
Tacrine-Based Hybrids: Past, Present, and Future

 ,
,  ,
,
Abstract
:1. Introduction
2. Summary of Tacrine-Based Hybrids Reported in 2006–2022
3. Tacrine Hybrids with Antioxidant Activity
3.1. Tacrine–Melatonin Hybrids
3.2. Other Hybrids with Antioxidant Activity
3.3. Tacrine–Ferulic Acid Hybrids
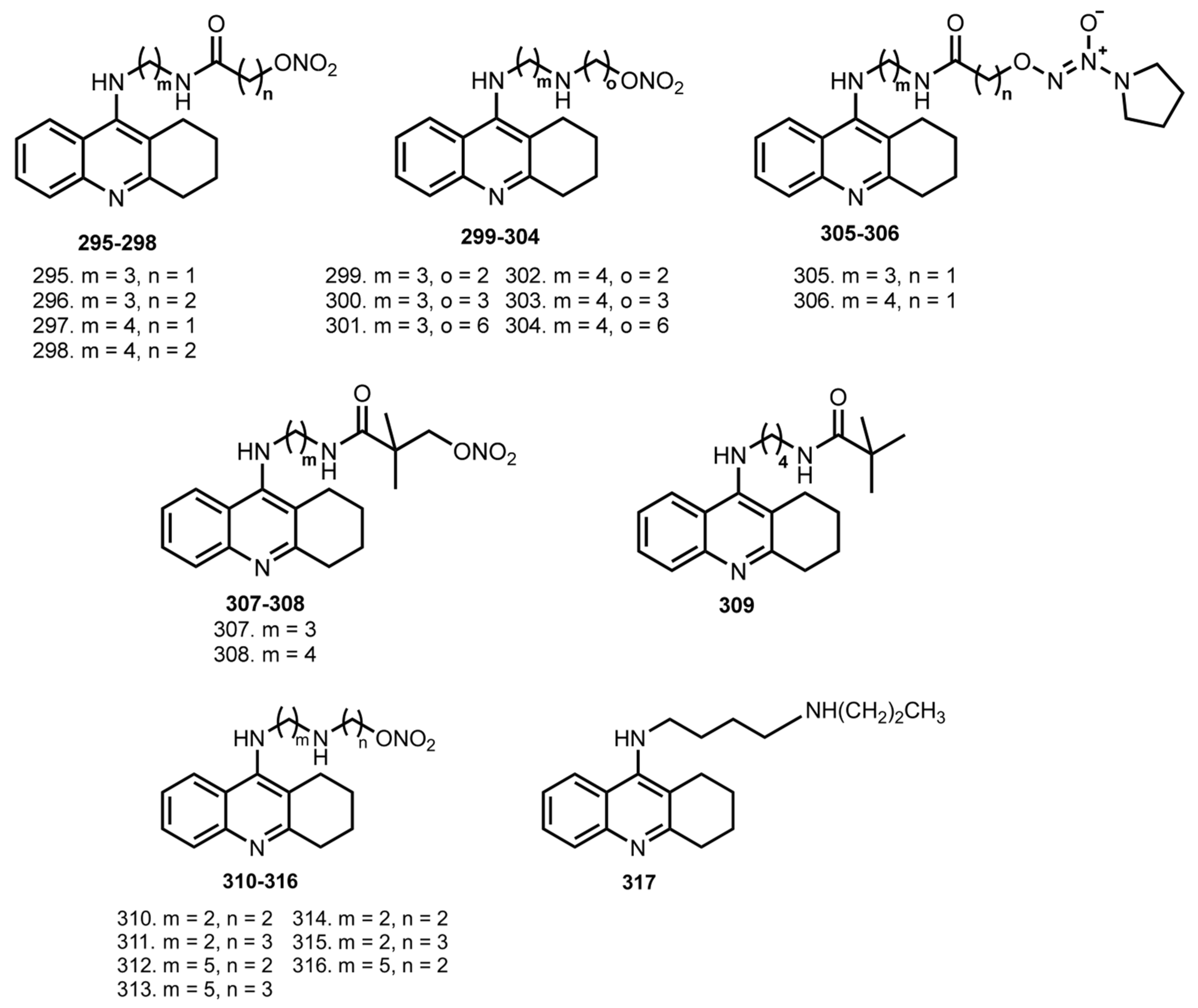
4. Tacrine Hybrids with NO-Donating Molecules
5. Tacrine Hybrids with Biological Active Organic Scaffolds
5.1. Tacrine–Phenothiazine Hybrids
5.2. Tacrine–Benzotiazole/Benzofuran Derivatives
5.3. Tacrine Hybrids with NSAIDS
5.4. Tacrine–Hupyridone Hybrids
5.5. Tacrine–Donepezil Hybrids
5.6. Tacrine–TPPU Hybrids
5.7. Tacrine–Huprine Hybrids
5.8. Tacrine–Bifendate Hybrids
5.9. Tacrine hybrids with HDAC Inhibitors
5.10. Tacrine Hybrids with Thio Derivatives
5.11. Tacrine Hybrids with Fluorescent Probes
5.12. Tacrine Hybrids with Ca2+ Channel Blocker
6. Tacrine Hybrids with Modulators of Cholinergic/Serotonergic System
6.1. Tacrine Hybrids with Modulators of Serotonin Receptors
6.2. Tacrine Hybrids with Modulator of Muscarinic Receptors
6.3. Tacrine Hybrids with Cannabinoid CB1 Receptor Antagonists
6.4. Tacrine Hybrids with Modulator of NMDA Receptors
6.5. Tacrine Hybrids with Modulators of Opioid Receptors
6.6. Tacrine Hybrids with MAO Inhibitors
7. Tacrine Hybrids with Natural Products
8. Tacrine Hybrids with Other Organic Scaffolds
9. Discussion
10. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Yan, R. Role of BACE1 in Alzheimer’s synaptic function. Transl. Neurodegener. 2017, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Ghouri, R.G.; Ans, A.H.; Akbar, A.; Toheed, A. Recommendations for Anti-inflammatory Treatments in Alzheimer’s Disease: A Comprehensive Review of the Literature. Cureus 2019, 11, e462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.-J.; Zhang, X.; Chen, W.-W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Arbel-Ornath, M.; Hudry, E.; Boivin, J.R.; Hashimoto, T.; Takeda, S.; Kuchibhotla, K.V.; Hou, S.; Lattarulo, C.R.; Belcher, A.M.; Shakerdge, N.; et al. Soluble oligomeric amyloid-β induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener. 2017, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Drews, A.; De, S.; Flagmeier, P.; Wirthensohn, D.C.; Chen, W.-H.; Whiten, D.R.; Rodrigues, M.; Vincke, C.; Muyldermans, S.; Paterson, R.W.; et al. Inhibiting the Ca2+ Influx Induced by Human CSF. Cell Rep. 2017, 21, 3310–3316. [Google Scholar] [CrossRef] [Green Version]
- Goodison, W.V.; Frisardi, V.; Kehoe, P.G. Calcium Channel Blockers and Alzheimer’s Disease: Potential Relevance in Treatment Strategies of Metabolic Syndrome. J. Alzheimer’s Dis. 2012, 30, S269–S282. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta—Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Gsell, W.; Jungkunz, G.; Riederer, P. Functional Neurochemistry of Alzheimer’s Disease. Cur. Pharm. Dis. 2004, 10, 265–293. [Google Scholar] [CrossRef]
- Werner, F.; Coveñas, R. Classical Neurotransmitters and Neuropeptides Involved in Major Depression in a Multi-neurotransmitter System: A Focus on Antidepressant Drugs. Curr. Med. Chem. 2013, 20, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Yamazaki, Y.; Miledi, R.; Sumikawa, K. Nicotinic and muscarinic agonists and acetylcholinesterase inhibitors stimulate a common pathway to enhance GluN2B-NMDAR responses. Proc. Natl. Acad. Sci. USA 2014, 111, 12538–12543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwomoh, L.; Tejeda, G.S.; Tobin, A.B. Targeting the M1 muscarinic acetylcholine receptor in Alzheimer’s disease. Neuronal Signal. 2022, 6, NS20210004. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [Green Version]
- Terry, A.V., Jr.; Callahan, P.; Hernandez, C.M. Nicotinic ligands as multifunctional agents for the treatment of neuropsychiatric disorders. Biochem. Pharmacol. 2015, 97, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Hoskin, J.L.; Al-Hasan, Y.; Sabbagh, M.N. Nicotinic Acetylcholine Receptor Agonists for the Treatment of Alzheimer’s Dementia: An Update. Nicotine Tob. Res. 2019, 21, 370–376. [Google Scholar] [CrossRef]
- Greenlee, W.; Clader, J.; Asberom, T.; McCombie, S.; Ford, J.; Guzik, H.; Kozlowski, J.; Li, S.; Liu, C.; Lowe, D.; et al. Muscarinic agonists and antagonists in the treatment of Alzheimer’s disease. Il Farm. 2001, 56, 247–250. [Google Scholar] [CrossRef]
- Furuie, H.; Yamada, K.; Ichitani, Y. MK-801-induced and scopolamine-induced hyperactivity in rats neonatally treated chronically with MK-801. Behav. Pharmacol. 2013, 24, 678–683. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Tayeb, H.O.; Yang, H.D.; Price, B.H.; Tarazi, F.I. Pharmacotherapies for Alzheimer’s disease: Beyond cholinesterase inhibitors. Pharmacol. Ther. 2012, 134, 8–25. [Google Scholar] [CrossRef] [PubMed]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Recanatini, M.; Cavalli, A.; Belluti, F.; Piazzi, L.; Rampa, A.; Bisi, A.; Gobbi, S.; Valenti, P.; Andrisano, V.; Bartolini, M.; et al. SAR of 9-Amino-1,2,3,4-tetrahydroacridine-Based Acetylcholinesterase Inhibitors: Synthesis, Enzyme Inhibitory Activity, QSAR, and Structure-Based CoMFA of Tacrine Analogues. J. Med. Chem. 2000, 43, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Soukup, O.; Jun, D.; Zdarova-Karasova, J.; Patocka, J.; Musilek, K.; Korabecny, J.; Krusek, J.; Kaniakova, M.; Sepsova, V.; Mandikova, J.; et al. A Resurrection of 7-MEOTA: A Comparison with Tacrine. Curr. Alzheimer Res. 2013, 10, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Musilek, K.; Holas, O.; Binder, J.; Zemek, F.; Marek, J.; Pohanka, M.; Opletalova, V.; Dohnal, V.; Kuca, K. Synthesis and in vitro evaluation of N-alkyl-7-methoxytacrine hydrochlorides as potential cholinesterase inhibitors in Alzheimer disease. Bioorg. Med. Chem. Lett. 2010, 20, 6093–6095. [Google Scholar] [CrossRef]
- Quintanova, C.; Keri, R.S.; Marques, S.M.; Fernandes, G.M.; Cardoso, S.M.; Luísa Serralheiro, M.; Amélia Santos, M. Design, synthesis and bioevaluation of tacrine hybrids with cinnamate and cinnamylidene acetate derivatives as potential anti-Alzheimer drugs. MedChemComm 2015, 6, 1969–1977. [Google Scholar] [CrossRef]
- Wu, W.-Y.; Dai, Y.-C.; Li, N.-G.; Dong, Z.-X.; Gu, T.; Shi, Z.-H.; Xue, X.; Tang, Y.-P.; Duan, J.-A. Novel multitarget-directed tacrine derivatives as potential candidates for the treatment of Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2017, 32, 572–587. [Google Scholar] [CrossRef] [Green Version]
- Carlier, P.R.; Chow, E.S.; Han, Y.; Liu, J.; El Yazal, J.; Pang, Y.-P. Heterodimeric Tacrine-Based Acetylcholinesterase Inhibitors: Investigating Ligand−Peripheral Site Interactions. J. Med. Chem. 1999, 42, 4225–4231. [Google Scholar] [CrossRef]
- Harel, M.; Sonoda, L.K.; Silman, I.; Sussman, J.L.; Rosenberry, T.L. Crystal Structure of Thioflavin T Bound to the Peripheral Site of Torpedo californica Acetylcholinesterase Reveals How Thioflavin T Acts as a Sensitive Fluorescent Reporter of Ligand Binding to the Acylation Site. J. Am. Chem. Soc. 2008, 130, 7856–7861. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sun, J.; Fang, L.; Liu, M.; Peng, S.; Liao, H.; Lehmann, J.; Zhang, Y. Tacrine–Ferulic Acid–Nitric Oxide (NO) Donor Trihybrids as Potent, Multifunctional Acetyl- and Butyrylcholinesterase Inhibitors. J. Med. Chem. 2012, 55, 4309–4321. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Inestrosa, N.C. Interactions of AChE with Aβ Aggregates in Alzheimer’s Brain: Therapeutic Relevance of IDN 5706. Front. Mol. Neurosci. 2011, 4, 19. [Google Scholar] [CrossRef]
- Pourshojaei, Y.; Abiri, A.; Eskandari, K.; Haghighijoo, Z.; Edraki, N.; Asadipour, A. Phenoxyethyl Piperidine/Morpholine Derivatives as PAS and CAS Inhibitors of Cholinesterases: Insights for Future Drug Design. Sci. Rep. 2019, 9, 19855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoobi, M.; Ghanoni, F.; Nadri, H.; Moradi, A.; Hamedani, M.P.; Homayouni Moghadam, F.; Emami, S.; Vosooghi, M.; Zadmard, R.; Foroumadi, A.; et al. New tetracyclic tacrine analogs containing pyrano[2,3-c]pyrazole: Efficient synthesis, biological assessment and docking simulation study. Eur. J. Med. Chem. 2015, 89, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Pourabdi, L.; Khoobi, M.; Nadri, H.; Moradi, A.; Moghadam, F.H.; Emami, S.; Mojtahedi, M.M.; Haririan, I.; Forootanfar, H.; Ameri, A.; et al. Synthesis and structure-activity relationship study of tacrine-based pyrano[2,3-c]pyrazoles targeting AChE/BuChE and 15-LOX. Eur. J. Med. Chem. 2016, 123, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roldán-Peña, J.M.; Alejandre-Ramos, D.; López, Ó.; Maya, I.; Lagunes, I.; Padrón, J.M.; Peña-Altamira, L.E.; Bartolini, M.; Monti, B.; Bolognesi, M.L.; et al. New tacrine dimers with antioxidant linkers as dual drugs: Anti-Alzheimer’s and antiproliferative agents. Eur. J. Med. Chem. 2017, 138, 761–773. [Google Scholar] [CrossRef]
- Sameem, B.; Saeedi, M.; Mahdavi, M.; Shafiee, A. A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 128, 332–345. [Google Scholar] [CrossRef]
- Girek, M.; Szymański, P. Phyto-Tacrine Hybrids as Promising Drugs to Treat Alzheimer’s Disease. ChemistrySelect 2019, 4, 5776–5790. [Google Scholar] [CrossRef]
- Eckroat, T.J.; Manross, D.L.; Cowan, S.C. Merged Tacrine-Based, Multitarget-Directed Acetylcholinesterase Inhibitors 2015–Present: Synthesis and Biological Activity. Int. J. Mol. Sci. 2020, 21, 5965. [Google Scholar] [CrossRef]
- Rodríguez-Franco, M.I.; Fernández-Bachiller, M.I.; Pérez, C.; Hernández-Ledesma, B.; Bartolomé, B. Novel Tacrine−Melatonin Hybrids as Dual-Acting Drugs for Alzheimer Disease, with Improved Acetylcholinesterase Inhibitory and Antioxidant Properties. J. Med. Chem. 2006, 49, 459–462. [Google Scholar] [CrossRef]
- Fernández-Bachiller, M.I.; Pérez, C.; Campillo, N.E.; Páez, J.A.; González-Muñoz, G.C.; Usán, P.; García-Palomero, E.; López, M.G.; Villarroya, M.; García, A.G.; et al. Tacrine-Melatonin Hybrids as Multifunctional Agents for Alzheimer’s Disease, with Cholinergic, Antioxidant, and Neuroprotective Properties. ChemMedChem 2009, 4, 828–841. [Google Scholar] [CrossRef]
- Benchekroun, M.; Romero, A.; Egea, J.; León, R.; Michalska, P.; Buendía, I.; Jimeno, M.L.; Jun, D.; Janockova, J.; Sepsova, V.; et al. The Antioxidant Additive Approach for Alzheimer’s Disease Therapy: New Ferulic (Lipoic) Acid Plus Melatonin Modified Tacrines as Cholinesterases Inhibitors, Direct Antioxidants, and Nuclear Factor (Erythroid-Derived 2)-Like 2 Activators. J. Med. Chem. 2016, 59, 9967–9973. [Google Scholar] [CrossRef]
- Fernández-Bachiller, M.I.; Pérez, C.; González-Muñoz, G.C.; Conde, S.; López, M.G.; Villarroya, M.; García, A.G.; Rodríguez-Franco, M.I. Novel Tacrine−8-Hydroxyquinoline Hybrids as Multifunctional Agents for the Treatment of Alzheimer’s Disease, with Neuroprotective, Cholinergic, Antioxidant, and Copper-Complexing Properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Li, Y.-P.; He, Y.; Huang, S.-L.; Tan, J.-H.; Ou, T.-M.; Li, D.; Gu, L.-Q.; Huang, Z.-S. Design, synthesis and evaluation of novel tacrine-multialkoxybenzene hybrids as dual inhibitors for cholinesterases and amyloid beta aggregation. Bioorg. Med. Chem. 2011, 19, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Li, Y.-P.; He, Y.; Huang, S.-L.; Li, D.; Gu, L.-Q.; Huang, Z.-S. Synthesis and evaluation of heterobivalent tacrine derivatives as potential multi-functional anti-Alzheimer agents. Eur. J. Med. Chem. 2011, 46, 2609–2616. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bachiller, M.I.; Pérez, C.; Monjas, L.; Rademann, J.; Rodríguez-Franco, M.I. New Tacrine–4-Oxo-4 H -chromene Hybrids as Multifunctional Agents for the Treatment of Alzheimer’s Disease, with Cholinergic, Antioxidant, and β-Amyloid-Reducing Properties. J. Med. Chem. 2012, 55, 1303–1317. [Google Scholar] [CrossRef] [Green Version]
- Chao, X.; He, X.; Yang, Y.; Zhou, X.; Jin, M.; Liu, S.; Cheng, Z.; Liu, P.; Wang, Y.; Yu, J.; et al. Design, synthesis and pharmacological evaluation of novel tacrine–caffeic acid hybrids as multi-targeted compounds against Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2012, 22, 6498–6502. [Google Scholar] [CrossRef]
- Chen, X.; Zenger, K.; Lupp, A.; Kling, B.; Heilmann, J.; Fleck, C.; Kraus, B.; Decker, M. Tacrine-Silibinin Codrug Shows Neuro- and Hepatoprotective Effects in Vitro and Pro-Cognitive and Hepatoprotective Effects in Vivo. J. Med. Chem. 2012, 55, 5231–5242. [Google Scholar] [CrossRef]
- Zenger, K.; Chen, X.; Decker, M.; Kraus, B. In-vitro stability and metabolism of a tacrine–silibinin codrug. J. Pharm. Pharmacol. 2013, 65, 1765–1772. [Google Scholar] [CrossRef]
- Mao, F.; Chen, J.; Zhou, Q.; Luo, Z.; Huang, L.; Li, X. Novel tacrine–ebselen hybrids with improved cholinesterase inhibitory, hydrogen peroxide and peroxynitrite scavenging activity. Bioorg. Med. Chem. Lett. 2013, 23, 6737–6742. [Google Scholar] [CrossRef]
- Lan, J.-S.; Xie, S.-S.; Li, S.-Y.; Pan, L.-F.; Wang, X.-B.; Kong, L.-Y. Design, synthesis and evaluation of novel tacrine-(β-carboline) hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2014, 22, 6089–6104. [Google Scholar] [CrossRef]
- Nepovimova, E.; Korabecny, J.; Dolezal, R.; Babkova, K.; Ondrejicek, A.; Jun, D.; Sepsova, V.; Horova, A.; Hrabinova, M.; Soukup, O.; et al. Tacrine–Trolox Hybrids: A Novel Class of Centrally Active, Nonhepatotoxic Multi-Target-Directed Ligands Exerting Anticholinesterase and Antioxidant Activities with Low In Vivo Toxicity. J. Med. Chem. 2015, 58, 8985–9003. [Google Scholar] [CrossRef]
- Luo, W.; Wang, T.; Hong, C.; Yang, Y.; Chen, Y.; Cen, J.; Xie, S.; Wang, C. Design, synthesis and evaluation of 4-dimethylamine flavonoid derivatives as potential multifunctional anti-Alzheimer agents. Eur. J. Med. Chem. 2016, 122, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Chand, K.; Alsoghier, H.M.; Chaves, S.; Santos, M.A. Tacrine-(hydroxybenzoyl-pyridone) hybrids as potential multifunctional anti-Alzheimer’s agents: AChE inhibition, antioxidant activity and metal chelating capacity. J. Inorg. Biochem. 2016, 163, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hong, G.; Li, X.; Zhang, Y.; Xu, Z.; Mao, L.; Feng, X.; Liu, T. Synthesis and activity towards Alzheimer’s disease in vitro: Tacrine, phenolic acid and ligustrazine hybrids. Eur. J. Med. Chem. 2018, 148, 238–254. [Google Scholar] [CrossRef]
- Li, K.; Jiang, Y.; Li, G.; Liu, T.; Yang, Z. Novel Multitarget Directed Tacrine Hybrids as Anti-Alzheimer’s Compounds Improved Synaptic Plasticity and Cognitive Impairment in APP/PS1 Transgenic Mice. ACS Chem. Neurosci. 2020, 11, 4316–4328. [Google Scholar] [CrossRef]
- Pérez-Areales, F.; Garrido, M.; Aso, E.; Bartolini, M.; De Simone, A.; Espargaró, A.; Ginex, T.; Sabate, R.; Pérez, B.; Andrisano, V.; et al. Centrally Active Multitarget Anti-Alzheimer Agents Derived from the Antioxidant Lead CR-6. J. Med. Chem. 2020, 63, 9360–9390. [Google Scholar] [CrossRef]
- Rani, A.; Singh, A.; Kaur, J.; Singh, G.; Bhatti, R.; Gumede, N.; Kisten, P.; Singh, P.; Sumanjit; Kumar, V. 1H-1,2,3-triazole grafted tacrine-chalcone conjugates as potential cholinesterase inhibitors with the evaluation of their behavioral tests and oxidative stress in mice brain cells. Bioorg. Chem. 2021, 114, 105053. [Google Scholar] [CrossRef]
- Viayna, E.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Cisternas, P.; Oliva, C.A.; Sánchez-López, E.; Ettcheto, M.; Bartolini, M.; De Simone, A.; Ricchini, M.; et al. Discovery of a Potent Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase with Antioxidant Activity that Alleviates Alzheimer-like Pathology in Old APP/PS1 Mice. J. Med. Chem. 2021, 64, 812–839. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Kraus, B.; Lehmann, J.; Heilmann, J.; Zhang, Y.; Decker, M. Design and synthesis of tacrine–ferulic acid hybrids as multi-potent anti-Alzheimer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 2905–2909. [Google Scholar] [CrossRef]
- Fleck, C.; Appenroth, D.; Fang, L.; Schott, Y.; Lehmann, J.; Decker, M. Investigation into the in vivo effects of five novel tacrine/ferulic acid and β-carboline derivatives on scopolamine-induced cognitive impairment in rats using radial maze paradigm. Arzneimittelforschung 2011, 60, 299–306. [Google Scholar] [CrossRef]
- Pi, R.; Mao, X.; Chao, X.; Cheng, Z.; Liu, M.; Duan, X.; Ye, M.; Chen, X.; Mei, Z.; Liu, P.; et al. Tacrine-6-Ferulic Acid, a Novel Multifunctional Dimer, Inhibits Amyloid-β-Mediated Alzheimer’s Disease-Associated Pathogenesis In Vitro and In Vivo. PLoS ONE 2012, 7, e31921. [Google Scholar] [CrossRef]
- Fu, Y.; Mu, Y.; Lei, H.; Wang, P.; Li, X.; Leng, Q.; Han, L.; Qu, X.; Wang, Z.; Huang, X. Design, Synthesis and Evaluation of Novel Tacrine-Ferulic Acid Hybrids as Multifunctional Drug Candidates against Alzheimer’s Disease. Molecules 2016, 21, 1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Yang, H.; Chen, Y.; Lin, H.; Li, Q.; Mo, J.; Bian, Y.; Pei, Y.; Sun, H. Synthesis, pharmacology and molecular docking on multifunctional tacrine-ferulic acid hybrids as cholinesterase inhibitors against Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2018, 33, 496–506. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Appenroth, D.; Decker, M.; Kiehntopf, M.; Roegler, C.; Deufel, T.; Fleck, C.; Peng, S.; Zhang, Y.; Lehmann, J. Synthesis and Biological Evaluation of NO-Donor-Tacrine Hybrids as Hepatoprotective Anti-Alzheimer Drug Candidates. J. Med. Chem. 2008, 51, 713–716. [Google Scholar] [CrossRef]
- Fang, L.; Appenroth, D.; Decker, M.; Kiehntopf, M.; Lupp, A.; Peng, S.; Fleck, C.; Zhang, Y.; Lehmann, J. NO-Donating Tacrine Hybrid Compounds Improve Scopolamine-Induced Cognition Impairment and Show Less Hepatotoxicity. J. Med. Chem. 2008, 51, 7666–7669. [Google Scholar] [CrossRef]
- Hui, A.; Chen, Y.; Zhu, S.; Gan, C.; Pan, J.; Zhou, A. Design and synthesis of tacrine-phenothiazine hybrids as multitarget drugs for Alzheimer’s disease. Med. Chem. Res. 2014, 23, 3546–3557. [Google Scholar] [CrossRef]
- Gorecki, L.; Uliassi, E.; Bartolini, M.; Janockova, J.; Hrabinova, M.; Hepnarova, V.; Prchal, L.; Muckova, L.; Pejchal, J.; Karasova, J.Z.; et al. Phenothiazine-Tacrine Heterodimers: Pursuing Multitarget Directed Approach in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 1698–1715. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Su, T.; Shan, W.; Luo, Z.; Sun, Y.; He, F.; Li, X. Inhibition of cholinesterase activity and amyloid aggregation by berberine-phenyl-benzoheterocyclic and tacrine-phenyl-benzoheterocyclic hybrids. Bioorg. Med. Chem. 2012, 20, 3038–3048. [Google Scholar] [CrossRef]
- Keri, R.S.; Quintanova, C.; Marques, S.M.; Esteves, A.R.; Cardoso, S.M.; Santos, M.A. Design, synthesis and neuroprotective evaluation of novel tacrine–benzothiazole hybrids as multi-targeted compounds against Alzheimer’s disease. Bioorg. Med. Chem. 2013, 21, 4559–4569. [Google Scholar] [CrossRef]
- Zha, X.; Lamba, D.; Zhang, L.; Lou, Y.; Xu, C.; Kang, D.; Chen, L.; Xu, Y.; Zhang, L.; De Simone, A.; et al. Novel Tacrine–Benzofuran Hybrids as Potent Multitarget-Directed Ligands for the Treatment of Alzheimer’s Disease: Design, Synthesis, Biological Evaluation, and X-ray Crystallography. J. Med. Chem. 2016, 59, 114–131. [Google Scholar] [CrossRef]
- Rajeshwari, R.; Chand, K.; Candeias, E.; Cardoso, S.; Chaves, S.; Santos, M. New Multitarget Hybrids Bearing Tacrine and Phenylbenzothiazole Motifs as Potential Drug Candidates for Alzheimer’s Disease. Molecules 2019, 24, 587. [Google Scholar] [CrossRef]
- Fancellu, G.; Chand, K.; Tomás, D.; Orlandini, E.; Piemontese, L.; Silva, D.F.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Novel tacrine–benzofuran hybrids as potential multi-target drug candidates for the treatment of Alzheimer’s Disease. J. Enzyme Inhib. Med. Chem. 2020, 35, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepovimova, E.; Svobodova, L.; Dolezal, R.; Hepnarova, V.; Junova, L.; Jun, D.; Korabecny, J.; Kucera, T.; Gazova, Z.; Motykova, K.; et al. Tacrine-Benzothiazoles: Novel class of potential multitarget anti-Alzheimeŕs drugs dealing with cholinergic, amyloid and mitochondrial systems. Bioorg. Chem. 2021, 107, 104596. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Peng, S.; Liao, H.; Zhang, Y.; Lehmann, J. Tacrine-Flurbiprofen Hybrids as Multifunctional Drug Candidates for the Treatment of Alzheimer’s Disease. Arch. Pharm. 2013, 346, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, J.; Huang, Z.; Liao, H.; Peng, S.; Lehmann, J.; Zhang, Y. NO-donating tacrine derivatives as potential butyrylcholinesterase inhibitors with vasorelaxation activity. Bioorg. Med. Chem. Lett. 2013, 23, 3162–3165. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, J.; Huang, Z.; Liao, H.; Peng, S.; Lehmann, J.; Zhang, Y. Design, synthesis and evaluation of tacrine–flurbiprofen–nitrate trihybrids as novel anti-Alzheimer’s disease agents. Bioorg. Med. Chem. 2013, 21, 2462–2470. [Google Scholar] [CrossRef]
- Zawada, K.; Czarnecka, K.; Girek, M.; Kręcisz, P.; Trejtnar, F.; Mandíková, J.; Jończyk, J.; Bajda, M.; Staśkiewicz, M.; Wójtowicz, P.; et al. New hybrids of tacrine and indomethacin as multifunctional acetylcholinesterase inhibitors. Chem. Pap. 2021, 75, 249–264. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, B.; Xia, S.; Fang, L.; Gou, S. ROS-responsive and multifunctional anti-Alzheimer prodrugs: Tacrine-ibuprofen hybrids via a phenyl boronate linker. Eur. J. Med. Chem. 2021, 212, 112997. [Google Scholar] [CrossRef]
- Chen, H.; Wu, X.; Gu, X.; Zhou, Y.; Ye, L.; Zhang, K.; Pan, H.; Wang, J.; Wei, H.; Zhu, B.; et al. Tacrine(10)-Hupyridone Prevents Post-operative Cognitive Dysfunction via the Activation of BDNF Pathway and the Inhibition of AChE in Aged Mice. Front. Cell. Neurosci. 2018, 12, 396. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Xiang, S.; Huang, L.; Lin, J.; Hu, S.; Mak, S.-H.; Wang, C.; Wang, Q.; Cui, W.; Han, Y. Tacrine(10)-hupyridone, a dual-binding acetylcholinesterase inhibitor, potently attenuates scopolamine-induced impairments of cognition in mice. Metab. Brain Dis. 2018, 33, 1131–1139. [Google Scholar] [CrossRef]
- Xuan, Z.; Gu, X.; Yan, S.; Xie, Y.; Zhou, Y.; Zhang, H.; Jin, H.; Hu, S.; Mak, M.S.H.; Zhou, D.; et al. Dimeric Tacrine(10)-hupyridone as a Multitarget-Directed Ligand To Treat Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 2462–2477. [Google Scholar] [CrossRef]
- Shao, D.; Zou, C.; Luo, C.; Tang, X.; Li, Y. Synthesis and evaluation of tacrine–E2020 hybrids as acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2004, 14, 4639–4642. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; Formosa, X.; Galdeano, C.; Gómez, T.; Muñoz-Torrero, D.; Scarpellini, M.; Viayna, E.; Badia, A.; Clos, M.V.; Camins, A.; et al. Novel Donepezil-Based Inhibitors of Acetyl- and Butyrylcholinesterase and Acetylcholinesterase-Induced β-Amyloid Aggregation. J. Med. Chem. 2008, 51, 3588–3598. [Google Scholar] [CrossRef] [PubMed]
- Codony, S.; Pont, C.; Griñán-Ferré, C.; Di Pede-Mattatelli, A.; Calvó-Tusell, C.; Feixas, F.; Osuna, S.; Jarné-Ferrer, J.; Naldi, M.; Bartolini, M.; et al. Discovery and In Vivo Proof of Concept of a Highly Potent Dual Inhibitor of Soluble Epoxide Hydrolase and Acetylcholinesterase for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2022, 65, 4909–4925. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C.; Viayna, E.; Sola, I.; Formosa, X.; Camps, P.; Badia, A.; Clos, M.V.; Relat, J.; Ratia, M.; Bartolini, M.; et al. Huprine–Tacrine Heterodimers as Anti-Amyloidogenic Compounds of Potential Interest against Alzheimer’s and Prion Diseases. J. Med. Chem. 2012, 55, 661–669. [Google Scholar] [CrossRef]
- Cen, J.; Guo, H.; Hong, C.; Lv, J.; Yang, Y.; Wang, T.; Fang, D.; Luo, W.; Wang, C. Development of tacrine-bifendate conjugates with improved cholinesterase inhibitory and pro-cognitive efficacy and reduced hepatotoxicity. Eur. J. Med. Chem. 2018, 144, 128–136. [Google Scholar] [CrossRef]
- Xu, A.; He, F.; Zhang, X.; Li, X.; Ran, Y.; Wei, C.; James Chou, C.; Zhang, R.; Wu, J. Tacrine-hydroxamate derivatives as multitarget-directed ligands for the treatment of Alzheimer’s disease: Design, synthesis, and biological evaluation. Bioorg. Chem. 2020, 98, 103721. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, X.-L.; Wu, P.-F.; Wang, C.-M.; Cao, H.; Li, L.; Guo, X.-J.; Wang, F.; Xie, N.; Jiang, F.-C.; et al. Multifunctional Mercapto-tacrine Derivatives for Treatment of Age-Related Neurodegenerative Diseases. J. Med. Chem. 2012, 55, 3588–3592. [Google Scholar] [CrossRef]
- Keri, R.S.; Quintanova, C.; Chaves, S.; Silva, D.F.; Cardoso, S.M.; Santos, M.A. New Tacrine Hybrids with Natural-Based Cysteine Derivatives as Multitargeted Drugs for Potential Treatment of Alzheimer’s Disease. Chem. Biol. Drug Des. 2016, 87, 101–111. [Google Scholar] [CrossRef]
- Cheng, X.; Gu, J.; Pang, Y.; Liu, J.; Xu, T.; Li, X.; Hua, Y.; Newell, K.A.; Huang, X.-F.; Yu, Y.; et al. Tacrine–Hydrogen Sulfide Donor Hybrid Ameliorates Cognitive Impairment in the Aluminum Chloride Mouse Model of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 3500–3509. [Google Scholar] [CrossRef]
- Camps, P.; Formosa, X.; Galdeano, C.; Muñoz-Torrero, D.; Ramírez, L.; Gómez, E.; Isambert, N.; Lavilla, R.; Badia, A.; Clos, M.V.; et al. Pyrano[3,2-c]quinoline−6-Chlorotacrine Hybrids as a Novel Family of Acetylcholinesterase- and β-Amyloid-Directed Anti-Alzheimer Compounds. J. Med. Chem. 2009, 52, 5365–5379. [Google Scholar] [CrossRef]
- Di Pietro, O.; Pérez-Areales, F.; Juárez-Jiménez, J.; Espargaró, A.; Clos, M.V.; Pérez, B.; Lavilla, R.; Sabaté, R.; Luque, F.J.; Muñoz-Torrero, D. Tetrahydrobenzo[h][1,6]naphthyridine-6-chlorotacrine hybrids as a new family of anti-Alzheimer agents targeting β-amyloid, tau, and cholinesterase pathologies. Eur. J. Med. Chem. 2014, 84, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Costa, J.S.; Lopes, J.P.B.; Russowsky, D.; Petzhold, C.L.; de Borges, A.C.; Ceschi, M.A.; Konrath, E.; Batassini, C.; Lunardi, P.S.; Gonçalves, C.A.S. Synthesis of tacrine-lophine hybrids via one-pot four component reaction and biological evaluation as acetyl- and butyrylcholinesterase inhibitors. Eur. J. Med. Chem. 2013, 62, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marco-Contelles, J.; León, R.; de los Ríos, C.; Guglietta, A.; Terencio, J.; López, M.; García, A.; Villarroya, M. Novel Multipotent Tacrine−Dihydropyridine Hybrids with Improved Acetylcholinesterase Inhibitory and Neuroprotective Activities as Potential Drugs for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2006, 49, 7607–7610. [Google Scholar] [CrossRef] [PubMed]
- Marco-Contelles, J.; León, R.; de los Ríos, C.; Samadi, A.; Bartolini, M.; Andrisano, V.; Huertas, O.; Barril, X.; Luque, F.J.; Rodríguez-Franco, M.; et al. Tacripyrines, the First Tacrine−Dihydropyridine Hybrids, as Multitarget-Directed Ligands for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2009, 52, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, M.; Pistolozzi, M.; Andrisano, V.; Egea, J.; López, M.G.; Iriepa, I.; Moraleda, I.; Gálvez, E.; Marco-Contelles, J.; Samadi, A. Chemical and Pharmacological Studies on Enantiomerically Pure p-Methoxytacripyrines, Promising Multi-Target-Directed Ligands for the Treatment of Alzheimer’s Disease. ChemMedChem 2011, 6, 1990–1997. [Google Scholar] [CrossRef]
- Wang, X.-L.; Xiong, Y.; Yang, Y.; Tuo, Q.; Wang, X.; Chen, R.; Tian, Q.; Zhang, Z.; Yan, X.; Yang, Z.; et al. A novel tacrine-dihydropyridine hybrid (-)SCR1693 induces tau dephosphorylation and inhibits Aβ generation in cells. Eur. J. Pharmacol. 2015, 754, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Chioua, M.; Buzzi, E.; Moraleda, I.; Iriepa, I.; Maj, M.; Wnorowski, A.; Giovannini, C.; Tramarin, A.; Portali, F.; Ismaili, L.; et al. Tacripyrimidines, the first tacrine-dihydropyrimidine hybrids, as multi-target-directed ligands for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 155, 839–846. [Google Scholar] [CrossRef]
- Sola, I.; Aso, E.; Frattini, D.; López-González, I.; Espargaró, A.; Sabaté, R.; Di Pietro, O.; Luque, F.J.; Clos, M.V.; Ferrer, I.; et al. Novel Levetiracetam Derivatives That Are Effective against the Alzheimer-like Phenotype in Mice: Synthesis, in Vitro, ex Vivo, and in Vivo Efficacy Studies. J. Med. Chem. 2015, 58, 6018–6032. [Google Scholar] [CrossRef]
- Więckowska, A.; Kołaczkowski, M.; Bucki, A.; Godyń, J.; Marcinkowska, M.; Więckowski, K.; Zaręba, P.; Siwek, A.; Kazek, G.; Głuch-Lutwin, M.; et al. Novel multi-target-directed ligands for Alzheimer’s disease: Combining cholinesterase inhibitors and 5-HT 6 receptor antagonists. Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2016, 124, 63–81. [Google Scholar] [CrossRef]
- Więckowska, A.; Wichur, T.; Godyń, J.; Bucki, A.; Marcinkowska, M.; Siwek, A.; Więckowski, K.; Zaręba, P.; Knez, D.; Głuch-Lutwin, M.; et al. Novel Multitarget-Directed Ligands Aiming at Symptoms and Causes of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1195–1214. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Xu, Y.; Liu, W.; Gong, Q.; Wang, W.; Qiu, X.; Zhu, J.; Mao, F.; Zhang, H.; et al. Novel Vilazodone–Tacrine Hybrids as Potential Multitarget-Directed Ligands for the Treatment of Alzheimer’s Disease Accompanied with Depression: Design, Synthesis, and Biological Evaluation. ACS Chem. Neurosci. 2017, 8, 2708–2721. [Google Scholar] [CrossRef] [PubMed]
- Elsinghorst, P.W.; Cieslik, J.S.; Mohr, K.; Tränkle, C.; Gütschow, M. First Gallamine−Tacrine Hybrid: Design and Characterization at Cholinesterases and the M 2 Muscarinic Receptor. J. Med. Chem. 2007, 50, 5685–5695. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Jumpertz, S.; Zhang, Y.; Appenroth, D.; Fleck, C.; Mohr, K.; Tränkle, C.; Decker, M. Hybrid Molecules from Xanomeline and Tacrine: Enhanced Tacrine Actions on Cholinesterases and Muscarinic M 1 Receptors. J. Med. Chem. 2010, 53, 2094–2103. [Google Scholar] [CrossRef]
- Hepnarova, V.; Korabecny, J.; Matouskova, L.; Jost, P.; Muckova, L.; Hrabinova, M.; Vykoukalova, N.; Kerhartova, M.; Kucera, T.; Dolezal, R.; et al. The concept of hybrid molecules of tacrine and benzyl quinolone carboxylic acid (BQCA) as multifunctional agents for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 150, 292–306. [Google Scholar] [CrossRef]
- Maspero, M.; Volpato, D.; Cirillo, D.; Chen, N.Y.; Messerer, R.; Sotriffer, C.; De Amici, M.; Holzgrabe, U.; Dallanoce, C. Tacrine-xanomeline and tacrine-iperoxo hybrid ligands: Synthesis and biological evaluation at acetylcholinesterase and M1 muscarinic acetylcholine receptors. Bioorg. Chem. 2020, 96, 103633. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.H.M.; Coolen, H.K.A.C.; van der Neut, M.A.W.; Borst, A.J.M.; Stork, B.; Verveer, P.C.; Kruse, C.G. Design, Synthesis, Biological Properties, and Molecular Modeling Investigations of Novel Tacrine Derivatives with a Combination of Acetylcholinesterase Inhibition and Cannabinoid CB 1 Receptor Antagonism. J. Med. Chem. 2010, 53, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Kral, J.; Horova, A.; Musilek, K.; Soukup, O.; Drtinova, L.; Gazova, Z.; Siposova, K.; Kuca, K. 7-Methoxytacrine-Adamantylamine Heterodimers as Cholinesterase Inhibitors in Alzheimer’s Disease Treatment—Synthesis, Biological Evaluation and Molecular Modeling Studies. Molecules 2013, 18, 2397–2418. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Areales, F.J.; Turcu, A.L.; Barniol-Xicota, M.; Pont, C.; Pivetta, D.; Espargaró, A.; Bartolini, M.; De Simone, A.; Andrisano, V.; Pérez, B.; et al. A novel class of multitarget anti-Alzheimer benzohomoadamantane chlorotacrine hybrids modulating cholinesterases and glutamate NMDA receptors. Eur. J. Med. Chem. 2019, 180, 613–626. [Google Scholar] [CrossRef] [Green Version]
- Ceschi, M.A.; da Costa, J.S.; Lopes, J.P.B.; Câmara, V.S.; Campo, L.F.; de Borges, A.C.; Gonçalves, C.; de Souza, D.; Konrath, E.L.; Karl, A.L.M.; et al. Novel series of tacrine-tianeptine hybrids: Synthesis, cholinesterase inhibitory activity, S100B secretion and a molecular modeling approach. Eur. J. Med. Chem. 2016, 121, 758–772. [Google Scholar] [CrossRef]
- Lu, C.; Zhou, Q.; Yan, J.; Du, Z.; Huang, L.; Li, X. A novel series of tacrine–selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2013, 62, 745–753. [Google Scholar] [CrossRef]
- Xie, S.-S.; Wang, X.; Jiang, N.; Yu, W.; Wang, K.D.G.; Lan, J.-S.; Li, Z.-R.; Kong, L.-Y. Multi-target tacrine-coumarin hybrids: Cholinesterase and monoamine oxidase B inhibition properties against Alzheimer’s disease. Eur. J. Med. Chem. 2015, 95, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.-S.; Wang, X.-B.; Li, J.-Y.; Yang, L.; Kong, L.-Y. Design, synthesis and evaluation of novel tacrine–coumarin hybrids as multifunctional cholinesterase inhibitors against Alzheimer’s disease. Eur. J. Med. Chem. 2013, 64, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Hamulakova, S.; Janovec, L.; Hrabinova, M.; Spilovska, K.; Korabecny, J.; Kristian, P.; Kuca, K.; Imrich, J. Synthesis and Biological Evaluation of Novel Tacrine Derivatives and Tacrine−Coumarin Hybrids as Cholinesterase Inhibitors. J. Med. Chem. 2014, 57, 7073–7084. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Wang, X.-B.; Xie, S.-S.; Jiang, N.; Wang, K.D.G.; Yao, H.-Q.; Sun, H.-B.; Kong, L.-Y. Multifunctional tacrine–flavonoid hybrids with cholinergic, β-amyloid-reducing, and metal chelating properties for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2013, 69, 632–646. [Google Scholar] [CrossRef]
- Viayna, E.; Sola, I.; Bartolini, M.; De Simone, A.; Tapia-Rojas, C.; Serrano, F.G.; Sabaté, R.; Juárez-Jiménez, J.; Pérez, B.; Luque, F.J.; et al. Synthesis and Multitarget Biological Profiling of a Novel Family of Rhein Derivatives As Disease-Modifying Anti-Alzheimer Agents. J. Med. Chem. 2014, 57, 2549–2567. [Google Scholar] [CrossRef] [Green Version]
- Thiratmatrakul, S.; Yenjai, C.; Waiwut, P.; Vajragupta, O.; Reubroycharoen, P.; Tohda, M.; Boonyarat, C. Synthesis, biological evaluation and molecular modeling study of novel tacrine–carbazole hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 75, 21–30. [Google Scholar] [CrossRef]
- Spilovska, K.; Korabecny, J.; Sepsova, V.; Jun, D.; Hrabinova, M.; Jost, P.; Muckova, L.; Soukup, O.; Janockova, J.; Kucera, T.; et al. Novel Tacrine-Scutellarin Hybrids as Multipotent Anti-Alzheimer’s Agents: Design, Synthesis and Biological Evaluation. Molecules 2017, 22, 1006. [Google Scholar] [CrossRef] [Green Version]
- Jeřábek, J.; Uliassi, E.; Guidotti, L.; Korábečný, J.; Soukup, O.; Sepsova, V.; Hrabinova, M.; Kuča, K.; Bartolini, M.; Peña-Altamira, L.E.; et al. Tacrine-resveratrol fused hybrids as multi-target-directed ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2017, 127, 250–262. [Google Scholar] [CrossRef]
- Lopes, J.P.B.; Silva, L.; da Costa Franarin, G.; Ceschi, M.A.; Lüdtke, D.S.; Dantas, R.F.; de Salles, C.; Silva, F.P., Jr.; Senger, M.R.; Guedes, I.A.; et al. Design, synthesis, cholinesterase inhibition and molecular modelling study of novel tacrine hybrids with carbohydrate derivatives. Bioorg. Med. Chem. 2018, 26, 5566–5577. [Google Scholar] [CrossRef]
- Chalupova, K.; Korabecny, J.; Bartolini, M.; Monti, B.; Lamba, D.; Caliandro, R.; Pesaresi, A.; Brazzolotto, X.; Gastellier, A.-J.; Nachon, F.; et al. Novel tacrine-tryptophan hybrids: Multi-target directed ligands as potential treatment for Alzheimer’s disease. Eur. J. Med. Chem. 2019, 168, 491–514. [Google Scholar] [CrossRef]
- Cheng, Z.-Q.; Zhu, K.-K.; Zhang, J.; Song, J.-L.; Muehlmann, L.A.; Jiang, C.-S.; Liu, C.-L.; Zhang, H. Molecular-docking-guided design and synthesis of new IAA-tacrine hybrids as multifunctional AChE/BuChE inhibitors. Bioorg. Chem. 2019, 83, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Freschi, M.; de Camargo Nascente, L.; Salerno, A.; de Melo Viana Teixeira, S.; Nachon, F.; Chantegreil, F.; Soukup, O.; Prchal, L.; Malaguti, M.; et al. Sustainable Drug Discovery of Multi-Target-Directed Ligands for Alzheimer’s Disease. J. Med. Chem. 2021, 64, 4972–4990. [Google Scholar] [CrossRef] [PubMed]
- Elsinghorst, P.W.; González Tanarro, C.M.; Gütschow, M. Novel Heterobivalent Tacrine Derivatives as Cholinesterase Inhibitors with Notable Selectivity Toward Butyrylcholinesterase. J. Med. Chem. 2006, 49, 7540–7544. [Google Scholar] [CrossRef]
- Chen, X.; Wehle, S.; Kuzmanovic, N.; Merget, B.; Holzgrabe, U.; König, B.; Sotriffer, C.A.; Decker, M. Acetylcholinesterase Inhibitors with Photoswitchable Inhibition of β-Amyloid Aggregation. ACS Chem. Neurosci. 2014, 5, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepovimova, E.; Uliassi, E.; Korabecny, J.; Peña-Altamira, L.E.; Samez, S.; Pesaresi, A.; Garcia, G.E.; Bartolini, M.; Andrisano, V.; Bergamini, C.; et al. Multitarget Drug Design Strategy: Quinone–Tacrine Hybrids Designed To Block Amyloid-β Aggregation and To Exert Anticholinesterase and Antioxidant Effects. J. Med. Chem. 2014, 57, 8576–8589. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Li, J.; Wei, H.; Huang, L.; Li, X. Tacrine-propargylamine derivatives with improved acetylcholinesterase inhibitory activity and lower hepatotoxicity as a potential lead compound for the treatment of Alzheimers disease. J. Enzyme Inhib. Med. Chem. 2015, 30, 995–1001. [Google Scholar] [CrossRef]
- Korabecny, J.; Andrs, M.; Nepovimova, E.; Dolezal, R.; Babkova, K.; Horova, A.; Malinak, D.; Mezeiova, E.; Gorecki, L.; Sepsova, V.; et al. 7-Methoxytacrine-p-Anisidine Hybrids as Novel Dual Binding Site Acetylcholinesterase Inhibitors for Alzheimer’s Disease Treatment. Molecules 2015, 20, 22084–22101. [Google Scholar] [CrossRef]
- Najafi, Z.; Mahdavi, M.; Saeedi, M.; Karimpour-Razkenari, E.; Asatouri, R.; Vafadarnejad, F.; Moghadam, F.H.; Khanavi, M.; Sharifzadeh, M.; Akbarzadeh, T. Novel tacrine-1,2,3-triazole hybrids: In vitro, in vivo biological evaluation and docking study of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 125, 1200–1212. [Google Scholar] [CrossRef]
- Riazimontazer, E.; Sadeghpour, H.; Nadri, H.; Sakhteman, A.; Tüylü Küçükkılınç, T.; Miri, R.; Edraki, N. Design, synthesis and biological activity of novel tacrine-isatin Schiff base hybrid derivatives. Bioorg. Chem. 2019, 89, 103006. [Google Scholar] [CrossRef]
- Yao, H.; Uras, G.; Zhang, P.; Xu, S.; Yin, Y.; Liu, J.; Qin, S.; Li, X.; Allen, S.; Bai, R.; et al. Discovery of Novel Tacrine–Pyrimidone Hybrids as Potent Dual AChE/GSK-3 Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2021, 64, 7483–7506. [Google Scholar] [CrossRef]
- Ozten, O.; Zengin Kurt, B.; Sonmez, F.; Dogan, B.; Durdagi, S. Synthesis, molecular docking and molecular dynamics studies of novel tacrine-carbamate derivatives as potent cholinesterase inhibitors. Bioorg. Chem. 2021, 115, 105225. [Google Scholar] [CrossRef] [PubMed]
- Przybyłowska, M.; Dzierzbicka, K.; Kowalski, S.; Demkowicz, S.; Daśko, M.; Inkielewicz-Stepniak, I. Design, synthesis and biological evaluation of novel N -phosphorylated and O -phosphorylated tacrine derivatives as potential drugs against Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2022, 37, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.C.; Liu, X.C.; Yang, S.H.; Song, L.L.; Zhou, S.J.; Deng, S.L.; Tian, L.; Cheng, L.Y. Melatonin Inhibits Oxidative Stress and Apoptosis in Cryopreserved Ovarian Tissues via Nrf2/HO-1 Signaling Pathway. Front. Mol. Biosci. 2020, 7, 163. [Google Scholar] [CrossRef]
- Rosini, M.; Andrisano, V.; Bartolini, M.; Bolognesi, M.L.; Hrelia, P.; Minarini, A.; Tarozzi, A.; Melchiorre, C. Rational Approach To Discover Multipotent Anti-Alzheimer Drugs. J. Med. Chem. 2005, 48, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Jia, X.; Wang, D.; Wei, C.; He, Y.; Chen, L.; Zhao, Y. Silibinin as a natural antioxidant for modifying polysulfone membranes to suppress hemodialysis-induced oxidative stress. J. Memb. Sci. 2019, 574, 86–99. [Google Scholar] [CrossRef]
- Schewe, T. Molecular actions of Ebselen—An antiinflammatory antioxidant. Gen. Pharmacol. Vasc. Syst. 1995, 26, 1153–1169. [Google Scholar] [CrossRef]
- Porciúncula, L.O.; Rocha, J.B.T.; Boeck, C.R.; Vendite, D.; Souza, D.O. Ebselen prevents excitotoxicity provoked by glutamate in rat cerebellar granule neurons. Neurosci. Lett. 2001, 299, 217–220. [Google Scholar] [CrossRef]
- Wlodek, S.T.; Antosiewicz, J.; McCammon, J.A.; Straatsma, T.P.; Gilson, M.K.; Briggs, J.M.; Humblet, C.; Sussman, J.L. Binding of tacrine and 6-chlorotacrine by acetylcholinesterase. Biopolymers 1996, 38, 109–117. [Google Scholar] [CrossRef]
- Tran, T.-D.; Nguyen, T.-C.-V.; Nguyen, N.-S.; Nguyen, D.-M.; Nguyen, T.-T.-H.; Le, M.-T.; Thai, K.-M. Synthesis of Novel Chalcones as Acetylcholinesterase Inhibitors. Appl. Sci. 2016, 6, 198. [Google Scholar] [CrossRef] [Green Version]
- Mezeiova, E.; Soukup, O.; Korabecny, J. Huprines—An insight into the synthesis and biological properties. Russ. Chem. Rev. 2020, 89, 999–1039. [Google Scholar] [CrossRef]
- Yang, G.; Wang, Y.; Tian, J.; Liu, J.-P. Huperzine A for Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. PLoS ONE 2013, 8, e74916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.Y.; Tang, X.C. Neuroprotective effects of huperzine A: New therapeutic targets for neurodegenerative disease. Trends Pharmacol. Sci. 2006, 27, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Torrero, D.; Camps, P. Huprines for Alzheimer’s disease drug development. Expert Opin. Drug Discov. 2008, 3, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Park, S.U. A recent overview on the biological and pharmacological activities of ferulic acid. Excli J. 2019, 18, 132–138. [Google Scholar] [CrossRef]
- Meng, G.; Meng, X.; Ma, X.; Zhang, G.; Hu, X.; Jin, A.; Zhao, Y.; Liu, X. Application of Ferulic Acid for Alzheimer’s Disease: Combination of Text Mining and Experimental Validation. Front. Neuroinform. 2018, 12, 31. [Google Scholar] [CrossRef] [Green Version]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-Hydroxydopamine model of parkinson’s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef]
- Tsikas, D. Analysis of nitrite and nitrate in biological fluids by assays based on the Griess reaction: Appraisal of the Griess reaction in the l-arginine/nitric oxide area of research. J. Chromatogr. B 2007, 851, 51–70. [Google Scholar] [CrossRef]
- Kerwin, J.F.; Heller, M. The arginine-nitric oxide pathway: A target for new drugs. Med. Res. Rev. 1994, 14, 23–74. [Google Scholar] [CrossRef]
- Esplugues, J.V. NO as a signalling molecule in the nervous system. Br. J. Pharmacol. 2002, 135, 1079–1095. [Google Scholar] [CrossRef] [Green Version]
- Balez, R.; Ooi, L. Getting to NO Alzheimer’s Disease: Neuroprotection versus Neurotoxicity Mediated by Nitric Oxide. Oxid. Med. Cell. Longev. 2016, 2016, 3806157. [Google Scholar] [CrossRef]
- Webb, D.J.; Megson, I.L. Nitric oxide donor drugs: Current status and future trends. Expert Opin. Investig. Drugs 2002, 11, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, G.; Bennett, B.; Reynolds, J. Nitric Oxide Mimetic Molecules as Therapeutic Agents in Alzheimers Disease. Curr. Alzheimer Res. 2005, 2, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Chegaev, K.; Federico, A.; Marini, E.; Rolando, B.; Fruttero, R.; Morbin, M.; Rossi, G.; Fugnanesi, V.; Bastone, A.; Salmona, M.; et al. NO-donor thiacarbocyanines as multifunctional agents for Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 4688–4698. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Fernandez-Enright, F.; Huang, X.-F. Structural contributions of antipsychotic drugs to their therapeutic profiles and metabolic side effects. J. Neurochem. 2012, 120, 371–384. [Google Scholar] [CrossRef]
- Krasnovskaya, O.; Spector, D.; Zlobin, A.; Pavlov, K.; Gorelkin, P.; Erofeev, A.; Beloglazkina, E.; Majouga, A. Metals in Imaging of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9190. [Google Scholar] [CrossRef]
- Ono, M.; Kawashima, H.; Nonaka, A.; Kawai, T.; Haratake, M.; Mori, H.; Kung, M.-P.; Kung, H.F.; Saji, H.; Nakayama, M. Novel Benzofuran Derivatives for PET Imaging of β-Amyloid Plaques in Alzheimer’s Disease Brains. J. Med. Chem. 2006, 49, 2725–2730. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Dokmeci, D. Ibuprofen and Alzheimer’s disease. Folia Med. 2004, 46, 5–10. [Google Scholar]
- Lim, G.P.; Yang, F.; Chu, T.; Chen, P.; Beech, W.; Teter, B.; Tran, T.; Ubeda, O.; Ashe, K.H.; Frautschy, S.A.; et al. Ibuprofen Suppresses Plaque Pathology and Inflammation in a Mouse Model for Alzheimer’s Disease. J. Neurosci. 2000, 20, 5709–5714. [Google Scholar] [CrossRef] [Green Version]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, B.L.; Cramer, P.E.; Varvel, N.H.; Reed-Geaghan, E.; Jiang, Q.; Szabo, A.; Herrup, K.; Lamb, B.T.; Landreth, G.E. Ibuprofen attenuates oxidative damage through NOX2 inhibition in Alzheimer’s disease. Neurobiol. Aging 2012, 33, e21–e197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pike, A. Pyridones in drug discovery: Recent advances. Bioorg. Med. Chem. Lett. 2021, 38, 127849. [Google Scholar] [CrossRef] [PubMed]
- Carlier, P.R.; Du, D.M.; Han, Y.; Liu, J.; Pang, Y.P. Potent, easily synthesized huperzine A-tacrine hybrid acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 1999, 9, 2335–2338. [Google Scholar] [CrossRef] [PubMed]
- Camps, P.; El Achab, R.; Görbig, D.M.; Morral, J.; Muñoz-Torrero, D.; Badia, A.; Baños, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; et al. Synthesis, in Vitro Pharmacology, and Molecular Modeling of Very Potent Tacrine−Huperzine A Hybrids as Acetylcholinesterase Inhibitors of Potential Interest for the Treatment of Alzheimer’s Disease. J. Med. Chem. 1999, 42, 3227–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Kan, K.; Carlier, P.; Pang, Y.; Han, Y. East Meets West in the Search for Alzheimers Therapeutics–Novel Dimeric Inhibitors from Tacrine and Huperzine A. Curr. Alzheimer Res. 2007, 4, 386–396. [Google Scholar] [CrossRef]
- Mak, S.; Li, W.; Fu, H.; Luo, J.; Cui, W.; Hu, S.; Pang, Y.; Carlier, P.R.; Tsim, K.W.; Pi, R.; et al. Promising tacrine/huperzine A-based dimeric acetylcholinesterase inhibitors for neurodegenerative disorders: From relieving symptoms to modifying diseases through multitarget. J. Neurochem. 2021, 158, 1381–1393. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, M.L.; Copani, A.; Rizzarelli, E. A promising connection between BDNF and Alzheimer’s disease. Aging 2018, 10, 1791–1792. [Google Scholar] [CrossRef]
- Knowles, J. Donepezil in Alzheimer’s disease: An evidence-based review of its impact on clinical and economic outcomes. Core Evid. 2006, 1, 195–219. [Google Scholar]
- Alonso, D.; Dorronsoro, I.; Rubio, L.; Muñoz, P.; García-Palomero, E.; Del Monte, M.; Bidon-Chanal, A.; Orozco, M.; Luque, F.J.; Castro, A.; et al. Donepezil–tacrine hybrid related derivatives as new dual binding site inhibitors of AChE. Bioorg. Med. Chem. 2005, 13, 6588–6597. [Google Scholar] [CrossRef]
- Rose, T.E.; Morisseau, C.; Liu, J.-Y.; Inceoglu, B.; Jones, P.D.; Sanborn, J.R.; Hammock, B.D. 1-Aryl-3-(1-acylpiperidin-4-yl)urea Inhibitors of Human and Murine Soluble Epoxide Hydrolase: Structure−Activity Relationships, Pharmacokinetics, and Reduction of Inflammatory Pain. J. Med. Chem. 2010, 53, 7067–7075. [Google Scholar] [CrossRef] [Green Version]
- Jonnalagadda, D.; Wan, D.; Chun, J.; Hammock, B.D.; Kihara, Y. A Soluble Epoxide Hydrolase Inhibitor, 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) Urea, Ameliorates Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2021, 22, 4650. [Google Scholar] [CrossRef]
- Camps, P.; El Achab, R.; Morral, J.; Muñoz-Torrero, D.; Badia, A.; Baños, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; Luque, F.J. New Tacrine−Huperzine A Hybrids (Huprines): Highly Potent Tight-Binding Acetylcholinesterase Inhibitors of Interest for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2000, 43, 4657–4666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Wang, Q.; Li, Y. Synthesis and Biological Activity of Wuweizisu C and Analogs. Curr. Top. Med. Chem. 2009, 9, 1660–1675. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y. Effect of dimethyl diphenyl bicarboxylate (DDB) on 9-amino-1,2,3,4-tetrahydroacridine-induced hepatotoxicity in mice. Yao Xue Xue Bao 2001, 36, 493–497. [Google Scholar] [PubMed]
- Xu, K.; Dai, X.-L.; Huang, H.-C.; Jiang, Z.-F. Targeting HDACs: A Promising Therapy for Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2011, 2011, 143269. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Li, Y.; Tang, P.; Yuan, Q. Design, synthesis and biological evaluation of novel 2-aminobenzamides containing dithiocarbamate moiety as histone deacetylase inhibitors and potent antitumor agents. Eur. J. Med. Chem. 2018, 143, 320–333. [Google Scholar] [CrossRef]
- Jeong, W.-H.; Kim, W.-I.; Lee, J.-W.; Park, H.-K.; Song, M.-K.; Choi, I.-S.; Han, J.-Y. Modulation of Long-Term Potentiation by Gamma Frequency Transcranial Alternating Current Stimulation in Transgenic Mouse Models of Alzheimer’s Disease. Brain Sci. 2021, 11, 1532. [Google Scholar] [CrossRef]
- Yang, Y.-J.; Wu, P.-F.; Long, L.-H.; Yu, D.-F.; Wu, W.-N.; Hu, Z.-L.; Fu, H.; Xie, N.; Jin, Y.; Ni, L.; et al. Reversal of aging-associated hippocampal synaptic plasticity deficits by reductants via regulation of thiol redox and NMDA receptor function. Aging Cell. 2010, 9, 709–721. [Google Scholar] [CrossRef]
- Eto, K.; Asada, T.; Arima, K.; Makifuchi, T.; Kimura, H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 293, 1485–1488. [Google Scholar] [CrossRef]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.-J.; He, L.; Zhang, W.; Liu, C.-L.; Ai, Y.-Q.; Zhang, Q. Hydrogen sulfide attenuates surgical trauma-induced inflammatory response and cognitive deficits in mice. J. Surg. Res. 2013, 183, 330–336. [Google Scholar] [CrossRef]
- Macdonald, I.R.; Martin, E.; Rosenberry, T.L.; Darvesh, S. Probing the Peripheral Site of Human Butyrylcholinesterase. Biochemistry 2012, 51, 7046–7053. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.P.B.; Silva, L.; Ceschi, M.A.; Lüdtke, D.S.; Zimmer, A.R.; Ruaro, T.C.; Dantas, R.F.; de Salles, C.M.C.; Silva, F.P., Jr.; Senger, M.R.; et al. Synthesis of new lophine–carbohydrate hybrids as cholinesterase inhibitors: Cytotoxicity evaluation and molecular modeling. MedChemComm 2019, 10, 2089–2101. [Google Scholar] [CrossRef] [PubMed]
- Sobrado, M.; López, M.; Carceller, F.; García, A.; Roda, J. Combined nimodipine and citicoline reduce infarct size, attenuate apoptosis and increase bcl-2 expression after focal cerebral ischemia. Neuroscience 2003, 118, 107–113. [Google Scholar] [CrossRef]
- Shi, J.-Q.; Wang, B.-R.; Tian, Y.-Y.; Xu, J.; Gao, L.; Zhao, S.-L.; Jiang, T.; Xie, H.-G.; Zhang, Y.-D. Antiepileptics Topiramate and Levetiracetam Alleviate Behavioral Deficits and Reduce Neuropathology in APPswe/PS1dE9 Transgenic Mice. CNS Neurosci. Ther. 2013, 19, 871–881. [Google Scholar] [CrossRef]
- Camps, P.; Cusack, B.; Mallender, W.D.; El Achab, R.E.; Morral, J.; Muñoz-Torrero, D.; Rosenberry, T.L. Huprine X is a novel high-affinity inhibitor of acetylcholinesterase that is of interest for treatment of Alzheimer’s disease. Mol. Pharmacol. 2000, 57, 409–417. [Google Scholar] [PubMed]
- Schneider, L.S.; Geffen, Y.; Rabinowitz, J.; Thomas, R.G.; Schmidt, R.; Ropele, S.; Weinstock, M. Low-dose ladostigil for mild cognitive impairment. Neurology 2019, 93, e1474–e1484. [Google Scholar] [CrossRef]
- Bender, A.M.; Jones, C.K.; Lindsley, C.W. Classics in Chemical Neuroscience: Xanomeline. ACS Chem. Neurosci. 2017, 8, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Yeatman, H.R.; Lane, J.R.; Choy, K.H.C.; Lambert, N.A.; Sexton, P.M.; Christopoulos, A.; Canals, M. Allosteric Modulation of M1 Muscarinic Acetylcholine Receptor Internalization and Subcellular Trafficking. J. Biol. Chem. 2014, 289, 15856–15866. [Google Scholar] [CrossRef] [Green Version]
- Shirey, J.K.; Brady, A.E.; Jones, P.J.; Davis, A.A.; Bridges, T.M.; Kennedy, J.P.; Jadhav, S.B.; Menon, U.N.; Xiang, Z.; Watson, M.L.; et al. A Selective Allosteric Potentiator of the M1 Muscarinic Acetylcholine Receptor Increases Activity of Medial Prefrontal Cortical Neurons and Restores Impairments in Reversal Learning. J. Neurosci. 2009, 29, 14271–14286. [Google Scholar] [CrossRef] [Green Version]
- Xin, R.; Chen, Z.; Fu, J.; Shen, F.; Zhu, Q.; Huang, F. Xanomeline Protects Cortical Cells From Oxygen-Glucose Deprivation via Inhibiting Oxidative Stress and Apoptosis. Front. Physiol. 2020, 11, 656. [Google Scholar] [CrossRef]
- Micale, V.; Drago, F.; Noerregaard, P.K.; Elling, C.E.; Wotjak, C.T. The Cannabinoid CB1 Antagonist TM38837 With Limited Penetrance to the Brain Shows Reduced Fear-Promoting Effects in Mice. Front. Pharmacol. 2019, 10, 207. [Google Scholar] [CrossRef] [Green Version]
- Wise, L.E.; Iredale, P.A.; Stokes, R.J.; Lichtman, A.H. Combination of Rimonabant and Donepezil Prolongs Spatial Memory Duration. Neuropsychopharmacology 2007, 32, 1805–1812. [Google Scholar] [CrossRef] [Green Version]
- Torres, E.; Duque, M.D.; López-Querol, M.; Taylor, M.C.; Naesens, L.; Ma, C.; Pinto, L.H.; Sureda, F.X.; Kelly, J.M.; Vázquez, S. Synthesis of benzopolycyclic cage amines: NMDA receptor antagonist, trypanocidal and antiviral activities. Bioorg. Med. Chem. 2012, 20, 942–948. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Gomes, C.M. S100 Proteins in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease (Review). Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Shahid Nadeem, M.; Azam Khan, J.; Kazmi, I.; Rashid, U. Design, Synthesis, and Bioevaluation of Indole Core Containing 2-Arylidine Derivatives of Thiazolopyrimidine as Multitarget Inhibitors of Cholinesterases and Monoamine Oxidase A/B for the Treatment of Alzheimer Disease. ACS Omega 2022, 7, 9369–9379. [Google Scholar] [CrossRef]
- Sterling, J.; Herzig, Y.; Goren, T.; Finkelstein, N.; Lerner, D.; Goldenberg, W.; Miskolczi, I.; Molnar, S.; Rantal, F.; Tamas, T.; et al. Novel dual inhibitors of AChE and MAO derived from hydroxy aminoindan and phenethylamine as potential treatment for Alzheimer’s disease. J. Med. Chem. 2002, 45, 5260–5279. [Google Scholar] [CrossRef]
- Fowler, J.S.; Logan, J.; Volkow, N.D.; Shumay, E.; McCall-Perez, F.; Jayne, M.; Wang, G.-J.; Alexoff, D.L.; Apelskog-Torres, K.; Hubbard, B.; et al. Evidence that Formulations of the Selective MAO-B Inhibitor, Selegiline, which Bypass First-Pass Metabolism, also Inhibit MAO-A in the Human Brain. Neuropsychopharmacology 2015, 40, 650–657. [Google Scholar] [CrossRef]
- Piazzi, L.; Rampa, A.; Bisi, A.; Gobbi, S.; Belluti, F.; Cavalli, A.; Bartolini, M.; Andrisano, V.; Valenti, P.; Recanatini, M. 3-(4-{[Benzyl(methyl)amino]methyl}phenyl)-6,7-dimethoxy-2 H -2-chromenone (AP2238) Inhibits Both Acetylcholinesterase and Acetylcholinesterase-Induced β-Amyloid Aggregation: A Dual Function Lead for Alzheimer’s Disease Therapy. J. Med. Chem. 2003, 46, 2279–2282. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Bizzarri, B.; Granese, A.; Carradori, S.; Yáñez,, M.; Orallo, F.; Ortuso, F.; et al. Synthesis, Molecular Modeling, and Selective Inhibitory Activity against Human Monoamine Oxidases of 3-Carboxamido-7-Substituted Coumarins. J. Med. Chem. 2009, 52, 1935–1942. [Google Scholar] [CrossRef]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bubley, A.; Erofeev, A.; Gorelkin, P.; Beloglazkina, E.; Majouga, A.; Krasnovskaya, O. Tacrine-Based Hybrids: Past, Present, and Future. Int. J. Mol. Sci. 2023, 24, 1717. https://doi.org/10.3390/ijms24021717
Bubley A, Erofeev A, Gorelkin P, Beloglazkina E, Majouga A, Krasnovskaya O. Tacrine-Based Hybrids: Past, Present, and Future. International Journal of Molecular Sciences. 2023; 24(2):1717. https://doi.org/10.3390/ijms24021717
Chicago/Turabian StyleBubley, Anna, Alexaner Erofeev, Peter Gorelkin, Elena Beloglazkina, Alexander Majouga, and Olga Krasnovskaya. 2023. "Tacrine-Based Hybrids: Past, Present, and Future" International Journal of Molecular Sciences 24, no. 2: 1717. https://doi.org/10.3390/ijms24021717
APA StyleBubley, A., Erofeev, A., Gorelkin, P., Beloglazkina, E., Majouga, A., & Krasnovskaya, O. (2023). Tacrine-Based Hybrids: Past, Present, and Future. International Journal of Molecular Sciences, 24(2), 1717. https://doi.org/10.3390/ijms24021717