
Post-Transcriptional Modification by Alternative Splicing and Pathogenic Splicing Variants in Cardiovascular Development and Congenital Heart Defects


Abstract
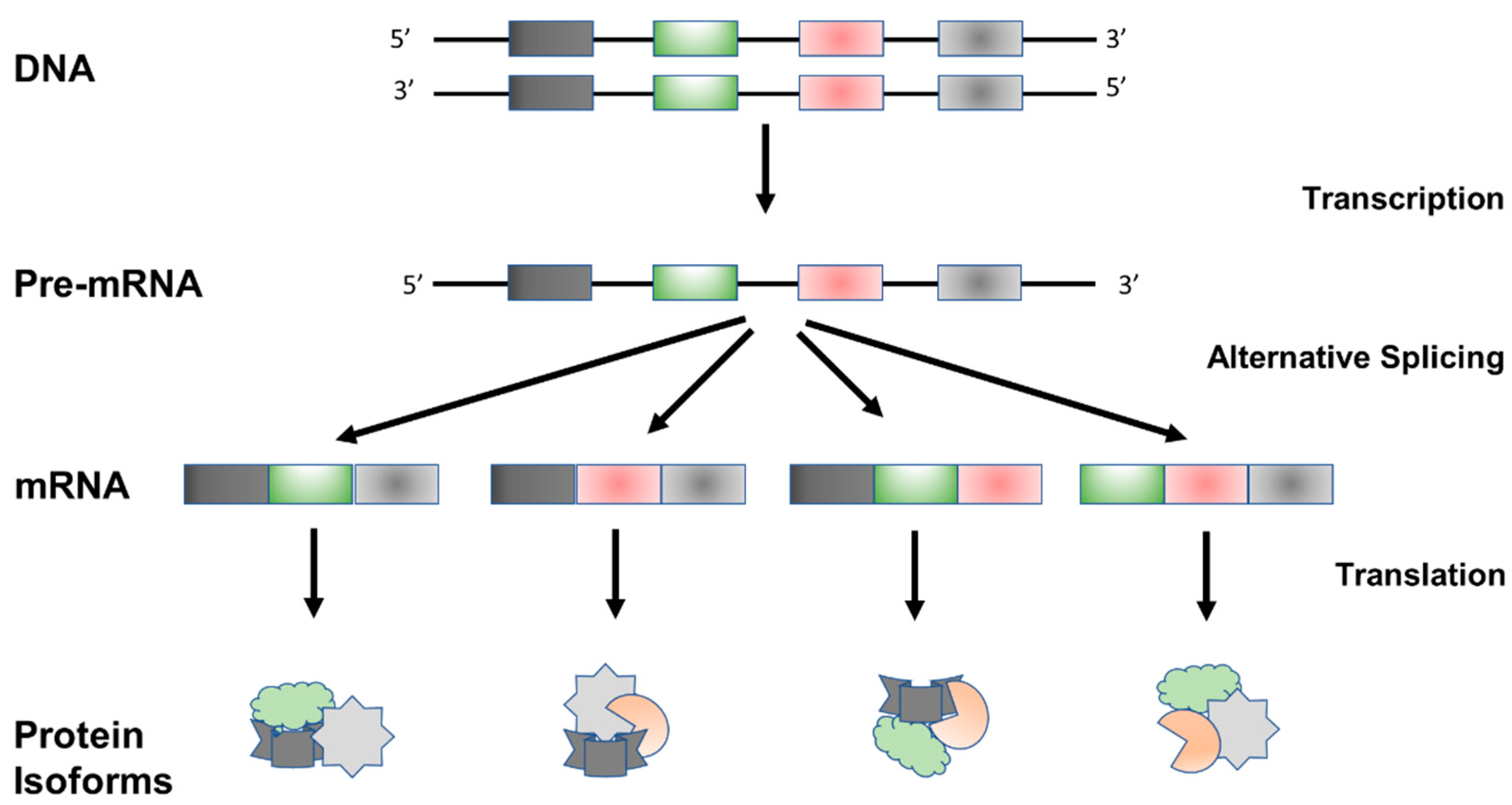
:1. Introduction
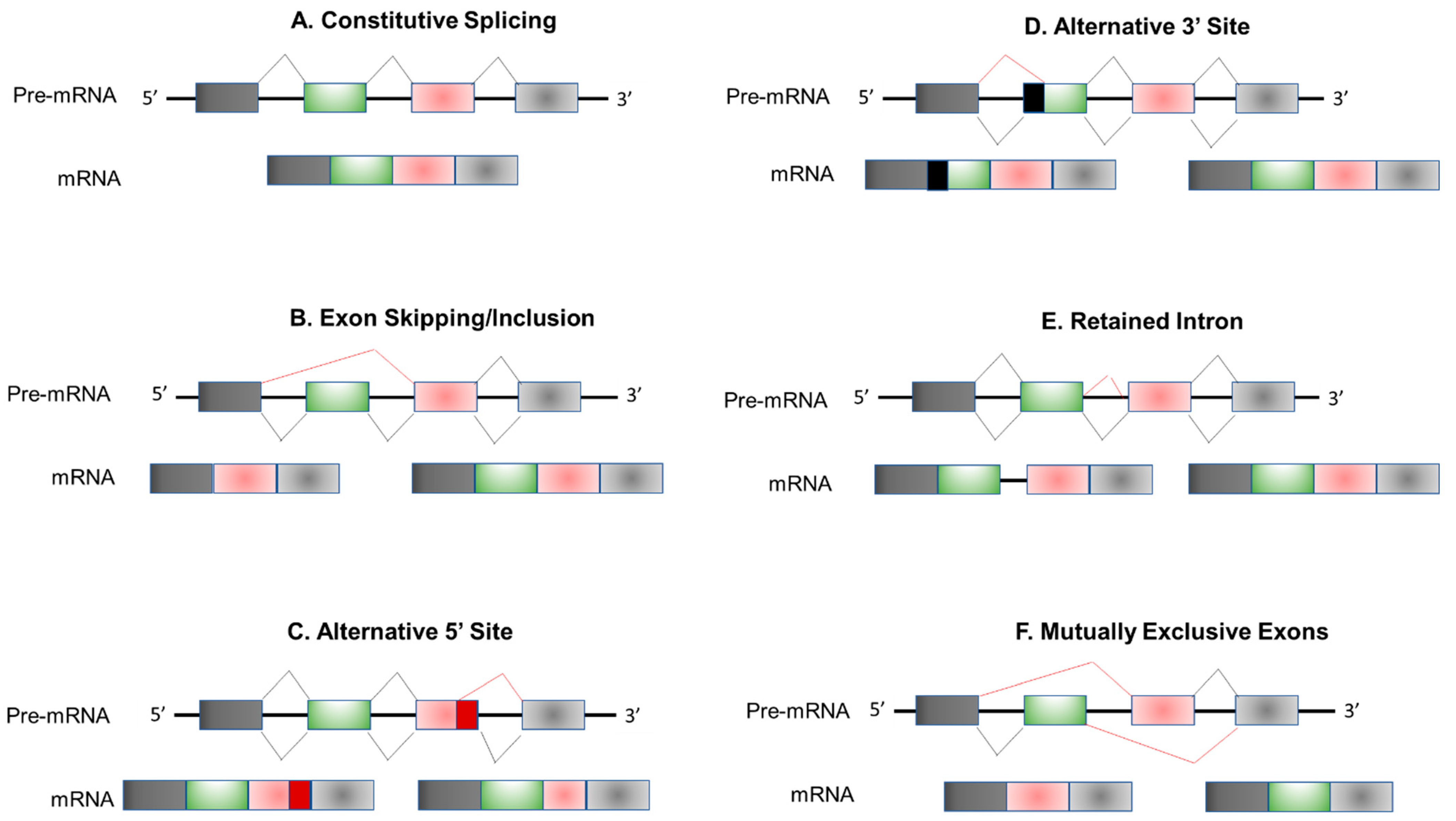
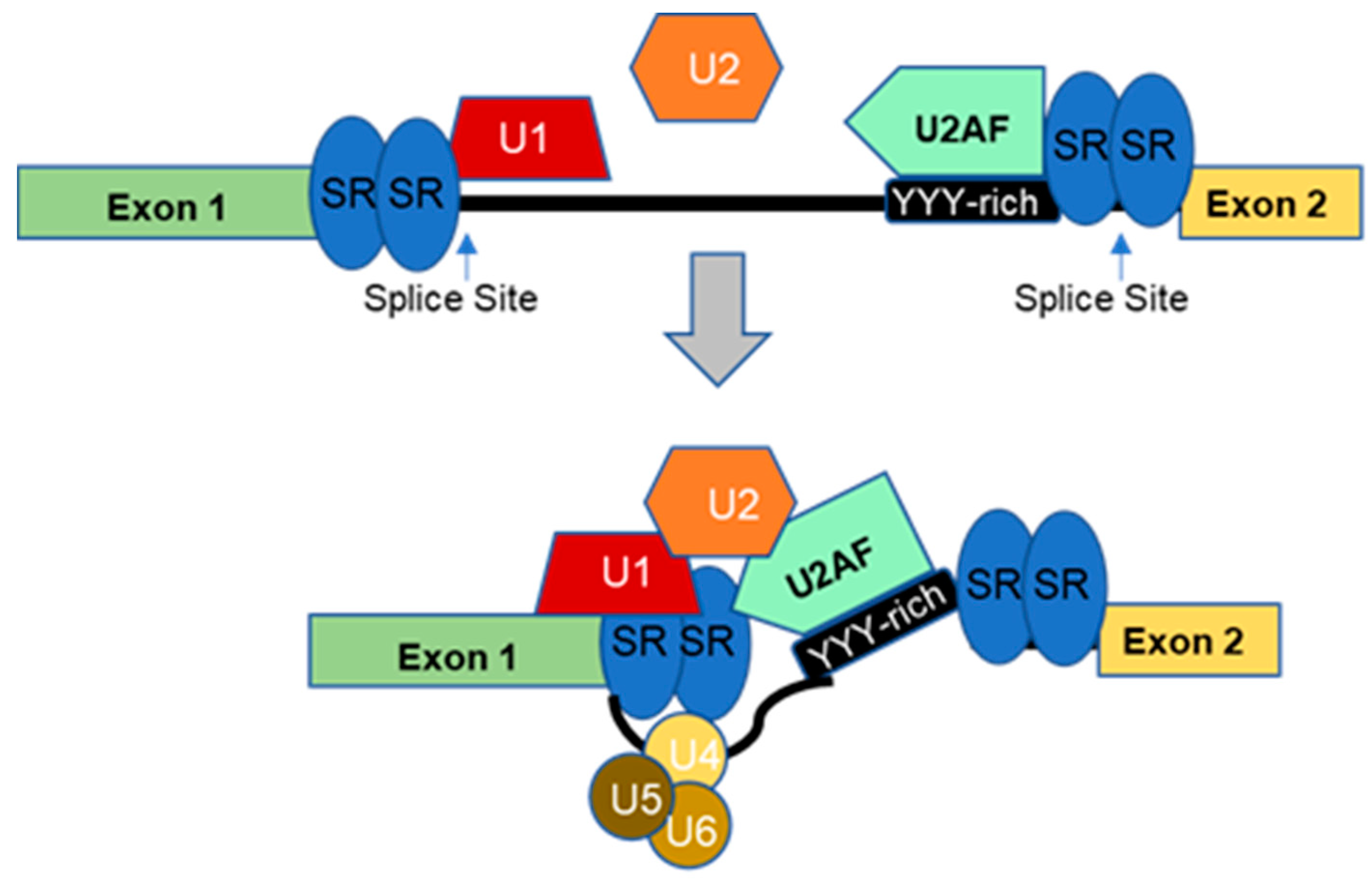
2. Regulation of Alternative Splicing
3. Alternative Splicing Transition during Heart Development
4. Dysregulated Alternative Splicing in Congenital Heart Defects (CHDs)
4.1. Splicing Transition in CHDs
4.2. Role of Pathogenic Variants in RBPs in CHDs
4.3. Role of Splicing Site Variants in CHDs
4.4. Splicing Variants Leading to Congenital Conduction Defects (Arrhythmias)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ARVD | Arrhythmogenic Right Ventricular Dysplasia |
| BAV | Bicuspid Aortic Valve |
| CHDs | Congenital Heart Defects |
| E–C | Excitation–Contraction |
| HLHS | Hypoplastic Left Heart Syndrome |
| LVNC | Left Ventricular Noncompaction |
| RBPs | RNA-Binding Proteins |
| RBPMS | RNA-Binding Protein with Multiple Splicing |
| RVOT | Right Ventricular Outflow Tract |
| TAV | Tricuspid Aortic Valve |
| TOF | Tetralogy of Fallot |
References
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell. 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, Y.; Li, X.; Chen, G.; Zhong, L.; Chen, G.; Liao, Y.; Liao, W.; Bin, J. Genome-wide analysis of alternative splicing during human heart development. Sci. Rep. 2016, 6, 35520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell. 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Kastner, B.; Will, C.L.; Stark, H.; Lührmann, R. Structural Insights into Nuclear pre-mRNA Splicing in Higher Eukaryotes. Cold Spring Harb. Perspect. Biol. 2019, 11, a032417. [Google Scholar] [CrossRef]
- Plaschka, C.; Lin, P.C.; Nagai, K. Structure of a pre-catalytic spliceosome. Nature 2017, 546, 617–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Wan, R.; Shi, Y. Molecular Mechanisms of pre-mRNA Splicing through Structural Biology of the Spliceosome. Cold Spring Harb. Perspect. Biol. 2019, 11, a032409. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhan, X.; Yan, C.; Zhang, W.; Liu, D.; Lei, J.; Shi, Y. Structures of the human spliceosomes before and after release of the ligated exon. Cell Res. 2019, 29, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Manning, K.S.; Cooper, T.A. The roles of RNA processing in translating genotype to phenotype. Nat. Rev. Mol. Cell Biol. 2017, 18, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Rimoldi, V.; Guella, I.; Soldà, G.; De Cristofaro, R.; Peyvandi, F.; Duga, S. Molecular characterization of in-frame and out-of-frame alternative splicings in coagulation factor XI pre-mRNA. Blood 2010, 115, 2065–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, J.; Huang, B.; Xu, Y.; Li, J.; Huang, L.; Lin, J.; Zhang, J.; Min, Q.-H.; Yang, W.-M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazin, P.V.; Khaitovich, P.; Cardoso-Moreira, M.; Kaessmann, H. Alternative splicing during mammalian organ development. Nat. Genet. 2021, 53, 925–934. [Google Scholar] [CrossRef]
- House, A.E.; Lynch, K.W. Regulation of alternative splicing: More than just the ABCs. J. Biol. Chem. 2008, 283, 1217–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Eldridge, A.G.; Li, Y.; Sharp, P.A.; Blencowe, B.J. The SRm160/300 splicing coactivator is required for exon-enhancer function. Proc. Natl. Acad. Sci. USA 1999, 96, 6125–6130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Biamonti, G.; Infantino, L.; Gaglio, D.; Amato, A. An Intricate Connection between Alternative Splicing and Phenotypic Plasticity in Development and Cancer. Cells 2019, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Cereda, M.; Pozzoli, U.; Rot, G.; Juvan, P.; Schweitzer, A.; Clark, T.; Ule, J. RNAmotifs: Prediction of multivalent RNA motifs that control alternative splicing. Genome Biol. 2014, 15, R20. [Google Scholar] [CrossRef]
- Gonatopoulos-Pournatzis, T.; Wu, M.; Braunschweig, U.; Roth, J.; Han, H.; Andrew, J.; Best, A.J.; Raj, B.; Aregger, M.; O’Hanlon, D.; et al. Genome-wide CRISPR-Cas9 Interrogation of Splicing Networks Reveals a Mechanism for Recognition of Autism-Misregulated Neuronal Microexons. Mol. Cell. 2018, 72, 510–524.e12. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, C.; Bangru, S.; Lin, F.; Lam, K.; Koenig, S.N.; Lubbers, E.R.; Hedhli, J.; Murphy, N.P.; Parker, D.J.; Dobrucki, L.W.; et al. Aberrant expression of a non-muscle RBFOX2 isoform triggers cardiac conduction defects in myotonic dystrophy. Dev. Cell. 2020, 52, 748–763.e6. [Google Scholar] [CrossRef]
- Anvar, S.Y.; Allard, G.; Tseng, E.; Sheynkman, G.M.; de Klerk, E.; Vermaat, M.; Yin, R.H.; Johansson, H.E.; Ariyurek, Y.; Dunnen, J.T.D.; et al. Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing. Genome Biol. 2018, 19, 46. [Google Scholar] [CrossRef] [Green Version]
- Ding, F.; Elowitz, M.B. Constitutive splicing and economies of scale in gene expression. Nat. Struct. Mol. Biol. 2019, 26, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Fiszbein, A.; Krick, K.S.; Begg, B.E.; Burge, C.B. Exon-Mediated Activation of Transcription Starts. Cell 2019, 179, 1551–1565.e17. [Google Scholar] [CrossRef]
- Manipur, I.; Granata, I.; Guarracino, M.R. Exploiting single-cell RNA sequencing data to link alternative splicing and cancer heterogeneity: A computational approach. Int. J. Biochem. Cell Biol. 2019, 108, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Lukacsovich, D.; Winterer, J.; Que, L.; Luo, W.; Lukacsovich, T.; Foldy, C. Single-Cell RNA-Seq Reveals Developmental Origins and Ontogenetic Stability of Neurexin Alternative Splicing Profiles. Cell Rep. 2019, 27, 3752–3759.e4. [Google Scholar] [CrossRef] [Green Version]
- Boti, M.A.; Adamopoulos, P.G.; Tsiakanikas, P.; Scorilas, A. Nanopore Sequencing Unveils Diverse Transcript Variants of the Epithelial Cell-Specific Transcription Factor Elf-3 in Human Malignancies. Genes 2021, 12, 839. [Google Scholar] [CrossRef]
- D’Antonio, M.; Nguyen, J.P.; Arthur, T.D.; Matsui, H.; Donovan, M.K.R.; D’Antonio-Chronowska, A.; Frazer, K.A. In heart failure reactivation of RNA-binding proteins is associated with the expression of 1,523 fetal-specific isoforms. PLoS Comput. Biol. 2022, 18, e1009918. [Google Scholar] [CrossRef] [PubMed]
- Bonnal, S.; López-Oreja Valcárcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Titus, M.B.; Wright, E.G.; Bono, J.M.; Poliakon, A.K.; Goldstein, B.R.; Super, M.K.; Young, L.A.; Manaj, M.; Litchford, M.; Reist, N.E.; et al. The conserved alternative splicing factor caper regulates neuromuscular phenotypes during development and aging. Dev. Biol. 2021, 473, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kimura, A.; Kuroyanagi, H. Alternative Splicing Regulator RBM20 and Cardiomyopathy. Front. Mol. Biosci. 2018, 5, 105. [Google Scholar] [CrossRef]
- Joly, A.L.; Andersson, J. Alternative splicing, FOXP3 and cardiovascular disease. Aging 2019, 11, 1905–1906. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Xu, C.; Ba, L.; Liu, Z.; Li, X.; Huang, J.; Simpson, E.; Gao, H.; Cao, D.; et al. QKI is a critical pre-mRNA alternative splicing regulator of cardiac myofibrillogenesis and contractile function. Nat. Commun. 2021, 12, 89. [Google Scholar] [CrossRef]
- Hasimbegovic, E.; Schweiger, V.; Kastner, N.; Spannbauer, A.; Traxler, D.; Lukovic, D.; Gyöngyösi, M.; Mester-Tonczar, J. Alternative Splicing in Cardiovascular Disease-A Survey of Recent Findings. Genes 2021, 12, 1457. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Wang, J.; Peng, J.; Zhou, Y.; Zhou, S.; Song, W.; Chen, S.; Wang, Q.; Bai, K.; Sun, K. Dynamic transcriptome profiling toward understanding the development of the human embryonic heart during different Carnegie stages. FEBS Lett. 2020, 594, 4307–4319. [Google Scholar] [CrossRef] [PubMed]
- Touma, M.; Kang, X.; Gao, F.; Zhao, Y.; Cass, A.; Biniwale, R.; Xiao, X.; Coppola, G.; Reemtsen, B.; Wang, Y. Wnt11 Regulates Neonatal Cardiac Chamber Development During Perinatal Maturation. JCI Insight 2017, 2, e94904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Lin, Y.; Liu, J.; Zhang, Z.G.; Fu, W.; Guo, L.Y.; Pan, L.; Kong, X.; Zhang, M.K.; Lu, Y.H.; et al. Rbm24 Regulates Alternative Splicing Switch in Embryonic Stem Cell Cardiac Lineage Differentiation. Stem Cells 2016, 34, 1776–1789. [Google Scholar] [CrossRef] [Green Version]
- Gabut, M.; Samavarchi-Tehrani, P.; Wang, X.; Slobodeniuc, V.; O’Hanlon, D.; Sung, H.K.; Alvarez, M.; Talukder, S.; Pan, Q.; Mazzoni, E.O.; et al. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 2011, 147, 132–146. [Google Scholar] [CrossRef]
- van den Hoogenhof, M.M.; Pinto, Y.M.; Creemers, E.E. RNA Splicing: Regulation and Dysregulation in the Heart. Circ. Res. 2016, 118, 454–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chothani, S.; Schäfer, S.; Adami, E.; Viswanathan, S.; Widjaja, A.A.; Langley, S.R.; Tan, J.; Wang, M.; Quaife, N.M.; Pua, C.J.; et al. Widespread Translational Control of Fibrosis in the Human Heart by RNA-Binding Proteins. Circulation 2019, 140, 937–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.X.; Mittman, S.; Colecraft, H.M. Distinctive modulatory effects of five human auxiliary beta2 subunit splice variants on L-type calcium channel gating. Biophys. J. 2003, 84, 3007–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooding, C.; Smith, C.W. Tropomyosin exons as models for alternative splicing. Adv. Exp. Med. Biol. 2008, 644, 27–42. [Google Scholar] [CrossRef]
- Cooper, T.A.; Ordahl, C.P. A single cardiac troponin T gene generates embryonic and adult isoforms via developmentally regulated alternate splicing. J. Biol. Chem. 1985, 260, 11140–11148. [Google Scholar] [CrossRef]
- McAuliffe, J.J.; Gao, L.Z.; Solaro, R.J. Changes in myofibrillar activation and troponin C Ca2+ binding associated with troponin T isoform switching in developing rabbit heart. Circ. Res. 1990, 66, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudice, J.; Xia, Z.; Wang, E.T.; Scavuzzo, M.A.; Ward, A.J.; Kalsotra, A.; Wang, W.; Wehrens, X.H.; Burge, C.B.; Li, W.; et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun. 2014, 5, 3603. [Google Scholar] [CrossRef] [Green Version]
- Martí-Gómez, C.; Larrasa-Alonso, J.; López-Olañeta, M.; Villalba-Orero, M.; García-Pavía, P.; Sánchez-Cabo, F.; Lara-Pezzi, E. Functional Impact and Regulation of Alternative Splicing in Mouse Heart Development and Disease. J. Cardiovasc. Transl. Res. 2022, 15, 1239–1255. [Google Scholar] [CrossRef]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar] [CrossRef] [Green Version]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Colak, R.; et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef]
- Ellis, J.D.; Barrios-Rodiles, M.; Colak, R.; Irimia, M.; Kim, T.; Calarco, J.A.; Wang, X.; Pan, Q.; O’Hanlon, D.; Kim, P.M.; et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol. Cell. 2012, 46, 884–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillman, A.A.; Hauser, D.N.; Gibbs, J.R.; Nalls, M.A.; McCoy, M.K.; Rudenko, I.N.; Galter, D.; Cookson, M.R. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat. Neurosci. 2013, 16, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Trabzuni, D.; Ramasamy, A.; Imran, S.; Walker, R.; Smith, C.; Weale, M.E.; Hardy, J.; Ryten, M.; North American Brain Expression Consortium. Widespread sex differences in gene expression and splicing in the adult human brain. Nat. Commun. 2013, 4, 2771. [Google Scholar] [CrossRef] [Green Version]
- Blech-Hermoni, Y.; Ladd, A.N. RNA binding proteins in the regulation of heart development. Int. J. Biochem. Cell Biol. 2013, 45, 2467–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudice, J.; Cooper, T.A. RNA-binding proteins in heart development. Adv. Exp. Med. Biol. 2014, 825, 389–429. [Google Scholar] [CrossRef]
- Ladd, A.N. New Insights Into the Role of RNA-Binding Proteins in the Regulation of Heart Development. Int. Rev. Cell Mol. Biol. 2016, 324, 125–185. [Google Scholar] [CrossRef] [PubMed]
- Blech-Hermoni, Y.; Sullivan, C.B.; Jenkins, M.W.; Wessely, O.; Ladd, A.N. CUG-BP, Elav-like family member 1 (CELF1) is required for normal myofibrillogenesis, morphogenesis, and contractile function in the embryonic heart. Dev. Dyn. 2016, 245, 854–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Ren, S.; Lee, J.H.; Qiu, J.; Chapski, D.J.; Rau, C.D.; Zhou, Y.; Abdellatif, M.; Nakano, A.; Vondriska, T.M.; et al. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J. Clin. Investig. 2016, 126, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.K.; Deshmukh, V.; Thatcher, K.; Belanger, K.K.; Rhyner, A.M.; Meng, S.; Holcomb, R.J.; Bressan, M.; Martin, J.F.; Cooke, J.P.; et al. RBFOX2 is required for establishing RNA regulatory networks essential for heart development. Nucleic Acids Res. 2022, 50, 2270–2286. [Google Scholar] [CrossRef]
- Wang, E.T.; Cody, N.A.L.; Joy, S.; Biancolella, M.; Wang, T.T.; Treasy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 2012, 150, 710–724. [Google Scholar] [CrossRef]
- Yang, J.; Hung, L.H.; Licht, T.; Kostin, S.; Looso, M.; Khrameeva, E.; Bindereif, A.; Schneider, S.; Braun, T. RBM24 is a major regulator of muscle-specific alternative splicing. Dev. Cell. 2014, 31, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeland, C.J.; van den Hoogenhof, M.M.; Beqqali, A.; Creemers, E.E. Insights into alternative splicing of sarcomeric genes in the heart. J. Mol. Cell Cardiol. 2015, 81, 107–113. [Google Scholar] [CrossRef]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.; Howell, N.; Pagano, D.; Andreka, P.; Vertesaljai, M.; Pecor, T.; Frenneaux, M.; Granzier, H. Titin isoform expression in aortic stenosis. Clin. Sci. 2009, 117, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Poon, K.L.; Tan, K.T.; Wei, Y.Y.; Ng, C.P.; Colman, A.; Korzh, V.; Xu, X.Q. RNA-binding protein RBM24 is required for sarcomere assembly and heart contractility. Cardiovasc. Res. 2012, 94, 418–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, P.; Wang, Z.; Morales, M.G.; Zhang, Y.; Bassel-Duby, R.; Liu, N.; Olson, E.N. RBPMS is an RNA-binding protein that mediates cardiomyocyte binucleation and cardiovascular development. Dev. Cell. 2022, 57, 959–973.e7. [Google Scholar] [CrossRef]
- Verma, S.K.; Deshmukh, V.; Nutter, C.A.; Jaworski, E.; Jin, W.; Wadhwa, L.; Abata, J.; Ricci, M.; Lincoln, J.; Martin, J.F.; et al. Rbfox2 function in RNA metabolism is impaired in hypoplastic left heart syndrome patient hearts. Sci. Rep. 2016, 6, 30896. [Google Scholar] [CrossRef]
- van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RMB20 Mutations Induce an Arrhythmognic Dilated Cardiomyopathy Related to Disturbed Calcium Handing. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef]
- Gu, Q.; Jin, N.; Sheng, H.; Yin, X.; Zhu, J. Cyclic AMP-dependent protein kinase A regulates the alternative splicing of CaMKIIδ. PLoS ONE 2011, 6, e25745. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Tan, K.T.; Liu, J.; Kong, X.; Huang, Z.; Xu, X.Q. Global profiling of Rbm24 bound RNAs uncovers a multi-tasking RNA binding protein. Int. J. Biochem. Cell Biol. 2018, 94, 10–21. [Google Scholar] [CrossRef]
- Feng, Y.; Valley, M.T.; Lazar, J.; Yang, A.L.; Bronson, R.T.; Firestein, S.; Coetzee, W.A.; Manley, J.L. SRp38 regulates alternative splicing and is required for Ca (2+) handling in the embryonic heart. Dev. Cell. 2009, 16, 528–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Yang, D.; Ding, J.H.; Wang, W.; Chu, P.-H.; Dalton, N.D.; Wang, H.-Y.; Bermingham, J.R.; Ye, Z.; Liu, F.; et al. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 2005, 120, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Paloschi, V.; Kurtovic, S.; Folkersen, L.; Gomez, D.; Wågsäter, D.; Roy, J.; Petrini, J.; Eriksson, M.J.; Caidahl, K.; Hamsten, A.; et al. Impaired splicing of fibronectin is associated with thoracic aortic aneurysm formation in patients with bicuspid aortic valve. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Ricci, M.; Xu, Y.; Hammond, H.L.; Willoughby, D.A.; Nathanson, L.; Rodriguez, M.M.; Vatta, M.; Lipshultz, S.E.; Lincoln, J. Myocardial alternative RNA splicing and gene expression profiling in early stage hypoplastic left heart syndrome. PLoS ONE 2012, 7, e29784. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, C.K.; Kibiryeva, N.; Marshall, J.E.; Bittel, D.C. scaRNA1 Levels Alter Pseudouridylation in Spliceosomal RNA U2 Affecting Alternative mRNA Splicing and Embryonic Development. Pediatr. Cardiol. 2020, 41, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Farazi, T.A.; Leonhardt, C.S.; Mukherjee, N.; Mihailovic, A.; Li, S.; Max, K.E.; Meyer, C.; Yamaji, M.; Cekan, P.; Jacobs, N.C.; et al. Identification of the RNA recognition element of the RBPMS family of RNA-binding proteins and their transcriptome-wide mRNA targets. RNA 2014, 20, 1090–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzuti, A.; Novelli, G.; Ratti, A.; Amati, F.; Bordoni, R.; Mandich, P.; Bellone, E.; Conti, E.; Bengala, M.; Mari, A.; et al. Isolation and characterization of a novel transcript embedded within HIRA, a gene deleted in DiGeorge syndrome. Mol. Genet. Metab. 1999, 67, 227–235. [Google Scholar] [CrossRef]
- Wang, Y.; Gogol-Döring, A.; Hu, H.; Fröhler, S.; Ma, Y.; Jens, M.; Maaskola, J. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol. Med. 2013, 5, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Tessier, S.J.; Loiselle, J.J.; McBain, A.; Pullen, C.; Koenderink, B.W.; Roy, J.G.; Sutherland, L.C. Insight into the role of alternative splicing within the RBM10v1 exon 10 tandem donor site. BMC Res. Notes 2015, 8, 46. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.J.; Teer, J.K.; Cherukuri, P.F.; Hansen, N.F.; Loftus, S.K.; NIH Intramural Sequencing Center (NISC); Chong, K.; Mullikin, J.C.; Biesecker, L.G. Massively Parallel Sequencing of Exons on the X Chromosome Identifies RBM10 as the Gene that Causes a Syndromic Form of Cleft Palate. Am. J. Hum. Genet. 2010, 86, 743–748. [Google Scholar] [CrossRef]
- Morris, S.A.; Ethen, M.K.; Penny, D.J.; Canfield, M.A.; Minard, C.G.; Fixler, D.E.; Nembhard, W.N. Prenatal diagnosis, birth location, surgical center, and neonatal mortality in infants with hypoplastic left heart syndrome. Circulation 2014, 129, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beqqali, A.; Bollen, I.A.; Rasmussen, T.B.; van den Hoogenhof, M.M.; van Deutekom, H.W.; Schafer, S.; Haas, J.; Meder, B.; Sørensen, K.E.; van Oort, R.J.; et al. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc Res. 2016, 112, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Chen, Q.; Wang, Q.K. Functional role of transcriptional factor TBX5 in pre-mRNA splicing and Holt-Oram syndrome via association with SC35. J. Biol. Chem. 2009, 284, 25653–25663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, D.; Vaigundan, D.; Shetty, M.; Krishnappa, J.; Kutty, A.V.M. Identification of intronic-splice site mutations in GATA4 gene in Indian patients with congenital heart disease. Mutat. Res. 2017, 803–805, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shi, L.; Xu, M.; Zheng, X.; Yu, Y.; Jin, J. RCAN1 Mutation and Functional Characterization in Children with Sporadic Congenital Heart Disease. Pediatr. Cardiol. 2018, 39, 226–235. [Google Scholar] [CrossRef]
- Fusco, C.; Morlino, S.; Micale, L.; Ferraris, A.; Grammatico, P.; Castori, M. Characterization of Two Novel Intronic Variants Affecting Splicing in FBN1-Related Disorders. Genes 2019, 10, 442. [Google Scholar] [CrossRef] [Green Version]
- Gong, Q.; Stump, M.R.; Deng, V.; Zhang, L.; Zhou, Z. Identification of Kv11.1 isoform switch as a novel pathogenic mechanism of long-QT syndrome. Circ. Cardiovasc. Genet. 2014, 7, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Awad, M.M.; Dalal, D.; Tichnell, C.; James, C.; Tucker, A.; Abraham, T.; Spevak, P.J.; Calkins, H.; Judge, D.P. Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum. Mutat. 2006, 27, 1157. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| RNA Binding Protein | Main Target(s) | CHD/Condition | References |
|---|---|---|---|
| RBFOX1 | MEF2 | Heart Failure/Fetal-like program | [44] |
| RBM24 | LDB4, CAMKIIδ, TPM, MyoM | Sarcomerogenesis | [51,54,55,56] |
| RBM20 | TTN | Ventricular Elasticity | [52,53] |
| CELF1 | Fetal-like program | Heart Development/ Myofibrinogenesis | [57,64,65] |
| MBNL1 | Fetal-like Program | Heart Development | [57,64,65] |
| SRp38 | Triadin | Excitation-Contraction Coupling | [62] |
| ASF/SF2 | CAMKIIδ | Excitation-Contraction Coupling | [63] |
| RBpms | Pdlim5 | Left Ventricle Noncompaction | [66] |
| RBFOX2 | Rho GTPases | Hypoplastic Left Ventricle | [67] |
| TBX5/SC35 | RNF | Holt-Oram Syndrome | [68] |
| Alternative Splicing in Congenital Heart Disease |
|---|
| RNA Binding Protein Genes |
| RBM10, RBM20, RBM24, RBFOX1, RBFOX2, SC35, SFB31, ASF/SF2, RBFOX2, RBpms, PUM1, CELF1, MBNL1, SRp38 |
| Splice Variants/Cardiac Transcription Regulators |
| TBX5, GATA4, HAY2 RCAN1 |
| Splice Variants/Cardiac Conduction Genes |
| PKP2, KV11.1 |
| Cardiac Structure/Sarcomere Genes |
| FBN1, TTN, TNNT2, TPM1, MyoM, LDB3 |
| Reference Citation | Affected Gene | Variant | Phenotypes |
|---|---|---|---|
| [63] Guo et al. Nat Med (2012) 18 (5), 766-773 | RBM20 | S635A | Dilated Cardiomyopathy |
| [78] Wang Y et al. EMBO Mol Med (2013) 5,1431-1442 | RBM10 | Del of 1292nucleotides (ChrX: 46929367-46930658 bp) | TARP Syndrome/CHD |
| [79] Tessier et al. BMB Research Notes (2015) 8,46 | RBM10 | Tandem donor splice site (GTGGTG) in RBM10 exon 10 | TARP Syndrome/CHD |
| [80] Johnston JJ et al. The American Journal of Human Genetics (2010) 86,743-748 | RBM10 | c.1235G>A; c.1893_1894insA | TARP Syndrome/CHD |
| [82] Beqqali A et al. Cardiovascular Research (2016) 112, 452-463 | RBM20 | c.2737G>A | Dilated Cardiomyopathy |
| [68] van den Hoogenhof et al. Circulation (2018) 138, 1330–1342. | RBM20 | Multiple Variants | Dilated Cardiomyopathy |
| [83] Fan C et al. The Journal of Biological Chemistry (2009) 284, 38, 25653-25663 | TBX5/SC35 | G80R | HOLT-Oram Syndrome/CHD |
| [84] Bose D. et al. Mutat Res Fund (2017) 803-805, 26-34 | GATA4 | g.83271C>A/M (intronic variant); g.86268A>R (intronic Variant) | Nonsyndromic ASD/VSD/AVSD |
| [85] Li X et al. Pediatr Cardiol (2018) 39, 226-235 | RCAN1 | g.482G>T (intronic variant) | Nonsyndromic ASD/VSD/AVSD |
| [86] Fusco C et al. Genes (2019) 10 (6), 442 | FBN1 | c.6872-24T>A; c. 7571-12T>A | Marfan’s Syndrome/CHD |
| [87] Gong Q et al. Circ Cardiovascular Gent (2014) 7 (4), 482-490 | KCNH2 | IVS9-2delA (a deletion of the A in AG dinucleotide of the 3’ acceptor site of KCNH2 intron 9) | Long QT Syndrome |
| [88] Awad MM et al. Human Mutat (2006) 27 (11), 1157. | PKP2 | c. 2484C>T + c.2484C>T | ARVD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, Z.; Touma, M. Post-Transcriptional Modification by Alternative Splicing and Pathogenic Splicing Variants in Cardiovascular Development and Congenital Heart Defects. Int. J. Mol. Sci. 2023, 24, 1555. https://doi.org/10.3390/ijms24021555
Mehta Z, Touma M. Post-Transcriptional Modification by Alternative Splicing and Pathogenic Splicing Variants in Cardiovascular Development and Congenital Heart Defects. International Journal of Molecular Sciences. 2023; 24(2):1555. https://doi.org/10.3390/ijms24021555
Chicago/Turabian StyleMehta, Zubin, and Marlin Touma. 2023. "Post-Transcriptional Modification by Alternative Splicing and Pathogenic Splicing Variants in Cardiovascular Development and Congenital Heart Defects" International Journal of Molecular Sciences 24, no. 2: 1555. https://doi.org/10.3390/ijms24021555
APA StyleMehta, Z., & Touma, M. (2023). Post-Transcriptional Modification by Alternative Splicing and Pathogenic Splicing Variants in Cardiovascular Development and Congenital Heart Defects. International Journal of Molecular Sciences, 24(2), 1555. https://doi.org/10.3390/ijms24021555