
Changes in Lipidomics, Metabolomics, and the Gut Microbiota in CDAA-Induced NAFLD Mice after Polyene Phosphatidylcholine Treatment

Abstract
:1. Introduction
2. Results and Discussion
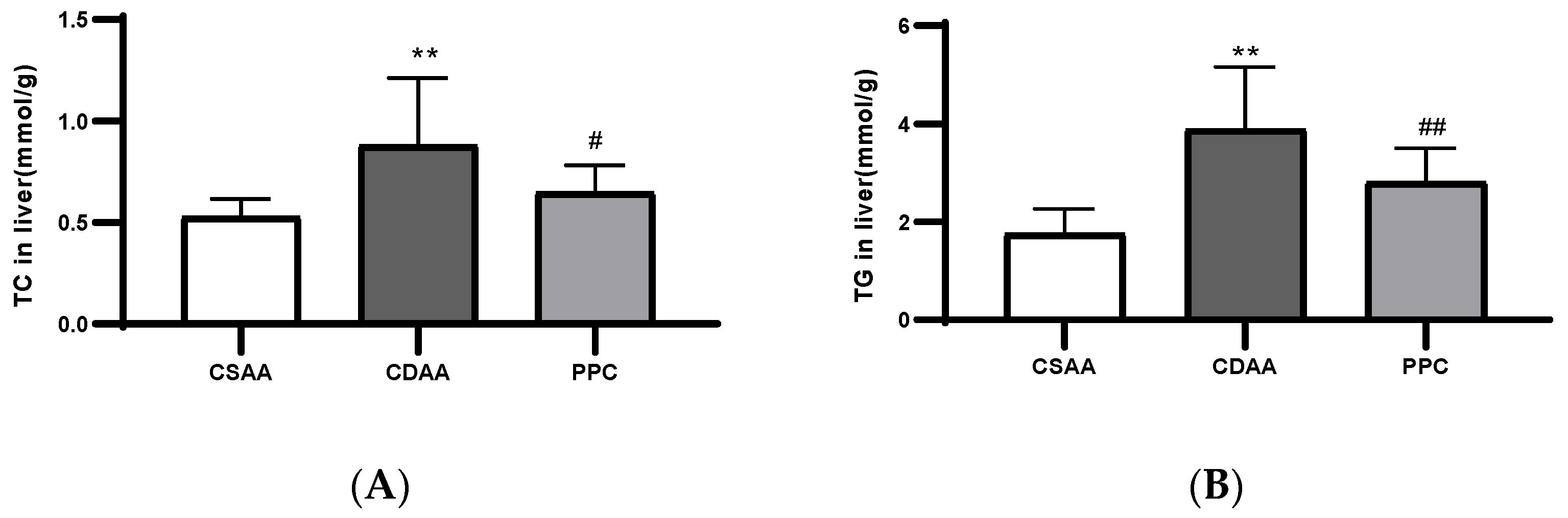
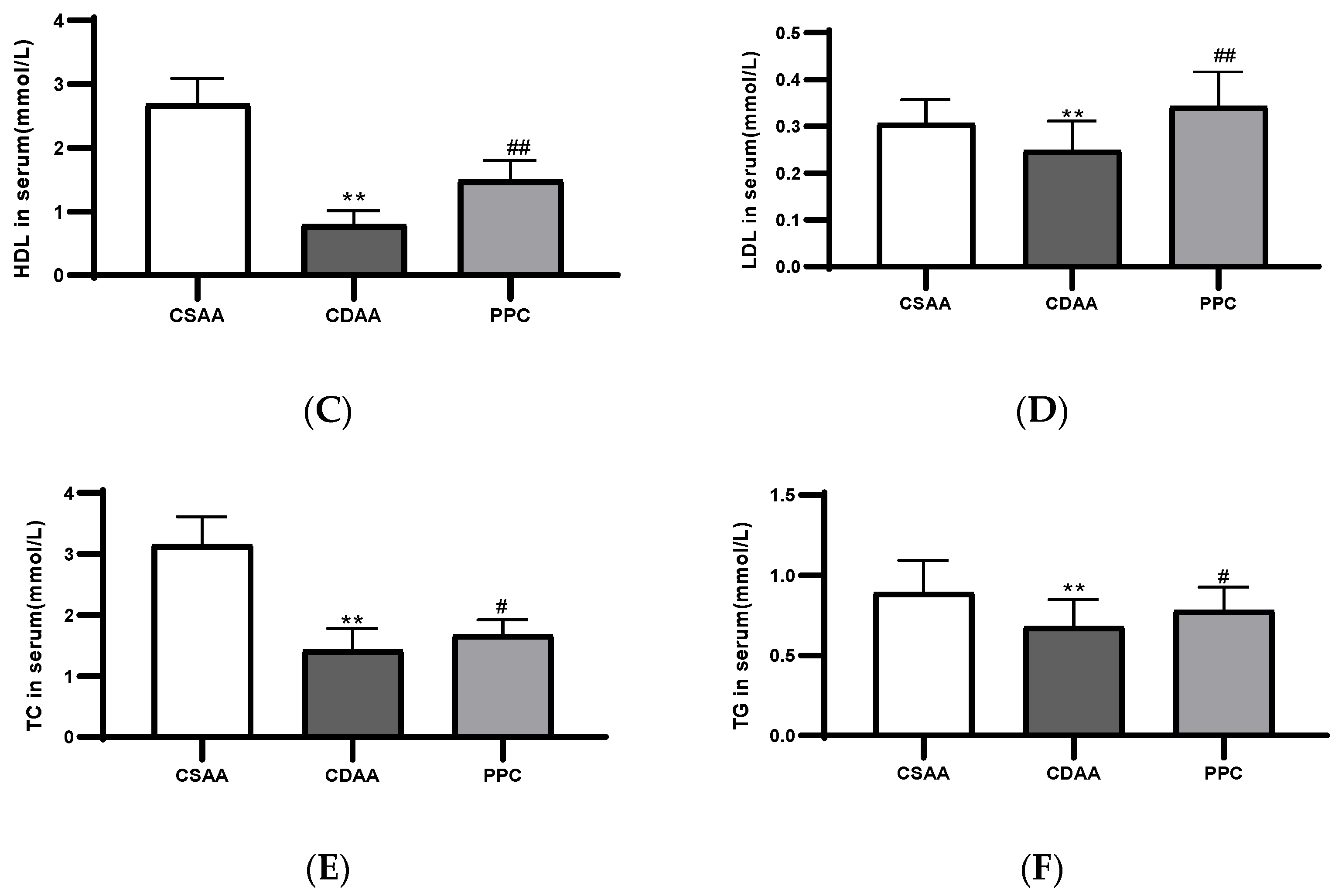
2.1. PPC Has Therapeutic Effects on a CDAA Diet-Induced Model of Fatty Liver in Mice
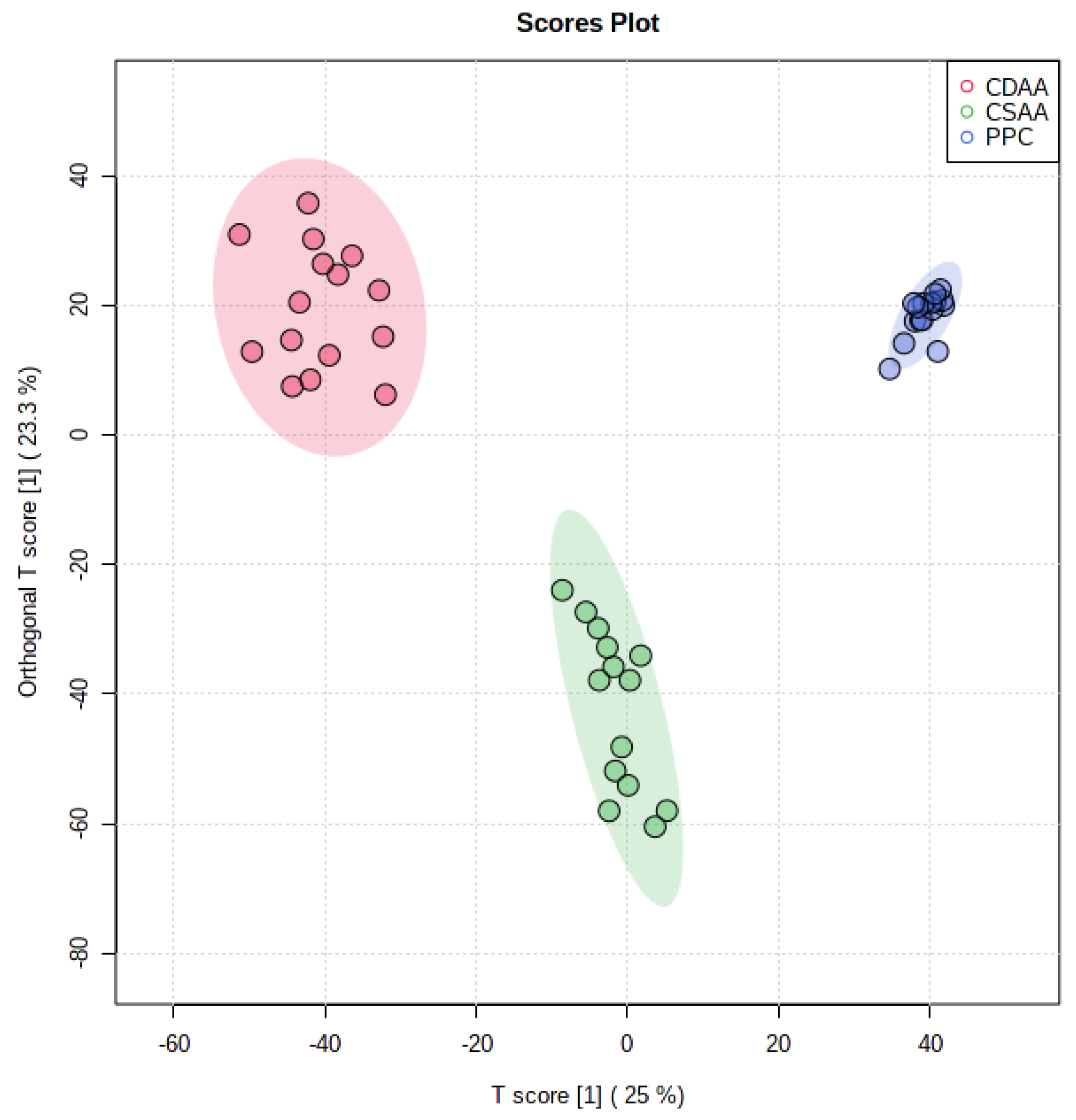
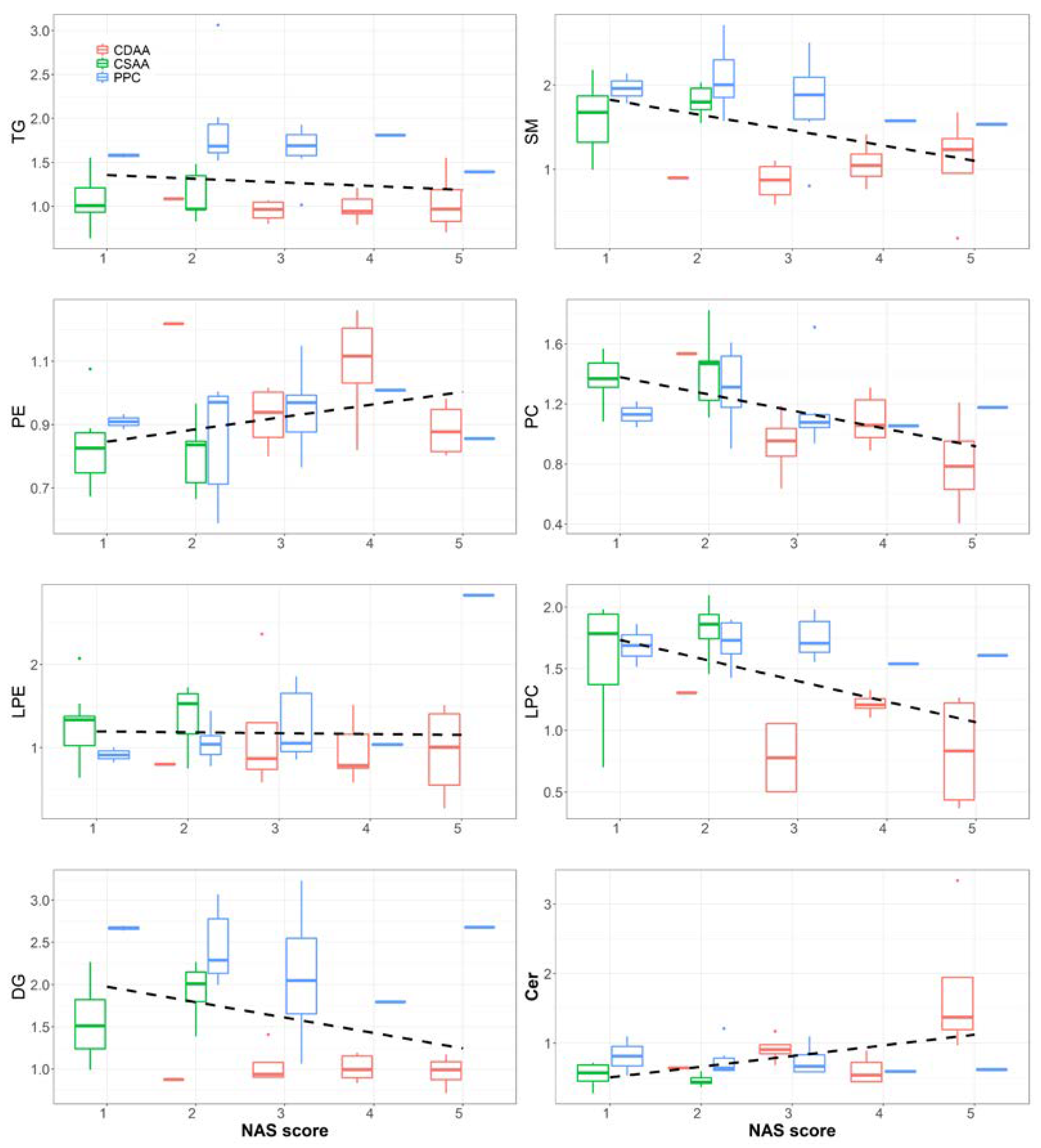
2.2. Effect of PPC on Serum Lipidome
2.3. Effect of PPC on Serum Metabolome
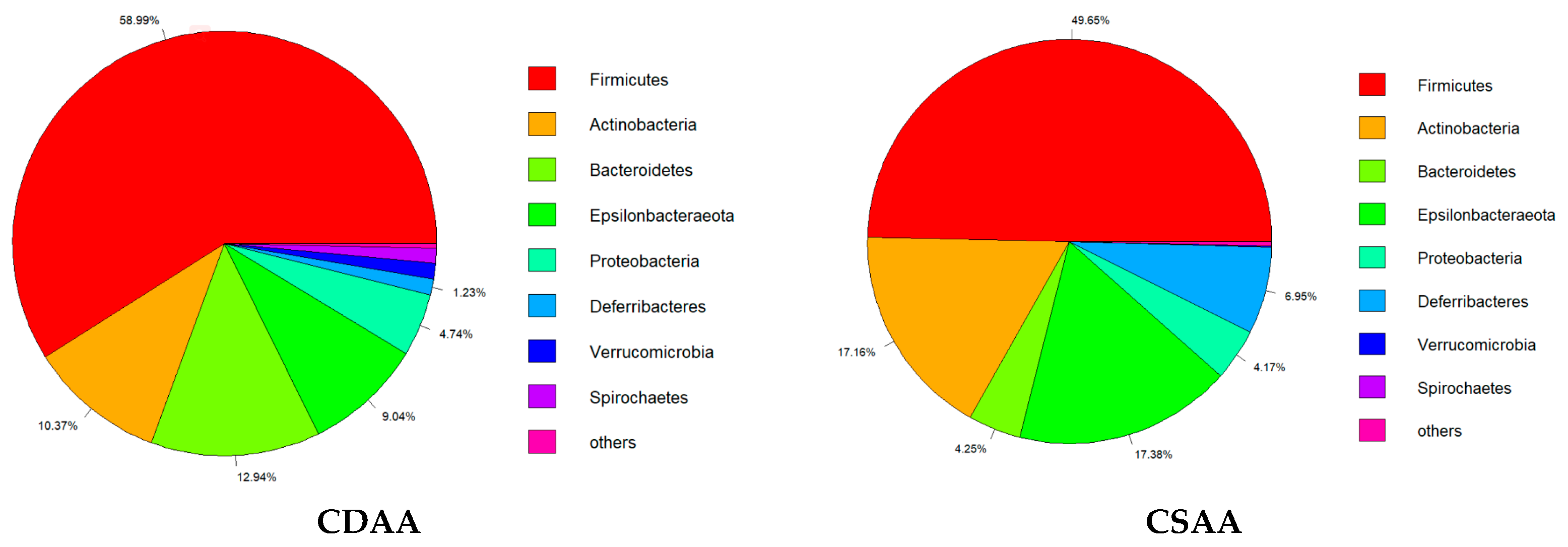
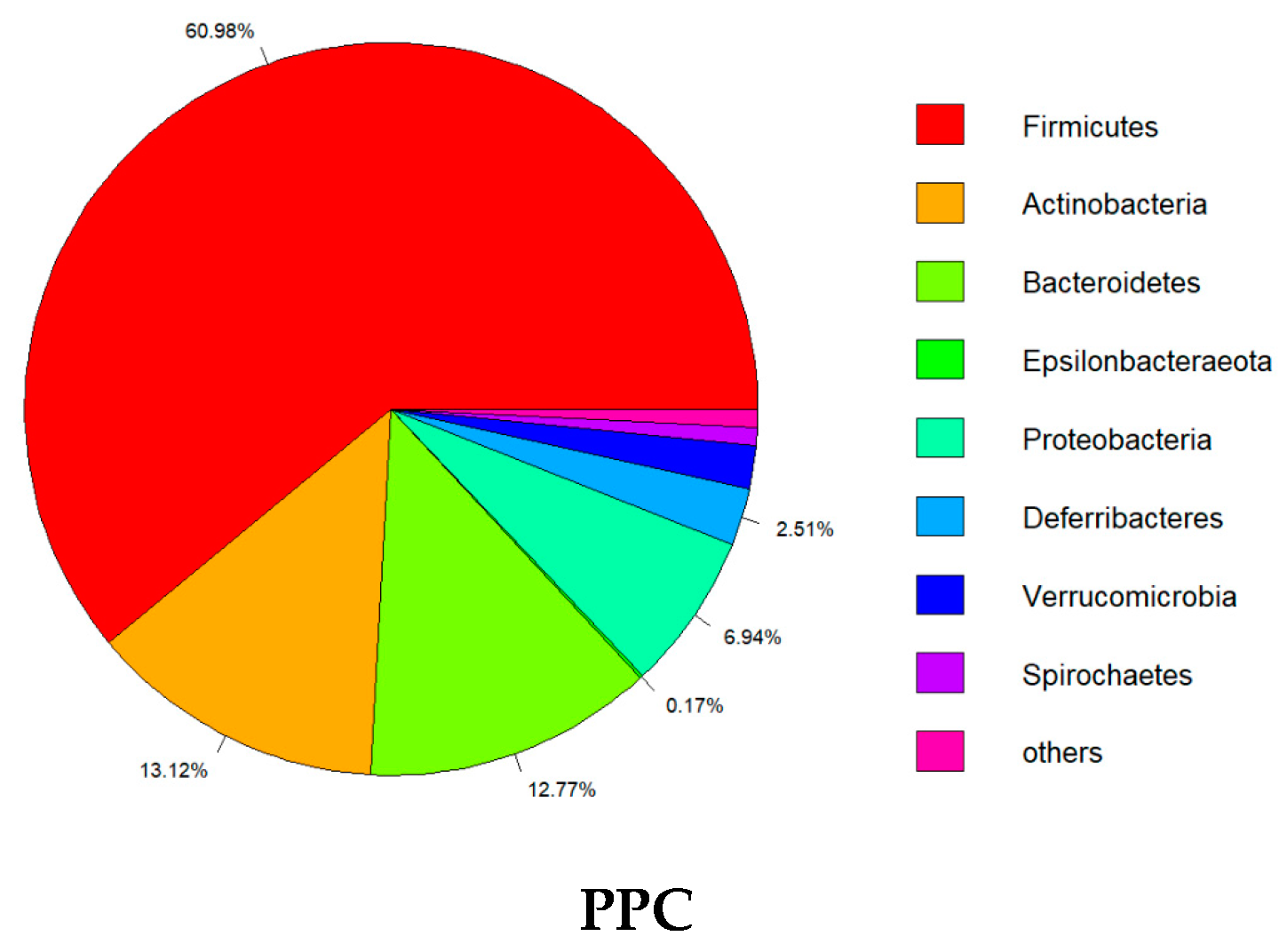
2.4. Effect of PPC on Gut Microbiota
3. Materials and Methods
3.1. Animal Model
3.2. Histological Analysis
3.3. Biochemical Indexes Analysis
3.4. Mice Liver Lipid Analysis
3.5. Lipidomics Analysis
3.6. Metabolomics Analysis
3.7. Gut Microbiota
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashtari, S.; Pourhoseingholi, M.A.; Zali, M.R. Non-alcohol fatty liver disease in Asia: Prevention and planning. World J. Hepatol. 2015, 7, 1788–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of nonalcoholic fatty liver disease in the United States: The Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Epidemiol. 2013, 178, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs. nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 39–40. [Google Scholar] [CrossRef] [Green Version]
- Tarantino, G.; Citro, V.; Capone, D. Nonalcoholic Fatty Liver Disease: A Challenge from Mechanisms to Therapy. J. Clin. Med. 2019, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Negi, C.K.; Babica, P.; Bajard, L.; Bienertova-Vasku, J.; Tarantino, G. Insights into the molecular targets and emerging pharmacotherapeutic interventions for nonalcoholic fatty liver disease. Metabolism 2022, 126, 154925. [Google Scholar] [CrossRef]
- Okiyama, W.; Tanaka, N.; Nakajima, T.; Tanaka, E.; Kiyosawa, K.; Gonzalez, F.J.; Aoyama, T. Polyenephosphatidylcholine prevents alcoholic liver disease in PPARalpha-null mice through attenuation of increases in oxidative stress. J. Hepatol. 2009, 50, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Karaman, A.; Demirbilek, S. Protective effect of polyunsaturated phosphatidylcholine on liver damage induced by biliary obstruction in rats. J. Pediatr. Surg. 2003, 38, 1341–1347. [Google Scholar] [CrossRef]
- Hiroko, I.; Kazuhiko, S.; Shunichi, M. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. Int. J. Clin. Exp. Pathol. 2013, 6, 2683–2696. [Google Scholar]
- Rinella, M.E.; Green, R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, D.; Wree, A.; Povero, D.; Solis, N.; Hernandez, A.; Pizarro, M.; Moshage, H.; Torres, J.; Feldstein, A.E.; Cabello-Verrugio, C.; et al. Andrographolide Ameliorates Inflammation and Fibrogenesis and Attenuates Inflammasome Activation in Experimental Non-Alcoholic Steatohepatitis. Sci. Rep. 2017, 7, 3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolbol, K.S.; Stierstorfer, B.; Rippmann, J.F.; Veidal, S.S.; Rigbolt, K.T.G.; Schonberger, T.; Gillum, M.P.; Hansen, H.H.; Vrang, N.; Jelsing, J.; et al. Disease Progression and Pharmacological Intervention in a Nutrient-Deficient Rat Model of Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2019, 64, 1238–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdollahi-Basir, M.H.; Shirini, F.; Tajik, H.; Ghasemzadeh, M.A. Zn (BDC)-(MOF): Introduction of a New Catalyst for the Synthesis Pyrimido[4,5-d]Pyrimidine Derivatives under Ultrasound Irradiation in the Absence of Solvent. Polycycl. Aromat. Compd. 2021, 41, 1580–1589. [Google Scholar] [CrossRef]
- Kulinski, A.; Vance, D.E.; Vance, J.E. A choline-deficient diet in mice inhibits neither the CDP-choline pathway for phosphatidylcholine synthesis in hepatocytes nor apolipoprotein B secretion. J. Biol. Chem. 2004, 279, 23916–23924. [Google Scholar] [CrossRef] [Green Version]
- Miyaki, T.; Nojiri, S.; Shinkai, N.; Kusakabe, A.; Matsuura, K.; Iio, E.; Takahashi, S.; Yan, G.; Ikeda, K.; Joh, T. Pitavastatin inhibits hepatic steatosis and fibrosis in non-alcoholic steatohepatitis model rats. Hepatol. Res. 2011, 41, 375–385. [Google Scholar] [CrossRef]
- Yu, Y.; Cai, J.; She, Z.; Li, H. Insights into the Epidemiology, Pathogenesis, and Therapeutics of Nonalcoholic Fatty Liver Diseases. Adv. Sci. 2019, 6, 1801585. [Google Scholar] [CrossRef]
- Lieber, C. Pathogenesis and treatment of alcoholic liver disease: Progress over the last 50 years. Rocz. Akad. Med. Bialymst. 2005, 5, 20. [Google Scholar]
- Seitz, H.K.; Lieber, C.S.; Stickel, F.; Salaspuro, M.; Schlemmer, H.P.; Horie, Y. Alcoholic liver disease: From pathophysiology to therapy. Alcohol. Clin. Exp. Res. 2005, 29, 1276–1281. [Google Scholar] [CrossRef]
- Li, Z.; Vance, D.E. Phosphatidylcholine and choline homeostasis. J. Lipid Res. 2008, 49, 1187–1194. [Google Scholar] [CrossRef] [Green Version]
- Houten, S.M.; Wanders, R.J.A.; Ranea-Robles, P. Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165720. [Google Scholar] [CrossRef] [PubMed]
- Savic, D.; Hodson, L.; Neubauer, S.; Pavlides, M. The Importance of the Fatty Acid Transporter L-Carnitine in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2020, 12, 2178. [Google Scholar] [CrossRef] [PubMed]
- El-Sheikh, H.; El-Haggar, S.; Elbedewy, T.J.D.; Syndrome, M. Comparative study to evaluate the effect of l-carnitine plus glimepiride versus glimepiride alone on insulin resistance in type 2 diabetic patients. Diabetes Metab. Syndr. 2019, 13, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jiang, W.; Chen, G.; Zhu, W.; Ding, W.; Ge, Z.; Tan, Y.; Ma, T.; Cui, G. L-carnitine treatment of insulin resistance: A systematic review and meta-analysis. Adv. Clin. Exp. Med. 2017, 26, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Leo, M.A.; Lieber, C.S. DLPC attenuates alcohol-induced cytotoxicity in HepG2 cells expressing CYP2E1. Alcohol Alcohol. 2005, 40, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, G.G.; Shear, N.H. Signaling for ethanol-induced apoptosis and repair in vitro. Clin. Biochem. 2001, 34, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Acierno, C.; Caturano, A.; Pafundi, P.C.; Nevola, R.; Adinolfi, L.E.; Sasso, F.C. Nonalcoholic fatty liver disease and type 2 diabetes: Pathophysiological mechanisms shared between the two faces of the same coin. Explor. Med. 2020, 1, 287–306. [Google Scholar] [CrossRef]
- Szendroedi, J.; Kaul, K.; Kloock, L.; Straßburger, K.; Schmid, A.I.; Chmelik, M.; Kacerovsky, M.; Kacerovsky-Bielesz, G.; Prikoszovich, T.; Brehm, A.; et al. Lower Fasting Muscle Mitochondrial Activity Relates to Hepatic Steatosis in Humans. Diabetes Care 2014, 37, 468–474. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Yokota, A.; Fukiya, S.; Islam, K.B.; Ooka, T.; Ogura, Y.; Hayashi, T.; Hagio, M.; Ishizuka, S. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes 2012, 3, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Joghataei, M.; Shahidi, F.; Pouladfar, G.; Mortazavi, S.A.; Ghaderi, A. Probiotic potential comparison of Lactobacillus strains isolated from Iranian traditional food products and human feces with standard probiotic strains. J. Sci. Food Agric. 2019, 99, 6680–6688. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, A.; Zhang, L.; Waqas, M.; Mehmood, K.; Iqbal, M.; Muyou, C.; Li, Z.; Lian, Y.; Sizhu, S.; et al. Probiotic potential of Lactobacillus on the intestinal microflora against Escherichia coli induced mice model through high-throughput sequencing. Microb. Pathog. 2019, 137, 103760. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Feng, N.; Zhang, C.; Liu, F.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Lactobacillus reuteri A9 and Lactobacillus mucosae A13 isolated from Chinese superlongevity people modulate lipid metabolism in a hypercholesterolemia rat model. FEMS Microbiol. Lett. 2019, 366, fnz254. [Google Scholar] [CrossRef] [PubMed]
- Vincent, W.-S.W.; Wong, G.L.-H. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann. Hepatol. 2013, 12, 256–262. [Google Scholar]
- Zhang, M.; Wang, C.; Wang, C.; Zhao, H.; Zhao, C.; Chen, Y.; Wang, Y.; McClain, C.; Feng, W. Enhanced AMPK phosphorylation contributes to the beneficial effects of Lactobacillus rhamnosus GG supernatant on chronic-alcohol-induced fatty liver disease. J. Nutr. Biochem. 2015, 26, 337–344. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef]
- Deng, P.H.J.; Petriello, M.C. Dietary inulin decreases circulating ceramides by suppressing neutral sphingomyelinase expression and activity in mice. J. Lipid Res. 2020, 61, 47–51. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAS Average Score | Variance | |
|---|---|---|
| CSAA | 1.44 | 0.60 |
| CDAA | 3.78 | 0.97 |
| PPC-treated CDAA | 2.56 | 0.96 |
| Lipid ID | m/z | RT (min) | Chemical Formula | Fold Change * | p-Value |
|---|---|---|---|---|---|
| LPC 18:0/0:0 | 524.37 | 3.17 | C26H54NO7P | −1.63 | 3.21 × 10−3 |
| LPC 18:1/0:0 | 522.36 | 1.85 | C26H52NO7P | −1.66 | 1.70 × 10−9 |
| LPC 20:0/0:0 | 552.40 | 4.20 | C28H58NO7P | −3.12 | 1.49 × 10−9 |
| LPC 22:0/0:0 | 580.44 | 5.31 | C30H62NO7P | −2.24 | 1.88 × 10−10 |
| LPC 22:1/0:0 | 578.42 | 3.73 | C30H60NO7P | −2.40 | 5.74 × 10−12 |
| LPC 22:6/0:0 | 568.34 | 1.43 | C30H50NO7P | −1.76 | 5.97 × 10−9 |
| LPC 24:0/0:0 | 608.47 | 6.78 | C32H66NO7P | −2.15 | 9.27 × 10−10 |
| PC 12:0_22:1 | 760.59 | 9.14 | C42H82NO8P | −3.28 | 4.18 × 10−5 |
| PC 12:0_26:5 | 808.58 | 8.45 | C46H82NO8P | −2.81 | 3.94 × 10−4 |
| PC 14:0_24:2 | 814.64 | 10.34 | C46H88NO8P | −3.47 | 2.26 × 10−13 |
| PC 14:1_22:4 | 780.56 | 8.50 | C44H78NO8P | −2.33 | 5.37 × 10−8 |
| PC 14:1_24:5 | 806.57 | 8.25 | C46H80NO8P | −1.78 | 3.94 × 10−8 |
| PC 16:0_20:1 | 788.62 | 10.01 | C44H86NO8P | −2.33 | 2.04 × 10−10 |
| PC 16:0_24:5 | 836.61 | 9.98 | C48H86NO8P | +5.36 | 8.29 × 10−4 |
| PC 17:1_17:1 | 758.57 | 8.73 | C42H80NO8P | −3.29 | 2.77 × 10−7 |
| PC 17:2_17:2 | 754.54 | 8.36 | C42H76NO8P | −3.12 | 2.00 × 10−6 |
| PC 18:2_18:2 | 782.57 | 8.40 | C44H80NO8P | −3.60 | 4.26 × 10−6 |
| PC 19:2_19:2 | 810.61 | 9.56 | C46H84NO8P | −1.96 | 4.74 × 10−4 |
| PC 20:2_18:3 | 808.58 | 8.78 | C46H82NO8P | +1.75 | 2.54 × 10−2 |
| PC 20:3_18:4 | 804.56 | 11.68 | C46H78NO8P | −3.05 | 1.91 × 10−9 |
| PC 24:0_14:1 | 816.65 | 11.68 | C46H90NO8P | −3.69 | 4.29 × 10−11 |
| PC O-35:3 | 756.59 | 7.92 | C43H82NO7P | −2.23 | 2.12 × 10−7 |
| PC O-38:2 | 800.66 | 11.11 | C46H90NO7P | −1.97 | 3.88 × 10−7 |
| SM 18:1;2O/14:0 | 675.55 | 7.91 | C37H75N2O6P | −2.01 | 1.12 × 10−6 |
| SM 18:2;2O/16:0 | 701.56 | 8.08 | C39H77N2O6P | −2.18 | 4.61 × 10−9 |
| SM 18:1;2O/22:0 | 787.67 | 10.77 | C45H91N2O6P | −2.52 | 3.60 × 10−8 |
| SM 18:1;2O/22:1 | 785.66 | 11.58 | C45H89N2O6P | −3.72 | 3.66 × 10−9 |
| SM 18:1;2O/23:0 | 801.69 | 13.39 | C46H93N2O6P | −1.63 | 1.15 × 10−3 |
| SM 18:1;2O/24:0 | 815.71 | 12.39 | C47H95N2O6P | −1.62 | 9.68 × 10−5 |
| Lipid ID | m/z | RT (min) | Chemical Formula | Fold Change * | p-Value |
|---|---|---|---|---|---|
| LPC 18:0/0:0 | 524.37 | 3.17 | C26H54NO7P | +1.65 | 8.89 × 10−5 |
| LPC 19:0/0:0 | 538.39 | 3.38 | C27H56NO7P | +1.91 | 1.19 × 10−6 |
| LPC 20:0/0:0 | 552.40 | 4.20 | C28H58NO7P | +1.66 | 1.66 × 10−6 |
| LPC 20:4/0:0 | 544.33 | 1.74 | C28H50NO7P | +2.51 | 4.60 × 10−5 |
| LPC 22:6/0:0 | 568.34 | 1.43 | C30H50NO7P | +1.55 | 6.80 × 10−7 |
| PC 12:0_22:1 | 760.59 | 9.14 | C44H82NO8P | +1.71 | 2.43 × 10−4 |
| PC 14:1_20:2 | 756.56 | 8.50 | C42H78NO8P | +1.76 | 8.47 × 10−4 |
| PC 14:1_24:5 | 806.57 | 8.25 | C46H78NO8P | +2.49 | 5.88 × 10−6 |
| PC 16:0_22:3 | 812.62 | 10.01 | C46H86NO8P | −2.51 | 6.82 × 10−5 |
| PC 16:0_24:5 | 836.62 | 9.98 | C48H86NO8P | −2.18 | 1.82 × 10−4 |
| PC 17:1_17:1 | 758.57 | 8.73 | C42H80NO8P | −3.96 | 4.88 × 10−6 |
| PC 17:2_17:2 | 754.54 | 8.36 | C42H76NO8P | +9.50 | 9.62 × 10−5 |
| PC 18:1_18:1 | 786.60 | 10.89 | C44H84NO8P | −4.16 | 1.12 × 10−3 |
| PC 18:2_18:2 | 782.58 | 8.40 | C44H80NO8P | +1.75 | 1.70 × 10−2 |
| PC 19:2_19:2 | 810.61 | 9.56 | C46H84NO8P | −1.93 | 3.97 × 10−3 |
| PC 24:0_14:1 | 816.65 | 11.68 | C46H90NO8P | +1.69 | 1.14 × 10−4 |
| PC O-35:1 | 760.62 | 11.95 | C43H86NO7P | +5.26 | 1.63 × 10−4 |
| PC O-35:3 | 756.59 | 11.10 | C43H82NO7P | +2.24 | 8.92 × 10−5 |
| PC O-36:1 | 774.64 | 11.44 | C44H88NO7P | +2.17 | 6.66 × 10−4 |
| PC O-37:1 | 788.65 | 13.09 | C45H90NO7P | +4.43 | 5.72 × 10−5 |
| PC O-37:5 | 780.58 | 8.33 | C45H82NO7P | +3.31 | 3.22 × 10−4 |
| PC O-38:1 | 802.67 | 12.32 | C46H92NO7P | +3.15 | 1.29 × 10−5 |
| PC O-38:2 | 800.66 | 11.63 | C46H90NO7P | +2.20 | 4.31 × 10−8 |
| PC O-39:3 | 812.66 | 13.46 | C47H90NO7P | +1.57 | 2.86 × 10−5 |
| PC O-39:4 | 810.62 | 9.71 | C47H88NO7P | −2.97 | 7.49 × 10−4 |
| PC O-40:1 | 830.70 | 13.13 | C48H96NO7P | +2.97 | 2.73 × 10−5 |
| SM 18:1;2O/14:0 | 675.55 | 7.91 | C37H75N2O6P | +1.52 | 1.40 × 10−6 |
| SM 18:1;2O/16:0 | 703.58 | 8.83 | C39H79N2O6P | +1.51 | 1.77 × 10−10 |
| SM 18:2;2O/16:0 | 701.56 | 8.08 | C39H77N2O6P | +1.56 | 1.96 × 10−6 |
| SM 18:1;2O/22:0 | 787.67 | 11.93 | C45H91N2O6P | +1.65 | 3.00 × 10−6 |
| SM 15:2;2O/25:0 | 785.66 | 11.17 | C45H89N2O6P | +1.86 | 1.37 × 10−4 |
| SM 18:1;2O/23:0 | 801.69 | 12.71 | C46H93N2O6P | +2.53 | 9.02 × 10−6 |
| SM 20:1;2O/21:0 | 701.56 | 11.66 | C46H93N2O6P | +3.29 | 1.09 × 10−5 |
| SM 22:2;2O/19:0 | 785.63 | 11.58 | C46H91N2O6P | +1.60 | 1.44 × 10−4 |
| SM 15:2;2O/26:6 | 787.67 | 10.77 | C46H79N2O6P | +2.59 | 1.38 × 10−2 |
| SM 18:1;2O/24:0 | 785.66 | 12.87 | C47H95N2O6P | +1.57 | 4.55 × 10−6 |
| SM 23:3;2O/20:4 | 801.69 | 13.39 | C48H85N2O6P | +1.77 | 1.26 × 10−7 |
| Metabolite Name | m/z | RT (min) | Chemical Formula | Fold Change * | p-Value |
|---|---|---|---|---|---|
| L-Phenylalanine | 166.08 | 2.8 | C9H11NO2 | +1.7254 | 0.0019 |
| Hexanoylglycine | 174.11 | 6.7 | C8H15NO3 | −1.5767 | 0.0477 |
| L-Carnitine | 184.09 | 0.8 | C7H15NO3 | −2.3805 | 0.0006 |
| Tryptophan | 227.07 | 4.5 | C11H12N2O2 | +1.8593 | 0.0181 |
| Hexanoylcarnitine | 260.18 | 6.3 | C13H25NO4 | −4.0906 | 0.0069 |
| Octadecenoylcarnitine | 426.35 | 10.88 | C25H47NO4 | −1.5700 | 0.0033 |
| Metabolite Name | m/z | RT (min) | Chemical Formula | Fold Change * | p-Value |
|---|---|---|---|---|---|
| L-Phenylalanine | 166.08 | 2.8 | C9H11NO2 | −1.4643 | 0.0307 |
| Hexanoylglycine | 174.11 | 6.7 | C8H15NO3 | +1.8401 | 0.0002 |
| L-Carnitine | 184.09 | 0.8 | C7H15NO3 | −2.8127 | 0.0354 |
| Hexanoylcarnitine | 260.18 | 6.3 | C13H25NO4 | +2.0028 | 0.0495 |
| Glu-Ile | 261.14 | 5.3 | C11H20N2O5 | −2.3478 | 0.0006 |
| 21-Deoxycortisol | 347.22 | 8.8 | C21H30O4 | +2.1645 | 0.0057 |
| Octadecenoylcarnitine | 426.35 | 10.88 | C25H47NO4 | +2.0037 | 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Zang, X.; Lv, J.; Zhang, Y.; Lv, Z.; Yu, M. Changes in Lipidomics, Metabolomics, and the Gut Microbiota in CDAA-Induced NAFLD Mice after Polyene Phosphatidylcholine Treatment. Int. J. Mol. Sci. 2023, 24, 1502. https://doi.org/10.3390/ijms24021502
Zhang J, Zang X, Lv J, Zhang Y, Lv Z, Yu M. Changes in Lipidomics, Metabolomics, and the Gut Microbiota in CDAA-Induced NAFLD Mice after Polyene Phosphatidylcholine Treatment. International Journal of Molecular Sciences. 2023; 24(2):1502. https://doi.org/10.3390/ijms24021502
Chicago/Turabian StyleZhang, Jiayuan, Xiaoling Zang, Jinxiao Lv, Yicong Zhang, Zhihua Lv, and Mingming Yu. 2023. "Changes in Lipidomics, Metabolomics, and the Gut Microbiota in CDAA-Induced NAFLD Mice after Polyene Phosphatidylcholine Treatment" International Journal of Molecular Sciences 24, no. 2: 1502. https://doi.org/10.3390/ijms24021502