Mitochondrial SSBP1 Protects Cells from Proteotoxic Stresses by Potentiating Stress-Induced HSF1 Transcriptional Activity. Nat. Commun. 2015, 6, 6580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münch, C.; Harper, J.W. Mitochondrial Unfolded Protein Response Controls Matrix Pre-RNA Processing and Translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münch, C. The Different Axes of the Mammalian Mitochondrial Unfolded Protein Response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, L.; Germain, D. Estrogen Receptor Mediates a Distinct Mitochondrial Unfolded Protein Response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, L.; Germain, D. SirT3 Regulates the Mitochondrial Unfolded Protein Response. Mol. Cell. Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD+ Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430. [Google Scholar] [CrossRef] [Green Version]
- Weng, H.; Ma, Y.; Chen, L.; Cai, G.; Chen, Z.; Zhang, S.; Ye, Q. A New Vision of Mitochondrial Unfolded Protein Response to the Sirtuin Family. Curr. Neuropharmacol. 2020, 18, 613–623. [Google Scholar] [CrossRef]
- Shao, L.W.; Niu, R.; Liu, Y. Neuropeptide Signals Cell Non-Autonomous Mitochondrial Unfolded Protein Response. Cell Res. 2016, 26, 1182–1196. [Google Scholar] [CrossRef] [Green Version]
- Durieux, J.; Wolff, S.; Dillin, A. The Cell-Non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef]
- Baqri, R.M.; Pietron, A.V.; Gokhale, R.H.; Turner, B.A.; Kaguni, L.S.; Shingleton, A.W.; Kunes, S.; Miller, K.E. Mitochondrial Chaperone TRAP1 Activates the Mitochondrial UPR and Extends Healthspan in Drosophila. Mech. Ageing Dev. 2014, 141–142, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byoung, H.K.; Plescia, J.; Ho, Y.S.; Meli, M.; Colombo, G.; Beebe, K.; Scroggins, B.; Neckers, L.; Altieri, D.C. Combinatorial Drug Design Targeting Multiple Cancer Signaling Networks Controlled by Mitochondrial Hsp90. J. Clin. Investig. 2009, 119, 454–464. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, S.H.; Venkatesh, S.; Li, M.; Lee, J.; Lu, B.; Hilchey, S.P.; Morse, K.M.; Metcalfe, H.M.; Skalska, J.; Andreeff, M.; et al. The Mitochondrial ATP-Dependent Lon Protease: A Novel Target in Lymphoma Death Mediated by the Synthetic Triterpenoid CDDO and Its Derivatives. Blood 2012, 119, 3321–3329. [Google Scholar] [CrossRef] [Green Version]
- Runkel, E.D.; Liu, S.; Baumeister, R.; Schulze, E. Surveillance-Activated Defenses Block the ROS-Induced Mitochondrial Unfolded Protein Response. PLoS Genet. 2013, 9, e1003346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial Protein Quality Control by the Proteasome Involves Ubiquitination and the Protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Liu, Z.; Qi, X. UPRmt Activation Protects against MPP+-Induced Toxicity in a Cell Culture Model of Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2021, 569, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Bora, S.; Vardhan, G.S.H.; Deka, N.; Khataniar, L.; Gogoi, D.; Baruah, A. Paraquat Exposure over Generation Affects Lifespan and Reproduction through Mitochondrial Disruption in C. Elegans. Toxicology 2021, 447, 152632. [Google Scholar] [CrossRef]
- Pharaoh, G.; Pulliam, D.; Hill, S.; Sataranatarajan, K.; van Remmen, H. Ablation of the Mitochondrial Complex IV Assembly Protein Surf1 Leads to Increased Expression of the UPRMT and Increased Resistance to Oxidative Stress in Primary Cultures of Fibroblasts. Redox Biol. 2016, 8, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, M.W.; Nargund, A.M.; Kirienko, N.V.; Gillis, R.; Fiorese, C.J.; Haynes, C.M. Mitochondrial UPR-Regulated Innate Immunity Provides Resistance to Pathogen Infection. Nature 2014, 516, 414–417. [Google Scholar] [CrossRef] [Green Version]
- Plumb, R.; Zhang, Z.R.; Appathurai, S.; Mariappan, M. A Functional Link between the Co-Translational Protein Translocation Pathway and the UPR. Elife 2015, 4, e07426. [Google Scholar] [CrossRef]
- Zhou, B.; Fang, L.; Dong, Y.; Yang, J.; Chen, X.; Zhang, N.; Zhu, Y.; Huang, T. Mitochondrial Quality Control Protects Photoreceptors against Oxidative Stress in the H2O2-Induced Models of Retinal Degeneration Diseases. Cell Death Dis. 2021, 12, 413. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.A.; Bennett, C.F.; Luo, C.; Balsa, E.; Jedrychowski, M.; O’Malley, K.E.; Latorre-Muro, P.; Ladley, R.P.; Reda, K.; Wright, P.M.; et al. Tetracyclines Promote Survival and Fitness in Mitochondrial Disease Models. Nat. Metab. 2021, 3, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Reche-López, D.; Cilleros-Holgado, P.; et al. UPRmt Activation Improves Pathological Alterations in Cellular Models of Mitochondrial Diseases. Orphanet J. Rare Dis. 2022, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kim, S.J.; Ryu, M.J.; Jang, Y.; Lee, M.J.; Ju, X.; Lee, Y.L.; Cui, J.; Shong, M.; Heo, J.Y.; et al. Chloramphenicol Mitigates Oxidative Stress by Inhibiting Translation of Mitochondrial Complex i in Dopaminergic Neurons of Toxin-Induced Parkinson’s Disease Model. Oxidative Med. Cell. Longev. 2019, 2019, 4174803. [Google Scholar] [CrossRef] [PubMed]
- Rauthan, M.; Pilon, M. A Chemical Screen to Identify Inducers of the Mitochondrial Unfolded Protein Response in C. Elegans. Worm 2015, 4, e1096490. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing Mitochondrial Proteostasis Reduces Amyloid-β Proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Xiong, Y.; Qian, J.; Xu, M.; Qiu, J.; Xia, J.; Ma, R.; Zhu, L.; Gao, J. Effects of Low Concentrations of Rotenone upon Mitohormesis in SH-SY5Y Cells. Dose Response 2013, 11, 270–280. [Google Scholar] [CrossRef]
- Heishima, K.; Sugito, N.; Soga, T.; Nishikawa, M.; Ito, Y.; Honda, R.; Kuranaga, Y.; Sakai, H.; Ito, R.; Nakagawa, T.; et al. Petasin Potently Inhibits Mitochondrial Complex I–Based Metabolism That Supports Tumor Growth and Metastasis. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Wang, Q.; Li, H.; Zhang, G.; Chen, X.; Wang, X. Itaconate Prolongs the Healthy Lifespan by Activating UPRmt in Caenorhabditis Elegans. Eur. J. Pharmacol. 2022, 923, 174951. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhu, L.; Qiu, W.; Liu, Y.; Yang, F.; Chen, W.; Xu, R. Nicotinamide Riboside Enhances Mitochondrial Proteostasis and Adult Neurogenesis through Activation of Mitochondrial Unfolded Protein Response Signaling in the Brain of ALS SOD1g93a Mice. Int. J. Biol. Sci. 2020, 16, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Romero-González, A.; Gómez-Fernandez, D.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; et al. Pterostilbene in Combination With Mitochondrial Cofactors Improve Mitochondrial Function in Cellular Models of Mitochondrial Diseases. Front. Pharmacol. 2022, 13, 862085. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lu, J.; Yang, M.; Cai, J.; Fu, Q.; Ma, J.; Zhu, L. The Mitochondrial Unfolded Protein Response (UPRmt) Protects against Osteoarthritis. Exp. Mol. Med. 2022, 54, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, X.; Qu, J.H.; Liu, B.; Zhang, P.; Zhang, T.; Fan, P.C.; Wang, X.M.; Xiao, G.Y.; Su, Y.; et al. Caloric Restriction Induces MicroRNAs to Improve Mitochondrial Proteostasis. iScience 2019, 17, 155–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oks, O.; Lewin, S.; Goncalves, I.L.; Sapir, A. The Uprmt Protects Caenorhabditis Elegans from Mitochondrial Dysfunction by Upregulating Specific Enzymes of the Mevalonate Pathway. Genetics 2018, 209, 457–473. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Jang, Y.; Zhu, J.; Namgung, E.; Go, D.; Seo, C.; Ju, X.; Cui, J.; Lee, Y.L.; Kang, H.; et al. Auraptene Enhances Junction Assembly in Cerebrovascular Endothelial Cells by Promoting Resilience to Mitochondrial Stress through Activation of Antioxidant Enzymes and Mtupr. Antioxidants 2021, 10, 475. [Google Scholar] [CrossRef]
- Xu, M.; Xue, R.Q.; Lu, Y.; Yong, S.Y.; Wu, Q.; Cui, Y.L.; Zuo, X.T.; Yu, X.J.; Zhao, M.; Zang, W.J. Choline Ameliorates Cardiac Hypertrophy by Regulating Metabolic Remodelling and UPRmt through SIRT3-AMPK Pathway. Cardiovasc. Res. 2019, 115, 530–545. [Google Scholar] [CrossRef]
- Romani, M.; Sorrentino, V.; Oh, C.M.; Li, H.; de Lima, T.I.; Zhang, H.; Shong, M.; Auwerx, J. NAD+ Boosting Reduces Age-Associated Amyloidosis and Restores Mitochondrial Homeostasis in Muscle. Cell Rep. 2021, 34, 108660. [Google Scholar] [CrossRef]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and Nuclear Accumulation of the Transcription Factor ATFS-1 Promotes OXPHOS Recovery during the UPRmt. Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Chen, D.; Gong, Q.; Xu, Q.; Pan, D.; Lu, F.; Tang, Q. Elucidation of SIRT-1/PGC-1α-Associated Mitochondrial Dysfunction and Autophagy in Nonalcoholic Fatty Liver Disease. Lipids Health Dis. 2021, 20, 40. [Google Scholar] [CrossRef]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naia, L.; Carmo, C.; Campesan, S.; Fão, L.; Cotton, V.E.; Valero, J.; Lopes, C.; Rosenstock, T.R.; Giorgini, F.; Rego, A.C. Mitochondrial SIRT3 Confers Neuroprotection in Huntington’s Disease by Regulation of Oxidative Challenges and Mitochondrial Dynamics. Free. Radic. Biol. Med. 2021, 163, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Kenny, T.C.; Germain, D. From Discovery of the CHOP Axis and Targeting ClpP to the Identification of Additional Axes of the UPRmt Driven by the Estrogen Receptor and SIRT3. J. Bioenerg. Biomembr. 2017, 49, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Papazoglou, A.; Valenzi, E.; Rojas, M.; Lafyatis, R.; Mora, A.L. Mitochondria, Aging, and Cellular Senescence: Implications for Scleroderma. Curr. Rheumatol. Rep. 2020, 22, 37. [Google Scholar] [CrossRef]
- Berendzen, K.M.; Durieux, J.; Shao, L.W.; Tian, Y.; Kim, H.; Wolff, S.; Liu, Y.; Dillin, A. Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell 2016, 166, 1553–1563.e10. [Google Scholar] [CrossRef] [Green Version]
- Ozkurede, U.; Miller, R.A. Improved Mitochondrial Stress Response in Long-Lived Snell Dwarf Mice. Aging Cell 2019, 18, e13030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owusu-Ansah, E.; Song, W.; Perrimon, N. Muscle Mitohormesis Promotes Longevity via Systemic Repression of Insulin Signaling. Cell 2013, 155, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Kozlov, A.V.; Camara, A.K.S. Mitochondria in Health and Diseases. Cells 2020, 9, 1177. [Google Scholar] [CrossRef]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic Proteins in Neurodegenerative Disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Burchell, V.S.; Gandhi, S.; Deas, E.; Wood, N.W.; Abramov, A.Y.; Plun-Favreau, H. Targeting Mitochondrial Dysfunction in Neurodegenerative Disease: Part i. Expert Opin. Ther. Targets 2010, 14, 369–385. [Google Scholar] [CrossRef]
- Hroudová, J.; Singh, N.; Fišar, Z. Mitochondrial Dysfunctions in Neurodegenerative Diseases: Relevance to Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial Dysfunction in Neurodegenerative Diseases and the Potential Countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial Energy Metabolism and Redox Signaling in Brain Aging and Neurodegeneration. Antioxid. Redox Signal. 2014, 20, 353–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Morciano, G.; Carinci, M.; Leo, S.; Pinton, P.; Rimessi, A. The “Mitochondrial Stress Responses”: The “Dr. Jekyll and Mr. Hyde” of Neuronal Disorders. Neural Regen. Res. 2022, 17, 2563–2575. [Google Scholar] [CrossRef]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.K.; Jucker, M.; et al. Loss of Function of the Mitochondrial Peptidase PITRM1 Induces Proteotoxic Stress and Alzheimer’s Disease-like Pathology in Human Cerebral Organoids. Mol. Psychiatry 2021, 26, 5733–5750. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Kelly, S.C.; Weinberg, R.B.; Beck, J.S. [P2–179]: Mitochondrial unfolded protein response (mtupr) dysfunction during the progression of alzheimer’s disease. Alzheimer’s Dement. 2017, 13, P674–P675. [Google Scholar] [CrossRef]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. Elegans Models of Parkinson’s Disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and Propagation of a Deleterious Mitochondrial Genome by the Mitochondrial Unfolded Protein Response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Yu, S.; Wang, J.; Qiao, J.; Liu, Y.; Wang, S.; Zhao, Y. Ginseng Protein Protects against Mitochondrial Dysfunction and Neurodegeneration by Inducing Mitochondrial Unfolded Protein Response in Drosophila Melanogaster PINK1 Model of Parkinson’s Disease. J. Ethnopharmacol. 2020, 247, 112213. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, Q.; Ren, Y.; Zhang, Y.; Feng, L. A53T α-Synuclein Induces Neurogenesis Impairment and Cognitive Dysfunction in Line M83 Transgenic Mice and Reduces the Proliferation of Embryonic Neural Stem Cells. Brain Res. Bull. 2022, 182, 118–129. [Google Scholar] [CrossRef]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal Huntingtin Function: An Alternative Approach to Huntington’s Disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Liu, F.; Liu, C.; Jin, B.; Jiang, Y.; Tang, C.M.; Qi, X.; Guo, X. Mutant Huntingtin Inhibits the Mitochondrial Unfolded Protein Response by Impairing ABCB10 MRNA Stability HHS Public Access. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial Unfolded Protein Response: An Emerging Pathway in Human Diseases. Free. Radic. Biol. Med. 2020, 163, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Riar, A.K.; Burstein, S.R.; Palomo, G.M.; Arreguin, A.; Manfredi, G.; Germain, D. Sex Specific Activation of the ERα Axis of the Mitochondrial UPR (UPRmt) in the G93A-SOD1 Mouse Model of Familial ALS. Hum. Mol. Genet. 2017, 26, 1318–1327. [Google Scholar] [CrossRef] [Green Version]
- Straub, I.R.; Weraarpachai, W.; Shoubridge, E.A. Multi-OMICS Study of a CHCHD10 Variant Causing ALS Demonstrates Metabolic Rewiring and Activation of Endoplasmic Reticulum and Mitochondrial Unfolded Protein Responses. Hum. Mol. Genet. 2021, 30, 687–705. [Google Scholar] [CrossRef]
- Pharaoh, G.; Sataranatarajan, K.; Street, K.; Hill, S.; Gregston, J.; Ahn, B.; Kinter, C.; Kinter, M.; van Remmen, H. Metabolic and Stress Response Changes Precede Disease Onset in the Spinal Cord of Mutant SOD1 ALS Mice. Front. Neurosci. 2019, 13, 487. [Google Scholar] [CrossRef]
- García-Díaz, L.; Coserria, F.; Antiñolo, G. Hypertrophic Cardiomyopathy Due to Mitochondrial Disease: Prenatal Diagnosis, Management, and Outcome. Case Rep. Obstet. Gynecol. 2013, 2013, 472356. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic Organization of Mitochondria in Human Heart and in Myocardial Disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956. [Google Scholar] [CrossRef] [Green Version]
- Victor, V.; Apostolova, N.; Herance, R.; Hernandez-Mijares, A.; Rocha, M. Oxidative Stress and Mitochondrial Dysfunction in Atherosclerosis: Mitochondria-Targeted Antioxidants as Potential Therapy. Curr. Med. Chem. 2009, 16, 4654–4667. [Google Scholar] [CrossRef]
- Ballinger, S.W. Mitochondrial Dysfunction in Cardiovascular Disease. Free. Radic. Biol. Med. 2005, 38, 1278–1295. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.S.; Manolis, A.A.; Manolis, T.A.; Apostolaki, N.E.; Apostolopoulos, E.J.; Melita, H.; Katsiki, N. Mitochondrial Dysfunction in Cardiovascular Disease: Current Status of Translational Research/Clinical and Therapeutic Implications. Med. Res. Rev. 2021, 41, 275–313. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Cardiovascular Disease: A Brief Review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; González, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.Y.; Chiu, Y.C.; Lee, A.Y.L.; Hwang, T.L. Mitochondrial Lon Protease Controls ROS-Dependent Apoptosis in Cardiomyocyte under Hypoxia. Mitochondrion 2015, 23, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Li, M.; Saito, T.; Tong, M.; Rashed, E.; Mareedu, S.; Zhai, P.; Bárcena, C.; López-Otín, C.; Yehia, G.; et al. Mitochondrial LonP1 Protects Cardiomyocytes from Ischemia/Reperfusion Injury in Vivo. J. Mol. Cell. Cardiol. 2019, 128, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Svaguša, T.; Martinić, M.; Martinić, M.; Kovačević, L.; Šepac, A.; Miličić, D.; Bulum, J.; Starčević, B.; Sirotković-Skerlev, M.; Seiwerth, F.; et al. Mitochondrial Unfolded Protein Response, Mitophagy and Other Mitochondrial Quality Control Mechanisms in Heart Disease and Aged Heart. Croat. Med. J. 2020, 61, 126–138. [Google Scholar] [CrossRef]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial Diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Li, Y.R.; Li, S.; Lin, C.C. Effect of Resveratrol and Pterostilbene on Aging and Longevity. BioFactors 2017, 44, 69–82. [Google Scholar] [CrossRef]
- Poveda-Huertes, D.; Taskin, A.A.; Dhaouadi, I.; Myketin, L.; Marada, A.; Habernig, L.; Büttner, S.; Vögtle, F.N. Increased Mitochondrial Protein Import and Cardiolipin Remodelling upon Early MtUPR. PLoS Genet. 2021, 17, e1009664. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Liu, F. Mitochondrial Stress: A Bridge between Mitochondrial Dysfunction and Metabolic Diseases? Cell. Signal. 2011, 23, 1528–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Metabolic Disorders—A Step towards Mitochondria Based Therapeutic Strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Sindhu, K.K. Oxidative Stress and Metabolic Syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Armutcu, F.; Ataymen, M.; Atmaca, H.; Gurel, A. Oxidative Stress Markers, C-Reactive Protein and Heat Shock Protein 70 Levels in Subjects with Metabolic Syndrome. Clin. Chem. Lab. Med. 2008, 46, 785–790. [Google Scholar] [CrossRef]
- Siasos, G.; Paschou, S.A.; Tousoulis, D. Mitochondria and Diabetes. Ann. Transl. Med. 2020, 8, 262. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; de la Mata, M.; Pavón, A.D.; Villanueva-Paz, M.; Povea-Cabello, S.; Cotán, D.; Álvarez-Córdoba, M.; Villalón-García, I.; Ybot-González, P.; Salas, J.J.; et al. Intracellular Cholesterol Accumulation and Coenzyme Q10 Deficiency in Familial Hypercholesterolemia. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3697–3713. [Google Scholar] [CrossRef]
- Fernandes, V.; Choudhary, M.; Kumar, A.; Singh, S.B. Proteotoxicity and Mitochondrial Dynamics in Aging Diabetic Brain. Pharmacol. Res. 2020, 159, 104948. [Google Scholar] [CrossRef]
- Wardelmann, K.; Blümel, S.; Rath, M.; Alfine, E.; Chudoba, C.; Schell, M.; Cai, W.; Hauffe, R.; Warnke, K.; Flore, T.; et al. Insulin Action in the Brain Regulates Mitochondrial Stress Responses and Reduces Diet-Induced Weight Gain. Mol. Metab. 2019, 21, 68–81. [Google Scholar] [CrossRef]
- Hauffe, R.; Rath, M.; Schell, M.; Ritter, K.; Kappert, K.; Deubel, S.; Ott, C.; Jähnert, M.; Jonas, W.; Schürmann, A.; et al. HSP60 Reduction Protects against Diet-Induced Obesity by Modulating Energy Metabolism in Adipose Tissue. Mol. Metab. 2021, 53, 101276. [Google Scholar] [CrossRef]
- Lee, H.J.; Chung, K.; Lee, H.; Lee, K.; Lim, J.H.; Song, J. Downregulation of Mitochondrial Lon Protease Impairs Mitochondrial Function and Causes Hepatic Insulin Resistance in Human Liver SK-HEP-1 Cells. Diabetologia 2011, 54, 1437–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinridders, A.; Lauritzen, H.P.M.M.; Ussar, S.; Christensen, J.H.; Mori, M.A.; Bross, P.; Kahn, C.R. Leptin Regulation of Hsp60 Impacts Hypothalamic Insulin Signaling. J. Clin. Investig. 2013, 123, 4667–4680. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; de Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, L.; Arbini, A.A.; Yao, J.L.; di Sant’Agnese, P.A.; Marra, E.; Greco, M. Mitochondrial DNA Depletion in Prostate Epithelial Cells Promotes Anoikis Resistance and Invasion through Activation of PI3K/Akt2. Cell Death Differ. 2009, 16, 571–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and Cancer Chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef]
- Girolimetti, G.; Guerra, F.; Iommarini, L.; Kurelac, I.; Vergara, D.; Maffia, M.; Vidone, M.; Amato, L.B.; Leone, G.; Dusi, S.; et al. Platinum-Induced Mitochondrial DNA Mutations Confer Lower Sensitivity to Paclitaxel by Impairing Tubulin Cytoskeletal Organization. Hum. Mol. Genet. 2017, 26, 2961–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guaragnella, N.; Giannattasio, S.; Moro, L. Mitochondrial Dysfunction in Cancer Chemoresistance. Biochem. Pharmacol. 2014, 92, 62–72. [Google Scholar] [CrossRef]
- Giannattasio, S.; Guaragnella, N.; Arbini, A.A.; Moro, L. Stress-Related Mitochondrial Components and Mitochondrial Genome as Targets of Anticancer Therapy. Chem. Biol. Drug Des. 2013, 81, 102–112. [Google Scholar] [CrossRef]
- Zhou, C.; Lyu, L.H.; Miao, H.K.; Bahr, T.; Zhang, Q.Y.; Liang, T.; Zhou, H.B.; Chen, G.R.; Bai, Y. Redox Regulation by SOD2 Modulates Colorectal Cancer Tumorigenesis through AMPK-Mediated Energy Metabolism. Mol. Carcinog. 2020, 59, 545–556. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Metabolism and ROS Generation Are Essential for Kras-Mediated Tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive Oxygen Species and Cancer Paradox: To Promote or to Suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive Oxygen Species in Cancer: A Dance with the Devil. Cancer Cell 2015, 27, 156–157. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Bermúdez, A.; Vallejo, C.G.; Vicente-Blanco, R.J.; Gallardo, M.E.; Fernández-Moreno, M.Á.; Quintanilla, M.; Garesse, R. Correction: Enhanced Tumorigenicity by Mitochondrial DNA Mild Mutations. Oncotarget 2020, 11, 1006. [Google Scholar] [CrossRef]
- Muñoz-Pinedo, C.; el Mjiyad, N.; Ricci, J.E. Cancer Metabolism: Current Perspectives and Future Directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S. Metabolism of Proliferating Cells. Cold Spring Harb. Perspect. Biol. 2021, 13, a040618. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Haga, H. ATF5: Development of Oncogenic Resistance to Radiotherapy. Aging 2015, 7, 453–454. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.; Zhang, J.; Tan, Z.; Shen, L.F.; Zhou, R.R.; Zhang, Y.Y. Maf1 Suppression of ATF5-Dependent Mitochondrial Unfolded Protein Response Contributes to Rapamycin-Induced Radio-Sensitivity in Lung Cancer Cell Line A549. Aging 2021, 13, 7300–7313. [Google Scholar] [CrossRef]
- Hu, M.; Wang, B.; Qian, D.; Li, L.; Zhang, L.; Song, X.; Liu, D.X. Interference with ATF5 Function Enhances the Sensitivity of Human Pancreatic Cancer Cells to Paclitaxel-Induced Apoptosis. Anticancer Res. 2012, 32, 4385–4394. [Google Scholar]
- Monaco, S.E.; Angelastro, J.M.; Szabolcs, M.; Greene, L.A. The Transcription Factor ATF5 Is Widely Expressed in Carcinomas, and Interference with Its Function Selectively Kills Neoplastic, but Not Nontransformed, Breast Cell Lines. Int. J. Cancer 2007, 120, 1883–1890. [Google Scholar] [CrossRef]
- Chen, A.; Qian, D.; Wang, B.; Hu, M.; Lu, J.; Qi, Y.; Liu, D.X. ATF5 Is Overexpressed in Epithelial Ovarian Carcinomas and Interference with Its Function Increases Apoptosis through the Downregulation of Bcl-2 in SKOV-3 Cells. Int. J. Gynecol. Pathol. 2012, 31, 532–537. [Google Scholar] [CrossRef]
- Kong, X.; Meng, W.; Zhou, Z.; Li, Y.; Zhou, B.; Wang, R.; Zhan, L. Overexpression of Activating Transcription Factor 5 in Human Rectal Cancer. Exp. Ther. Med. 2011, 2, 827–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, L.A.; Lee, H.Y.; Angelastro, J.M. The Transcription Factor ATF5: Role in Neurodevelopment and Neural Tumors. J. Neurochem. 2009, 108, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Hua, Z.Y.; Hansen, J.N.; He, M.; Dai, S.K.; Choi, Y.; Fulton, M.D.; Lloyd, S.M.; Szemes, M.; Sen, J.; Ding, H.F.; et al. PRMT1 Promotes Neuroblastoma Cell Survival through ATF5. Oncogenesis 2020, 9, 50. [Google Scholar] [CrossRef]
- He, F.; Xiao, H.; Cai, Y.; Zhang, N. ATF5 and HIF1α Cooperatively Activate HIF1 Signaling Pathway in Esophageal Cancer. Cell Commun. Signal. 2021, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Feldheim, J.; Kessler, A.F.; Schmitt, D.; Wilczek, L.; Linsenmann, T.; Dahlmann, M.; Monoranu, C.M.; Ernestus, R.I.; Hagemann, C.; Löhr, M. Expression of Activating Transcription Factor 5 (ATF5) Is Increased in Astrocytomas of Different WHO Grades and Correlates with Survival of Glioblastoma Patients. Onco Targets Ther. 2018, 11, 8673. [Google Scholar] [CrossRef] [Green Version]
- Nukuda, A.; Endoh, H.; Yasuda, M.; Mizutani, T.; Kawabata, K.; Haga, H. Role of ATF5 in the Invasive Potential of Diverse Human Cancer Cell Lines. Biochem. Biophys. Res. Commun. 2016, 474, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; O’Malley, J.; Chaudhary, A.K.; Inigo, J.R.; Yadav, N.; Kumar, R.; Chandra, D. Hsp60 and IL-8 Axis Promotes Apoptosis Resistance in Cancer. Br. J. Cancer 2019, 121, 934–943. [Google Scholar] [CrossRef]
- Chalmers, S.A.; Eidelman, A.S.; Ewer, J.C.; Ricca, J.M.; Serrano, A.; Tucker, K.C.; Vail, C.M.; Kurt, R.A. A Role for HMGB1, HSP60 and Myd88 in Growth of Murine Mammary Carcinoma in Vitro. Cell. Immunol. 2013, 282, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Li, X.; Zhang, W.; Chen, Y.; Zhu, S.; Chen, L.; Xu, R.; Lv, Y.; Wu, D.; Guo, M.; et al. HSP60-Regulated Mitochondrial Proteostasis and Protein Translation Promote Tumor Growth of Ovarian Cancer. Sci. Rep. 2019, 9, 6792. [Google Scholar] [CrossRef] [PubMed]
- Goard, C.A.; Schimmer, A.D. Mitochondrial Matrix Proteases as Novel Therapeutic Targets in Malignancy. Oncogene 2014, 33, 2690–2699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Li, J.; Liu, X.; Wang, G.; Luo, M.; Deng, H. Down-Regulation of HSP60 Suppresses the Proliferation of Glioblastoma Cells via the ROS/AMPK/MTOR Pathway. Sci. Rep. 2016, 6, 28388. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Ryu, J.; Kim, J.E. CCAR2/DBC1 and Hsp60 Positively Regulate Expression of Survivin in Neuroblastoma Cells. Int. J. Mol. Sci. 2019, 20, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vocka, M.; Langer, D.; Fryba, V.; Petrtyl, J.; Hanus, T.; Kalousova, M.; Zima, T.; Petruzelka, L. Novel Serum Markers HSP60, CHI3L1, and IGFBP-2 in Metastatic Colorectal Cancer. Oncol. Lett. 2019, 18, 6284–6292. [Google Scholar] [CrossRef] [Green Version]
- Li, X.S.; Xu, Q.; Fu, X.Y.; Luo, W.S. Heat Shock Protein 60 Overexpression Is Associated with the Progression and Prognosis in Gastric Cancer. PLoS ONE 2014, 9, e107507. [Google Scholar] [CrossRef]
- Zhou, C.; Sun, H.; Zheng, C.; Gao, J.; Fu, Q.; Hu, N.; Shao, X.; Zhou, Y.; Xiong, J.; Nie, K.; et al. Oncogenic HSP60 Regulates Mitochondrial Oxidative Phosphorylation to Support Erk1/2 Activation during Pancreatic Cancer Cell Growth Article. Cell Death Dis. 2018, 9, 161. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of Mortalin/Mthsp70/Grp75 Contributes to Human Carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef]
- Liu, L.X.; Lu, J.C.; Zeng, H.Y.; Cai, J.B.; Zhang, P.F.; Guo, X.J.; Huang, X.Y.; Dong, R.Z.; Zhang, C.; Kang, Q.; et al. Mortalin Stabilizes CD151-Depedent Tetraspanin-Enriched Microdomains and Implicates in the Progression of Hepatocellular Carcinoma. J. Cancer 2019, 10, 6199–6206. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, L.; Yang, Y.; Han, Y.; Wang, Y.; Liu, W.; Zuo, J. Oncogenic Role of Mortalin Contributes to Ovarian Tumorigenesis by Activating the MAPK–ERK Pathway. J. Cell. Mol. Med. 2016, 20, 2111–2121. [Google Scholar] [CrossRef]
- Starenki, D.; Sosonkina, N.; Hong, S.K.; Lloyd, R.V.; Park, J.I. Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 2069. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Che, S.L.; Piao, J.J.; Xu, M.; Chen, L.Y.; Lin, Z.H. Mortalin Overexpression Predicts Poor Prognosis in Early Stage of Non–Small Cell Lung Cancer. Tumor Biol. 2017, 39, 1010428317695918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, X.; Luk, J.M.; Lee, N.P.; Peng, J.; Leng, X.; Guan, X.Y.; Lau, G.K.; Beretta, L.; Fan, S.T. Association of Mortalin (HSPA9) with Liver Cancer Metastasis and Prediction for Early Tumor Recurrence. Mol. Cell. Proteom. 2008, 7, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, A.R.; Wadhwa, R.; Kaul, S.C.; Yun, C.O. Why Is Mortalin a Potential Therapeutic Target for Cancer? Front. Cell Dev. Biol. 2022, 10, 1219. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.Y.; Chiu, Y.C.; Fang, W.C.; Cheng, C.W.; Kuo, C.Y.; Juan, H.F.; Wu, S.H.; Lee, A.Y.L. Mitochondrial Lon Regulates Apoptosis through the Association with Hsp60-MtHsp70 Complex. Cell Death Dis. 2015, 6, e1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Zeng, B.; Tao, C.; Lu, M.; Ren, G. ClpP Regulates Breast Cancer Cell Proliferation, Invasion and Apoptosis by Modulating the Src/PI3K/Akt Signaling Pathway. PeerJ 2020, 8, e8754. [Google Scholar] [CrossRef]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef] [Green Version]
- Gibellini, L.; Losi, L.; de Biasi, S.; Nasi, M.; Tartaro, D.L.; Pecorini, S.; Patergnani, S.; Pinton, P.; de Gaetano, A.; Carnevale, G.; et al. LonP1 Differently Modulates Mitochondrial Function and Bioenergetics of Primary versus Metastatic Colon Cancer Cells. Front. Oncol. 2018, 8, 254. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, J.C.; Seo, J.H.; Agarwal, E.; Wang, Y.; Kossenkov, A.V.; Tang, H.Y.; Speicher, D.W.; Altieri, D.C. Akt Phosphorylation of Mitochondrial Lonp1 Protease Enables Oxidative Metabolism and Advanced Tumor Traits. Oncogene 2019, 38, 6926–6939. [Google Scholar] [CrossRef]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Kenny, T.C.; Craig, A.J.; Villanueva, A.; Germain, D. Mitohormesis Primes Tumor Invasion and Metastasis. Cell Rep. 2019, 27, 2292–2303.e6. [Google Scholar] [CrossRef] [PubMed]


| mtUPR Factor Activated | Type of Cancer | Reference |
|---|---|---|
| ATF5 | Lung | [169] |
| Pancreatic | [170] | |
| Carcinoma | [171] | |
| Ovary | [172] | |
| Rectal | [173] | |
| Leukemia | [174] | |
| Neural tumors | [175,176] | |
| Esophageal | [177] | |
| Astrocytoma | [178] | |
| Hsp60 | Mammary | [181] |
| Ovary | [182] | |
| Prostate | [183] | |
| Glioblastoma | [184] | |
| Neuroblastoma | [185] | |
| Colorectal | [186] | |
| Gastric | [187] | |
| Pancreatic | [188] | |
| Hsp70 | Liver | [193] |
| Ovary | [191] | |
| Thyroid | [192] | |
| LonP1 | Colon | [199] |
| ClpP | Mammary | [197] |
| Leukemia | [201] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cilleros-Holgado, P.; Gómez-Fernández, D.; Piñero-Pérez, R.; Reche-López, D.; Álvarez-Córdoba, M.; Munuera-Cabeza, M.; Talaverón-Rey, M.; Povea-Cabello, S.; Suárez-Carrillo, A.; Romero-González, A.; et al. mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2023, 24, 1482. https://doi.org/10.3390/ijms24021482
Cilleros-Holgado P, Gómez-Fernández D, Piñero-Pérez R, Reche-López D, Álvarez-Córdoba M, Munuera-Cabeza M, Talaverón-Rey M, Povea-Cabello S, Suárez-Carrillo A, Romero-González A, et al. mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. International Journal of Molecular Sciences. 2023; 24(2):1482. https://doi.org/10.3390/ijms24021482
Chicago/Turabian StyleCilleros-Holgado, Paula, David Gómez-Fernández, Rocío Piñero-Pérez, Diana Reche-López, Mónica Álvarez-Córdoba, Manuel Munuera-Cabeza, Marta Talaverón-Rey, Suleva Povea-Cabello, Alejandra Suárez-Carrillo, Ana Romero-González, and et al. 2023. "mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases" International Journal of Molecular Sciences 24, no. 2: 1482. https://doi.org/10.3390/ijms24021482