
Mining TCGA Database for Genes with Prognostic Value in Breast Cancer


Abstract
1. Introduction
2. Results
2.1. Demographics, Staging, and Tumor Subtypes
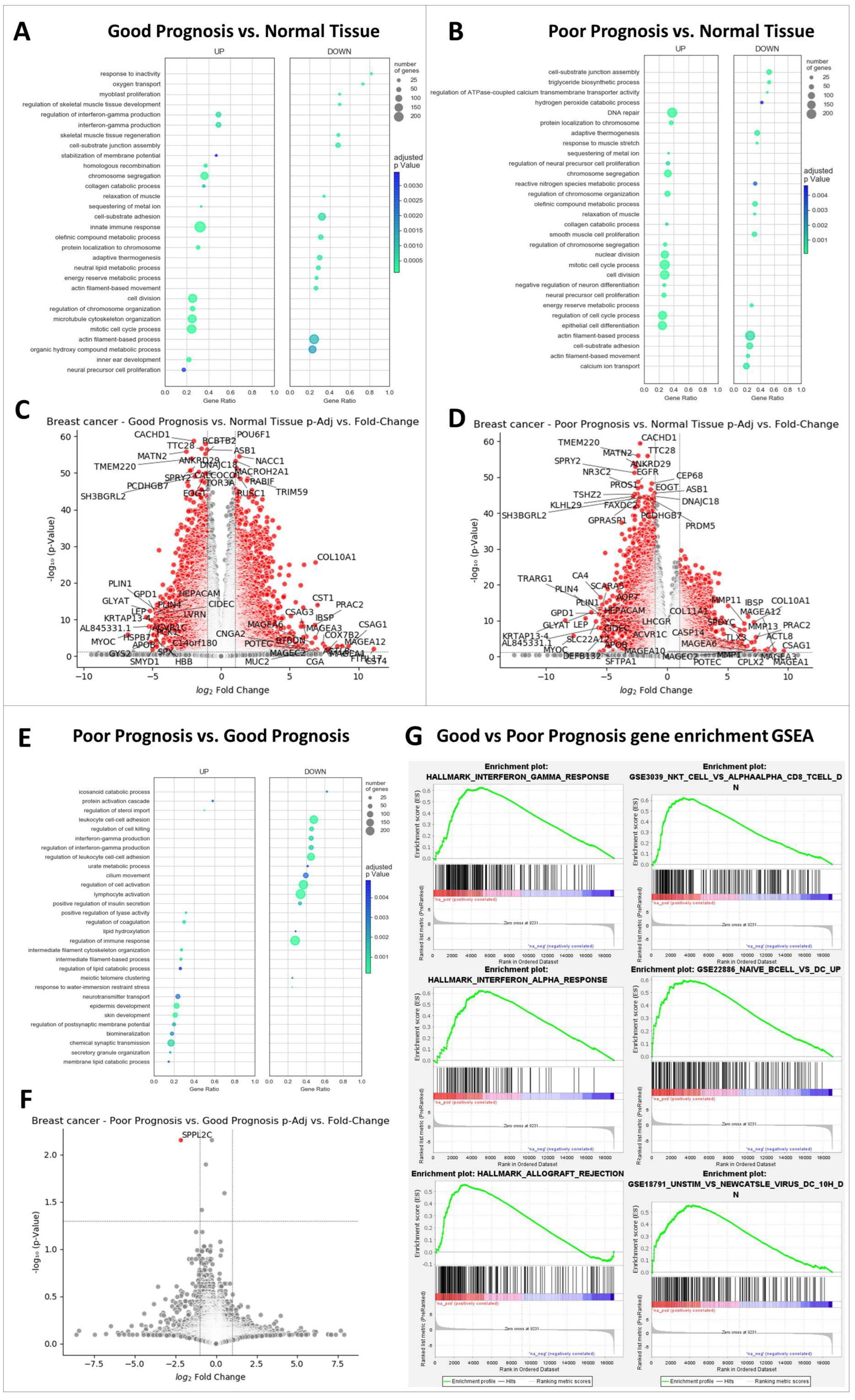
2.2. The Most Significant Differences between GP and PP Samples Are Interferon Gamma Signaling and Anti-Tumor Immune Response
2.3. Highly Expression of MAGEA Family Members, PRAC2, CSAG1, and COL10A Gene Profiles in Breast Cancer Samples
2.4. Gene Signature and Survival in Patients with Breast Cancer
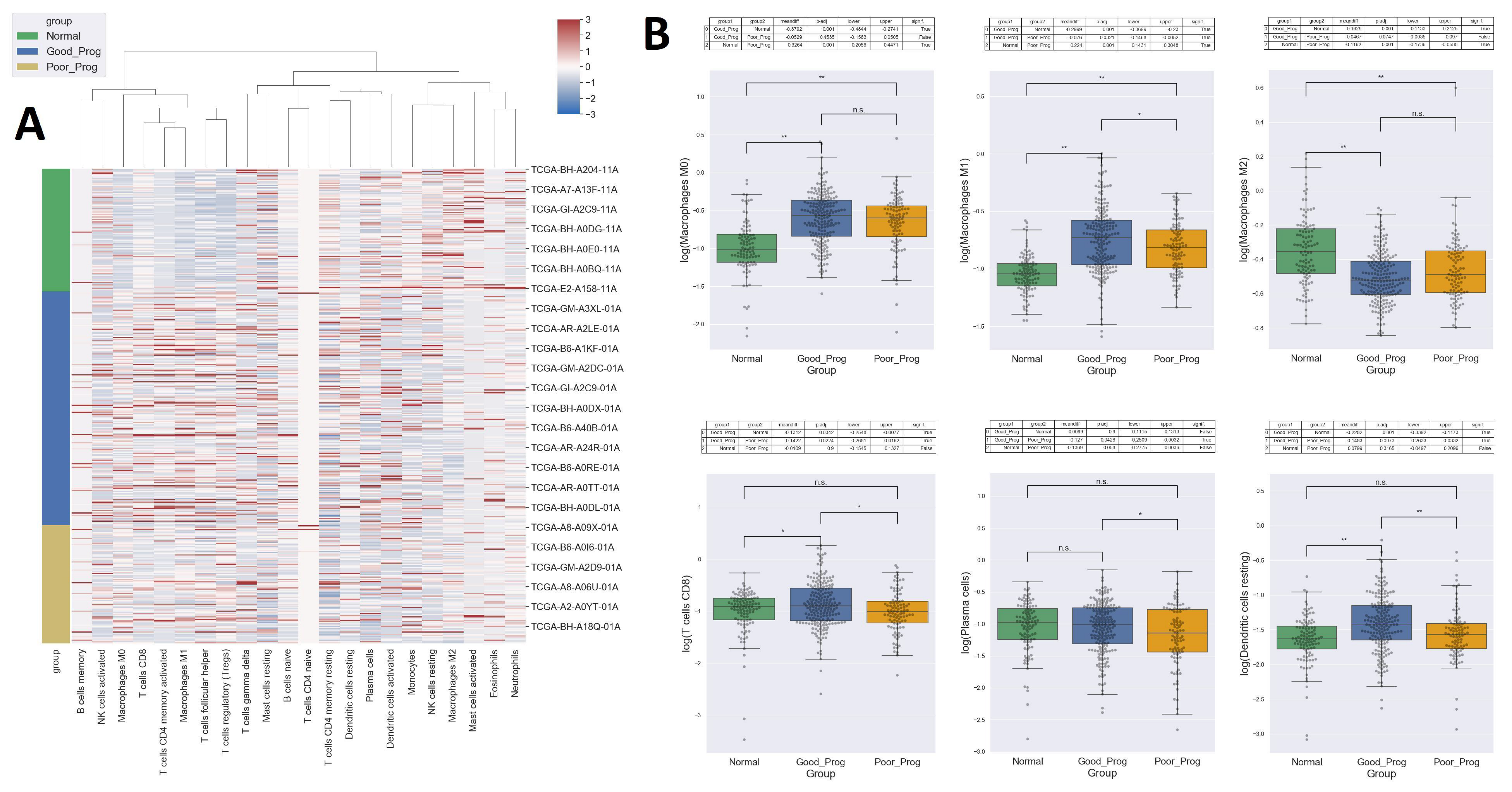
2.5. High Levels of Pro-Inflammatory Macrophages and Cytotoxic T Cells, While Lower Levels of Anti-Inflammatory Macrophages Are Found in Tumor Samples Compared to Normal Tissue
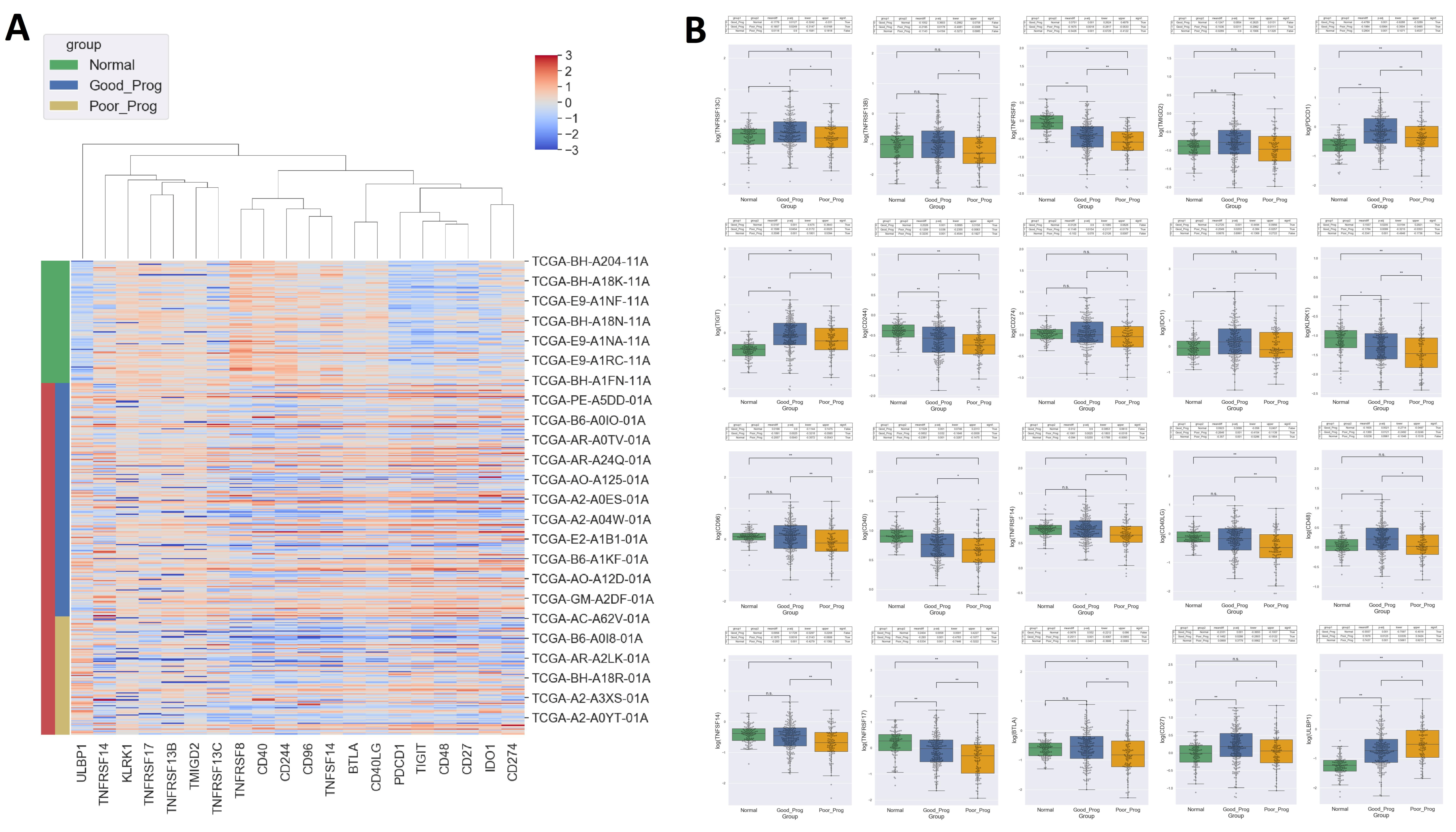
2.6. Identification of Immune Checkpoint Molecules Associated with Breast Cancer
- (i)
- 6 genes encoding for immune-inhibitors had lower values in PP: programmed cell death protein 1 (PDCD1), B and T lymphocytes associated protein (BTLA), T cell immunoreceptor with Ig and ITIM domains (TIGIT), indoleamine 2,3-dioxygenase 1 (IDO1), clusters of differentiation 96, 244 (CD96, CD244),
- (ii)
- 13 genes encoding for immune-stimulators had lower values in PP: clusters of differentiation 27, 48, 40, 40LG, 274 (CD27, CD48, CD40, CD40LG, CD274), killer cell lectin like receptor K1 (KLRK1), transmembrane and immunoglobulin domanin containing protein 2 (TMIGD2), TNF receptor superfamily members 8, 13B, 13C, 14, 17 (TNFRSF8, TNFRSF13B, TNFRSF13C, TNFRSF14, TNFRSF17), TNF superfamily member 14 (TNFSF14), and
- (iii)
- 1 gene encoding for an immune-stimulator had greater expression in PP: UL16 Binding Protein 1 (ULBP1).
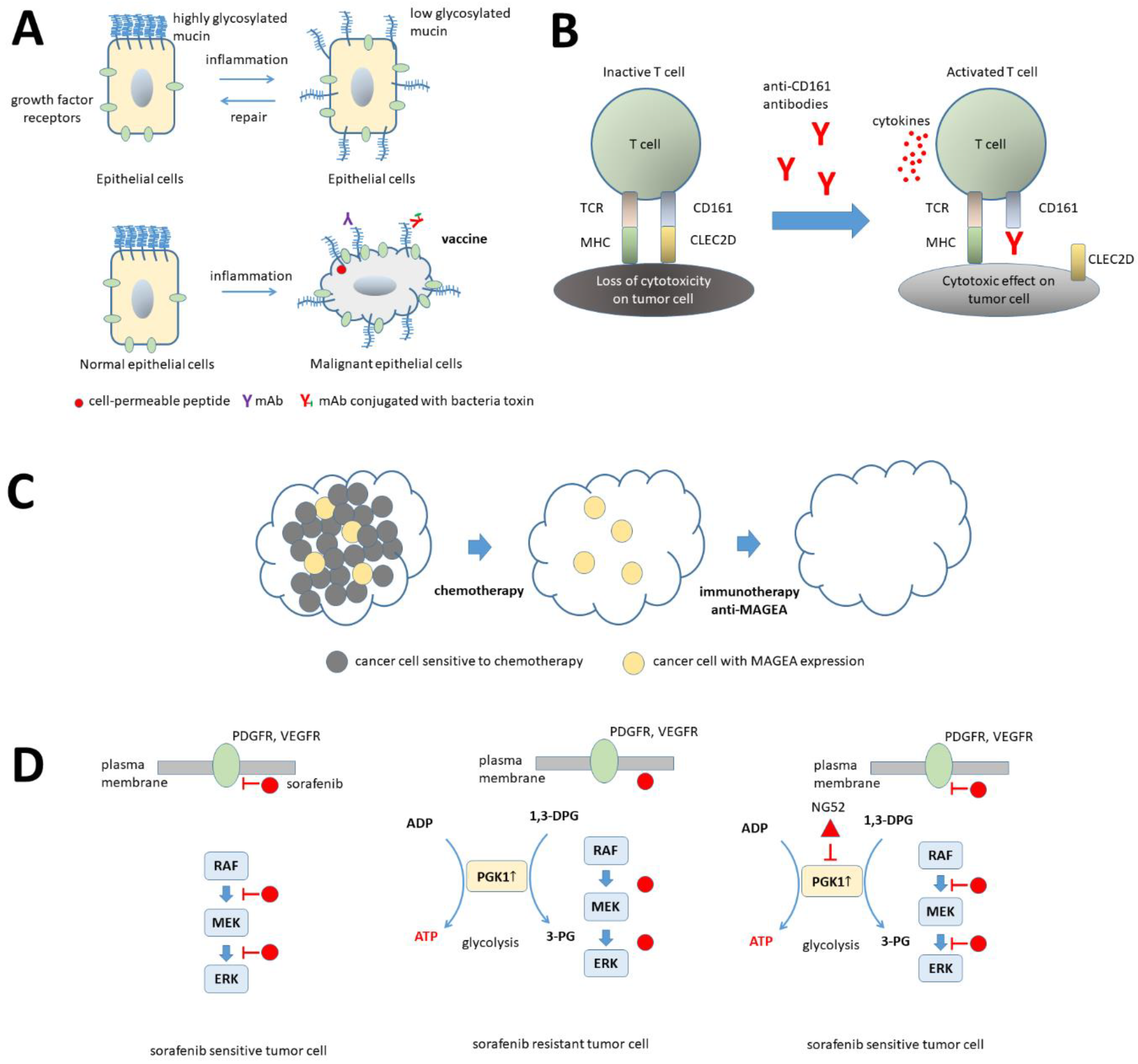
3. Discussion
4. Materials and Methods
4.1. Data Used
4.2. Identification of Breast Cancer Subtypes
4.3. Differential Gene Expression (DEG) and Gene Ontology (GO)
4.4. Cell Type Enrichment Analysis
4.5. Survival Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 66. [Google Scholar] [CrossRef]
- Zagami, P.; Carey, L.A. Triple Negative Breast Cancer: Pitfalls and Progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast Cancer Intrinsic Subtype Classification, Clinical Use and Future Trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast Cancer Development and Progression: Risk Factors, Cancer Stem Cells, Signaling Pathways, Genomics, and Molecular Pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef] [PubMed]
- Luond, F.; Tiede, S.; Christofori, G. Breast Cancer as an Example of Tumour Heterogeneity and Tumour Cell Plasticity during Malignant Progression. Br. J. Cancer 2021, 125, 164–175. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Sharpe, A.H. Introduction to Checkpoint Inhibitors and Cancer Immunotherapy. Immunol. Rev. 2017, 276, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. Pd-1/Pd-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Muenst, S.; Soysal, S.D.; Gao, F.; Obermann, E.C.; Oertli, D.; Gillanders, W.E. The Presence of Programmed Death 1 (Pd-1)-Positive Tumor-Infiltrating Lymphocytes is Associated with Poor Prognosis in Human Breast Cancer. Breast Cancer Res. Treat. 2013, 139, 667–676. [Google Scholar] [CrossRef]
- Liu, L.; Shen, Y.; Zhu, X.; Lv, R.; Li, S.; Zhang, Z.; Shi, Y.G.; Tan, L. Eralpha Is a Negative Regulator of Pd-L1 Gene Transcription in Breast Cancer. Biochem. Biophys. Res. Commun. 2018, 505, 157–161. [Google Scholar] [CrossRef]
- Liu, S.; Chen, S.; Yuan, W.; Wang, H.; Chen, K.; Li, D.; Li, D. Pd-1/Pd-L1 Interaction up-Regulates Mdr1/P-Gp Expression in Breast Cancer Cells Via Pi3k/Akt and Mapk/Erk Pathways. Oncotarget 2017, 8, 99901–99912. [Google Scholar] [CrossRef] [PubMed]
- Lumachi, F.; Santeufemia, D.A.; Basso, S.M. Current Medical Treatment of Estrogen Receptor-Positive Breast Cancer. World J. Biol. Chem. 2015, 6, 231–239. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, Y.; He, L.; Imani, S.; Wen, Q. Clinical Features of Patients with Her2-Positive Breast Cancer and Development of a Nomogram for Predicting Survival. ESMO Open 2021, 6, 100232. [Google Scholar] [CrossRef]
- Onitilo, A.A.; Engel, J.; Joseph, A.O.; Li, Y.H. Is Oestrogen Receptor-Negative/Progesterone Receptor-Positive (ER−/PR+) a Real Pathological Entity? Ecancermedicalscience 2021, 15, 1278. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-Cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Beltjens, F.; Molly, D.; Bertaut, A.; Richard, C.; Desmoulins, I.; Loustalot, C.; Charon-Barra, C.; Courcet, E.; Bergeron, A.; Ladoire, S.; et al. ER−/PR+ Breast Cancer: A Distinct Entity, Which Is Morphologically and Molecularly Close to Triple-Negative Breast Cancer. Int. J. Cancer 2021, 149, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Hefti, M.M.; Hu, R.; Knoblauch, N.W.; Collins, L.C.; Haibe-Kains, B.; Tamimi, R.M.; Beck, A.H. Estrogen Receptor Negative/Progesterone Receptor Positive Breast Cancer Is Not a Reproducible Subtype. Breast Cancer Res. BCR 2013, 15, R68. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Gorski, J. Estrogen-Induced Transcription of the Progesterone Receptor Gene Does Not Parallel Estrogen Receptor Occupancy. Proc. Natl. Acad. Sci. USA 1996, 93, 15180–15184. [Google Scholar] [CrossRef]
- Kufe, D.W. Mucins in Cancer: Function, Prognosis and Therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. Muc1: A Multifaceted Oncoprotein with a Key Role in Cancer Progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W. Muc1-C Oncoprotein as a Target in Breast Cancer: Activation of Signaling Pathways and Therapeutic Approaches. Oncogene 2013, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Choi, S.; Park, Y.; Jin, H.S. Mucin1 and Mucin16: Therapeutic Targets for Cancer Therapy. Pharmaceuticals 2021, 14, 1053. [Google Scholar] [CrossRef]
- Schooten, E.; Di Maggio, A.; van Bergen En Henegouwen, P.M.P.; Kijanka, M.M. Mage-a Antigens as Targets for Cancer Immunotherapy. Cancer Treat. Rev. 2018, 67, 54–62. [Google Scholar] [CrossRef]
- Weon, J.L.; Potts, P.R. The Mage Protein Family and Cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef]
- Wong, P.P.; Yeoh, C.C.; Ahmad, A.S.; Chelala, C.; Gillett, C.; Speirs, V.; Jones, J.L.; Hurst, H.C. Identification of Magea Antigens as Causal Players in the Development of Tamoxifen-Resistant Breast Cancer. Oncogene 2014, 33, 4579–4588. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Fabian, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argaez, V.; Lara-Riegos, J.; Ramirez-Camacho, M.A.; Alvarez-Sanchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Tang, R.; Zhang, X.; Madushi, W.M.; Luo, D.; Dang, Y.; Li, Z.; Wei, K.; Chen, G. Overexpression of Mmp Family Members Functions as Prognostic Biomarker for Breast Cancer Patients: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0135544. [Google Scholar] [CrossRef]
- Gjerstorff, M.F.; Andersen, M.H.; Ditzel, H.J. Oncogenic Cancer/Testis Antigens: Prime Candidates for Immunotherapy. Oncotarget 2015, 6, 15772–15787. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, Z.A.; Whitehurst, A.W. Emerging Contributions of Cancer/Testis Antigens to Neoplastic Behaviors. Trends Cancer 2018, 4, 701–712. [Google Scholar] [CrossRef]
- He, Y.; Wang, X.; Lu, W.; Zhang, D.; Huang, L.; Luo, Y.; Xiong, L.; Li, H.; Zhang, P.; Li, Q.; et al. Pgk1 Contributes to Tumorigenesis and Sorafenib Resistance of Renal Clear Cell Carcinoma Via Activating Cxcr4/Erk Signaling Pathway and Accelerating Glycolysis. Cell Death Dis. 2022, 13, 118. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New Knowledge of the Mechanisms of Sorafenib Resistance in Liver Cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Wang, W.L.; Jiang, Z.R.; Hu, C.; Chen, C.; Hu, Z.Q.; Wang, A.L.; Wang, L.; Liu, J.; Wang, W.C.; Liu, Q.S. Pharmacologically Inhibiting Phosphoglycerate Kinase 1 for Glioma with Ng52. Acta Pharmacol. Sin. 2021, 42, 633–640. [Google Scholar] [CrossRef]
- Li, T.; Huang, H.; Shi, G.; Zhao, L.; Li, T.; Zhang, Z.; Liu, R.; Hu, Y.; Liu, H.; Yu, J.; et al. Tgf-Beta1-Sox9 Axis-Inducible Col10a1 Promotes Invasion and Metastasis in Gastric Cancer Via Epithelial-to-Mesenchymal Transition. Cell Death Dis. 2018, 9, 849. [Google Scholar] [CrossRef]
- Huang, H.; Li, T.; Ye, G.; Zhao, L.; Zhang, Z.; Mo, D.; Wang, Y.; Zhang, C.; Deng, H.; Li, G.; et al. High Expression of Col10a1 Is Associated with Poor Prognosis in Colorectal Cancer. OncoTargets Ther. 2018, 11, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; He, C.; Wei, J.; Zhang, Z.; Luo, Y.; Tan, H.; Ren, C. Pgk1 Is a Potential Survival Biomarker and Invasion Promoter by Regulating the Hif-1alpha-Mediated Epithelial-Mesenchymal Transition Process in Breast Cancer. Cell. Physiol. Biochem. 2018, 51, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Chan, M.H.; Li, C.H.; Yang, C.J.; Tseng, Y.W.; Tsai, H.F.; Chiou, J.; Hsiao, M. Metabolic Protein Phosphoglycerate Kinase 1 Confers Lung Cancer Migration by Directly Binding Hiv Tat Specific Factor 1. Cell Death Discov. 2021, 7, 135. [Google Scholar] [CrossRef]
- Gou, R.; Hu, Y.; Liu, O.; Dong, H.; Gao, L.; Wang, S.; Zheng, M.; Li, X.; Lin, B. Pgk1 Is a Key Target for Anti-Glycolytic Therapy of Ovarian Cancer: Based on the Comprehensive Analysis of Glycolysis-Related Genes. Front. Oncol. 2021, 11, 682461. [Google Scholar] [CrossRef]
- Braud, V.M.; Meghraoui-Kheddar, A.; Elaldi, R.; Petti, L.; Germain, C.; Anjuere, F. Llt1-Cd161 Interaction in Cancer: Promises and Challenges. Front. Immunol. 2022, 13, 847576. [Google Scholar] [CrossRef]
- Cheng, X.; Cao, Y.; Wang, X.; Cheng, L.; Liu, Y.; Lei, J.; Peng, W.; Shi, D. Systematic Pan-Cancer Analysis of Klrb1 with Prognostic Value and Immunological Activity across Human Tumors. J. Immunol. Res. 2022, 2022, 5254911. [Google Scholar] [CrossRef]
- Sun, Y.; Malaer, J.D.; Mathew, P.A. Lectin-Like Transcript 1 as a Natural Killer Cell-Mediated Immunotherapeutic Target for Triple Negative Breast Cancer and Prostate Cancer. J. Cancer Metastasis Treat. 2019, 2019, 80. [Google Scholar] [CrossRef]
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.R.; et al. Inhibitory Cd161 Receptor Identified in Glioma-Infiltrating T Cells by Single-Cell Analysis. Cell 2021, 184, 1281–1298. [Google Scholar] [CrossRef]
- Prat, A.; Fan, C.; Fernandez, A.; Hoadley, K.A.; Martinello, R.; Vidal, M.; Viladot, M.; Pineda, E.; Arance, A.; Munoz, M.; et al. Response and Survival of Breast Cancer Intrinsic Subtypes Following Multi-Agent Neoadjuvant Chemotherapy. BMC Med. 2015, 13, 303. [Google Scholar] [CrossRef]
- Hwang, K.T.; Kim, E.K.; Jung, S.H.; Lee, E.S.; Kim, S.I.; Lee, S.; Park, H.K.; Kim, J.; Oh, S.; Kim, Y.A.; et al. Tamoxifen Therapy Improves Overall Survival in Luminal a Subtype of Ductal Carcinoma in Situ: A Study Based on Nationwide Korean Breast Cancer Registry Database. Breast Cancer Res. Treat. 2018, 169, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Lippman, S.M.; Brown, P.H. Tamoxifen Prevention of Breast Cancer: An Instance of the Fingerpost. J. Natl. Cancer Inst. 1999, 91, 1809–1819. [Google Scholar] [CrossRef]
- Tovey, S.M.; Brown, S.; Doughty, J.C.; Mallon, E.A.; Cooke, T.G.; Edwards, J. Poor Survival Outcomes in Her2-Positive Breast Cancer Patients with Low-Grade, Node-Negative Tumours. Br. J. Cancer 2009, 100, 680–683. [Google Scholar] [CrossRef]
- Goncalves, H., Jr.; Guerra, M.R.; Duarte Cintra, J.R.; Fayer, V.A.; Brum, I.V.; Bustamante Teixeira, M.T. Survival Study of Triple-Negative and Non-Triple-Negative Breast Cancer in a Brazilian Cohort. Clin. Med. Insights Oncol. 2018, 12, 1179554918790563. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.Y.; Chang, C.J.; Cheng, J.S. Survival, Treatment Regimens and Medical Costs of Women Newly Diagnosed with Metastatic Triple-Negative Breast Cancer. Sci. Rep. 2022, 12, 729. [Google Scholar] [CrossRef]
- Xu, Y.H.; Deng, J.L.; Wang, L.P.; Zhang, H.B.; Tang, L.; Huang, Y.; Tang, J.; Wang, S.M.; Wang, G. Identification of Candidate Genes Associated with Breast Cancer Prognosis. DNA Cell Biol. 2020, 39, 1205–1227. [Google Scholar] [CrossRef]
- Lu, Z.N.; Song, J.; Sun, T.H.; Sun, G. Ube2c Affects Breast Cancer Proliferation through the Akt/Mtor Signaling Pathway. Chin. Med. J. 2021, 134, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.Y.; Gu, Y.L.; Zhu, Q.; Li, X.H.; Jiang, G.Q. The Role of Ferroptosis-Related Genes for Overall Survival Prediction in Breast Cancer. J. Clin. Lab. Anal. 2021, 35, e24094. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I Interferons in Anticancer Immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Zeng, S.; Vitiello, G.A.; Seifert, A.M.; Medina, B.D.; Beckman, M.J.; Loo, J.K.; Santamaria-Barria, J.; Maltbaek, J.H.; Param, N.J.; et al. Macrophages and CD8(+) T Cells Mediate the Antitumor Efficacy of Combined CD40 Ligation and Imatinib Therapy in Gastrointestinal Stromal Tumors. Cancer Immunol. Res. 2018, 6, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Todorović-Raković, N.; Milovanović, J.; Greenman, J.; Radulovic, M. The Prognostic Significance of Serum Interferon-Gamma (Ifn-Gamma) in Hormonally Dependent Breast Cancer. Cytokine 2022, 152, 155836. [Google Scholar] [CrossRef] [PubMed]
- Skeate, J.G.; Otsmaa, M.E.; Prins, R.; Fernandez, D.J.; Da Silva, D.M.; Kast, W.M. Tnfsf14: Lighting the Way for Effective Cancer Immunotherapy. Front. Immunol. 2020, 11, 922. [Google Scholar] [CrossRef]
- Kim, H.R.; Ha, S.J.; Hong, M.H.; Heo, S.J.; Koh, Y.W.; Choi, E.C.; Kim, E.K.; Pyo, K.H.; Jung, I.; Seo, D.; et al. Pd-L1 Expression on Immune Cells, but Not on Tumor Cells, Is a Favorable Prognostic Factor for Head and Neck Cancer Patients. Sci. Rep. 2016, 6, 36956. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. Revigo Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal | All Tumors | Good Prognosis | Poor Prognosis | Good vs. Poor Prognosis p Value | |
|---|---|---|---|---|---|
| Number | 105 | 301 | 200 | 101 | |
| Age | |||||
| Mean (StDev) | 58.2 (14) | 57.5 (13.5) | 54.3 (11.7) | 63.7 (14.5) | <0.0001 *** (t Test) |
| Median (Min:Max) | 58 (30:90) | 56 (27:90) | 54 (27:85) | 62 (31:90) | |
| Stage | |||||
| Stage I a | - | 43 (14.6%) | 36 (18.2%) | 7 (7.2%) | 0.001 ** (Fisher exact test) |
| Stage IA | - | 13 (4.4%) | 11 (5.6%) | 2 (2.1%) | |
| Stage IB | - | 2 (0.7%) | 2 (1%) | 0 (0%) | |
| Stage II a | - | 2 (0.7%) | 2 (1%) | 0 (0%) | 0.461 (Fisher exact test) |
| Stage IIA | - | 83 (28.1%) | 61 (30.8%) | 22 (22.7%) | |
| Stage IIB | - | 64 (21.7%) | 46 (23.2%) | 18 (18.6%) | |
| Stage III a | - | 2 (0.7%) | 0 (0%) | 2 (2.1%) | 0.0047 ** (Fisher exact test) |
| Stage IIIA | - | 51 (17.3%) | 32 (16.2%) | 19 (19.6%) | |
| Stage IIIB | - | 12 (4.1%) | 4 (2%) | 8 (8.2%) | |
| Stage IIIC | - | 11 (3.7%) | 4 (2%) | 7 (7.2%) | |
| Stage IV | - | 12 (4.1%) | 0 (0%) | 12 (12.4%) | <0.0001 ** (Fisher exact test) |
| Subtype | |||||
| Luminal A | - | 206 (68.4%) | 142 (71%) | 64 (63.4%) | 0.191 (Fisher exact test) |
| Luminal B | - | 18 (6%) | 9 (4.5%) | 9 (8.9%) | 0.196 (Fisher exact test) |
| HER2 enriched | - | 15 (5%) | 11 (5.5%) | 4 (4%) | 0.780 (Fisher exact test) |
| Triple Negative | - | 62 (20.6%) | 38 (19%) | 24 (23.8%) | 0.366 (Fisher exact test) |
| Race | |||||
| Caucasian | 98 (93.3%) | 235 (78.1%) | 160 (80%) | 75 (74.3%) | 0.302 (Fisher exact test) |
| African/African American | 5 (4.8%) | 48 (15.9%) | 30 (15%) | 18 (17.8%) | 0.617 (Fisher exact test) |
| Asian | 1 (1%) | 10 (3.3%) | 7 (3.5%) | 3 (3%) | 1 (Fisher exact test) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippi, A.; Mocanu, M.-M. Mining TCGA Database for Genes with Prognostic Value in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 1622. https://doi.org/10.3390/ijms24021622
Filippi A, Mocanu M-M. Mining TCGA Database for Genes with Prognostic Value in Breast Cancer. International Journal of Molecular Sciences. 2023; 24(2):1622. https://doi.org/10.3390/ijms24021622
Chicago/Turabian StyleFilippi, Alexandru, and Maria-Magdalena Mocanu. 2023. "Mining TCGA Database for Genes with Prognostic Value in Breast Cancer" International Journal of Molecular Sciences 24, no. 2: 1622. https://doi.org/10.3390/ijms24021622
APA StyleFilippi, A., & Mocanu, M.-M. (2023). Mining TCGA Database for Genes with Prognostic Value in Breast Cancer. International Journal of Molecular Sciences, 24(2), 1622. https://doi.org/10.3390/ijms24021622