Abstract
A new coordination compound of copper(II) with a tolfenamate ligand of the paddle-wheel-like structure [CuII2(Tolf)4(MeOH)2]∙2MeOH was obtained and structurally characterized. Chemical bonds of Cu(II)∙∙∙Cu(II) and Cu(II)–O were theoretically analyzed and compared with the results for selected similar structures from the CSD database. QTAIM analysis showed that the Cu(II)∙∙∙Cu(II) interaction has a strength comparable to a hydrogen bond, as indicated by the electron density at a critical point. The remaining QTAIM parameters indicate stability of the Cu(II)∙∙∙Cu(II) interaction. Other methods, such as NCI and NBO, also indicate a significant strength of this interaction. Thus, the Cu(II)∙∙∙Cu(II) interaction can be treated as one of the noncovalent interactions that affects the structure of the coordination compound, the packing of molecules in the crystal, and the general properties of the compound.
1. Introduction
Tolfenamic acid belongs to the group of fenamic acids (N-arylanthranilic acids), which are very popular compounds among the non-steroidal anti-inflammatory drugs (NSAIDs) that have long been used to treat inflammation, pain, and fever. Many NSAIDs exhibit chemopreventive and chemosuppressive effects [1,2,3,4,5] in different types of cancer [6,7]. The fenamic acids commonly used in pharmacy are mefenamic, tolfenamic, flufenamic, and meclofenamic acid. The clinical action and effectiveness of tolfenamic acid are known in the treatment of migraines [8], dysmenorrhea [9], as well as in rheumatic diseases [10,11]. All fenamic acids have undesired side effects; therefore, the simplest compound in this group—fenamic acid—is out of therapeutic application. Apart from side effects, the problem limiting the application of fenamic acids is their low water solubility, which a determinant of their low bioavailability. One of the best methods of improving the water solubility and bioavailability is the synthesis of fenamic acid salts with metal cations [12,13,14]. An additional advantage of using salts with metal cations is the supply of cations that play an important role in biological systems [15,16]. Among metal cations which can be used to produce salts or coordination compounds with organic drugs, Cu2+ plays an essential role due to its relatively low toxicity in comparison to other metal cation compounds [17,18], synergistic inflammatory properties [19], and potential anticancer properties [20,21,22].
The physical, biological, and therapeutic drug properties are related to the molecular structure; therefore, the crystal structure of a new drug is crucial in predicting and explaining properties, and determining the crystal structure is the first step in research leading to the design of a new drug.
In this paper we have undertaken determination of the crystal structure of a new dinuclear copper(II) coordination compound with tolfenamate ligands of the formula [CuII2(Tolf)4(MeOH)2]∙2MeOH (Tolf = tolfenamate anion). The most characteristic feature of this structure is the paddle-wheel entity with the bond linking two Cu2+ cations and bonds linking the cations with the carboxylate groups.
It is known that apart from typical covalent bonds, there are many noncovalent bonds, which, despite being weaker than covalent ones, very often determine the packing of molecules in the crystal, as well as their physicochemical properties. Commonly known noncovalent interactions that are fundamental in natural compounds are hydrogen bonds, ion–ion, van der Waals, and dispersive interaction [23] The range of typical noncovalent interactions known for a very long time has recently expanded to include pnictogen interactions [24,25,26]. Therefore, the question arises whether the interaction of Cu(II)∙∙∙Cu(II) is also one of the noncovalent interactions. An interesting approach seems to be to consider what type of bond Cu(II)∙∙∙Cu(II) is in coordination compounds of drugs, in terms of currently used theoretical methods, in order to broaden the knowledge of chemical bonds. Research into the nature of chemical bonds, in particular metal–metal bonds in multinuclear coordination compounds, may be crucial in the analysis of clusters and multinuclear compounds occurring in biological systems. Therefore, the search for answers to fundamental questions about the nature and strength of chemical bonds, including metal–metal bonds, using current theoretical methods may in the future contribute to the clarification of their properties and find correlations between structure and function.
On the basis of a Cambridge Structural Database (CSD, Version 5.43) [27] search, two other representative structures were selected for which the Cu2+∙∙∙Cu2+ and Cu2+–O bond lengths were also analyzed. The proposed Cu(II) paddle-wheel-like structures for which theoretical studies were performed are [Cu2(CH3COO)4(Hbet)2] (CSD refcode: QEBQIX) [28], the structure of a Cu(II) coordination compound with betaine, and [Cu2(CH3COO)4(CH4N2O)2]∙H2O (CSD refcode: ACURCU01) [29], a Cu(II) compound with urea.
To analyze the character of copper–copper bonds, much theoretical research has been done. The first investigations for metal–metal bonds were carried out by Hoffmann and co-workers [30,31] and Cotton et al. [32]. Copper(I) dinuclear coordination compounds were extensively studied by Pyykkӧ and co-workers [33,34] and Carvajal et al. [35]. Recent research by Dinda and Samuelson presented the AIM analysis for Cu(I)∙∙∙Cu(I) bonds in copper(I) compounds [36].
In this work, the theoretical analysis for Cu(II)∙∙∙Cu(II) and Cu(II)–O bonds using the quantum theory of atoms in molecules (QTAIM) [37] has been carried out. According to this theory, two atoms are bonded if they are located at a common bond path with a bond critical point (BCP), which is a saddle point of the electron density on the gradient path. Electron density at the BCP is directly related to the bond strength [38,39], and the sign of the Laplacian of the electron density at the BCP indicates the interaction type. Concentration of electron density between atoms is connected with the negative sign of the Laplacian characteristic for the shared shell interaction—covalent and polarized bonds and depletion of the electron density at the BCP are connected to the positive sign of the Laplacian characteristic for the open-shell interaction, such as the hydrogen bond, van der Waals, and ionic interaction [40]. Stability of the bond is described by ellipticity (ε) of the electron clouds at the BCP [41,42] and the nonlinearity of the bond path, which cannot be too bent to avoid bond instability. The chemical bond and intermolecular interaction can be also characterized with the atomic net charge—q(A), the electron population of a particular atom N(A), %δ(A,B)—the average number of electrons in atom A that are delocalized between atom A and other atoms in the molecule, Bond(A,A’)/2—the number of electron pairs in atom A that are delocalized (shared) between atom A and atoms to which atom A is bonded, δ(A,B)—the electron delocalization index for atoms A and B, i.e., the average number of electrons delocalized (shared) between atoms A and B obtained by numerical integration of the charge density over the basin of the atom.
In the studied crystals, weak interactions were also analyzed by the Noncovalent Interaction (NCI) approach [43]. So far, the presented work is the first to discuss the nature of interactions between copper(II) ions in polynuclear coordination compounds using theoretical methods for characterizing noncovalent interactions, the QTAIM and NCI methods.
2. Results
To date, the Cambridge Structural Database [27] presents 1613 hits corresponding to the search for paddle-wheel-like double-core Cu(II) deposited structures with carboxylate ligands. The structures were searched for using the following criteria: the range for Cu(II)∙∙∙Cu(II) distance at 2.40 to 3.27 Å. Among this, 778 hits are the structures of compounds in which, in addition to carboxylates, there are also other ligands coordinating to copper(II) by an oxygen atom, while in 672 hits, non-carboxylate ligands are linked to Cu(II) via a nitrogen atom. These two groups of compounds containing, in addition to the carboxylate ligands, O- or N-donor ligands, respectively, differ somewhat in the length of the Cu(II)∙∙∙Cu(II) bonds. Compounds with O-donor ligands tend to form slightly shorter Cu(II)∙∙∙Cu(II) bonds compared to compounds with N-donor ligands, as shown by histograms generated in the CSD [27] (upper plot for structures with only O-donor ligands, bottom plot for structures with N-donor ligands in addition to carboxylates) (Figure 1, Figure S7).
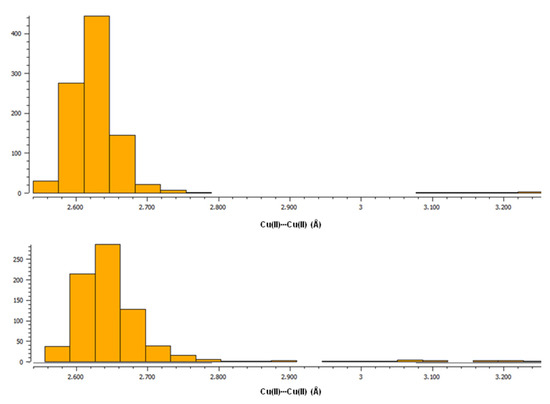
Figure 1.
Histograms from CSD [27] showing the tendency of Cu(II)∙∙∙Cu(II) bond lengths in paddle-wheel-like double-core Cu(II) compounds with carboxylate ligands. (Top): a plot for structures with only O-donor ligands; (Bottom): a plot for structures with N-donor ligands apart from carboxylates.
In this paper, the crystal structure of a new paddle-wheel-like double-core Cu(II) coordination compound with tolfenamate and methanol ligands is presented, tetrakis(μ2-2-((3-Chloro-2-methylphenyl)amino)benzoato-O,O’)-bis(methanol)-dicopper(II)(Cu–Cu)–methanol(1/1) (Table 1). So far, the analogous structure of copper(II) compound with tolfenamate ligands and DMF molecules, tetrakis(μ2-2-((3-Chloro-2-methylphenyl)amino)benzoato- O,O’)-bis(N,N-dimethylformamide-O)-dicopper(II)(Cu–Cu) has been reported previously (CSD refcode: ARALUA) [44]. According to the data, fourteen paddle-wheel-like structures with fenamate ligands have been published [44,45,46,47,48,49,50,51,52]. The Cu(II)∙∙∙Cu(II) bond length for the studied [CuII2(Tolf)4(MeOH)2]∙2MeOH equals 2.5880(4) Å and is in the range of bond lengths typical for this group of compounds, ranging between 2.572 Å (for EDUBOX [45] —a compound with flufenamate and water ligands) and 2.627 Å (for SUTPIG [46]—a compound with mefenamate and DMSO ligands). For the aforementioned compound with tolfenamate and DMF ligands (ARALUA [44]), the copper(II)∙∙∙copper(II) bond equals 2.6074(18) A. In the case of structures coordinated with fenamates and ethanol or methanol molecules, the bond lengths are about 2.58 Å (POMHOP [48], EDUBUD [45]) and 2.59 Å (MADTES [49]).

Table 1.
Crystallographic data and refinement parameters for [CuII2(Tolf)4(MeOH)2]∙2MeOH.
Moreover, in the Cambridge Structural Database, there are also two paddle-wheel structures with a fenamate ligand and N-coordinated ligands as well, viz., with vinylpyridine (DIFQUG [53]) and with 4-methylpyridin-2-amine (LIFBAF [54]), for which the Cu(II)∙∙∙Cu(II) bond lengths equal 2.63 and 2.64 Å, respectively; this is in line with the observed trends in Figure 1. In our studies we compared the theoretical analysis for [CuII2(Tolf)4(MeOH)2]∙2MeOH with the results obtained for two selected structures from the CSD database of [Cu2(CH3COO)4(Hbet)2] (Hbet = betaine) (QEBQIX) [28] and [Cu2(CH3COO)4(CH4N2O)2]∙H2O (CH4N2O2—urea) (CSD refcode ACURCU01) [29]. When selecting structures, we took into account low R-factors, the simplicity of structures with a low molecular weight, as well as the Cu(II)∙∙∙Cu(II) bond lengths from the typical range for paddle-wheel-like structures with O-donor ligands and from the range of the largest distances. For QEBQIX, the Cu2+∙∙∙Cu2+ distance equals 2.6897(7) Å, and for ACURCU01, it is 2.624(1) Å; these distances are significantly longer compared to the investigated compound (2.5844(4) Å).
2.1. Crystal Structure of [CuII2(Tolf)4(MeOH)2]∙2MeOH
The studied compound tetrakis(μ2-2-((3-chloro-2-methylphenyl)amino)benzoate -O,O’)-bis(methanol)-dicopper(II)(Cu–Cu)–methanol(1/1) of the formula [CuII2(Tolf)4(MeOH)2]∙2MeOH (Tolf = tolfenamate anion) is a dinuclear copper(II) coordination compound. The compound is composed of four tolfenamate anions and two methanol molecules coordinating to two copper(II) ions (Figure 2). The compound adopts the geometry of a so-called “paddle-wheel structure” in which each of the tolfenamate ions coordinates to both copper(II) centers in dimer, and each of the oxygen atoms of the carboxylate group is bonded to one copper(II) cation. The coordination sphere of each of the Cu2+ ions is a distorted octahedron. The interplanar angle between carboxylate groups is 89.59(4) Å. The studied compound is centrosymmetric.
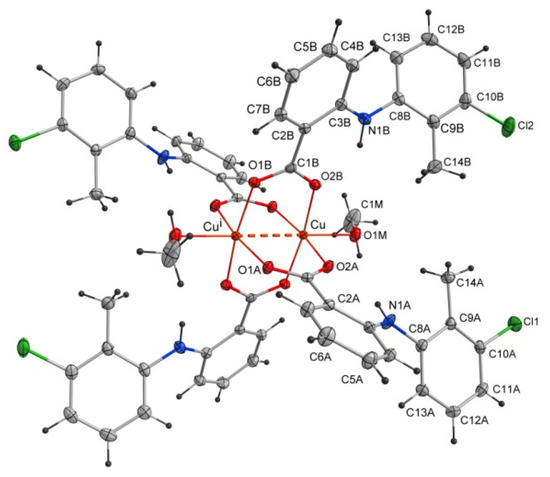
Figure 2.
Structure of the coordination compound of [CuII2(Tolf)4(MeOH)2]∙2MeOH. For clarity, uncoordinated methanol molecules were removed. The dashed line shows the distance between copper(II) ions. Symmetry code: i = −x + 1, −y + 1, −z + 1.
Selected bond lengths and angles are presented in Table 2 and Table 3. The structure reveals the characteristic for fenamate’s intramolecular hydrogen bonds: N1A–H1A∙∙∙O2A and N1B–H1B∙∙∙O2B (Table 4). The intermolecular hydrogen bonds with methanol molecules are observed in which the oxygen atom from the uncoordinated methanol molecule (O2M) is an acceptor (O1M–H1M∙∙∙O2M) and a proton donor (O2M–H2M∙∙∙O1Bi, i = −x + 1, −y + 1, −z + 1) as well (Table 4). The crystal is stabilized by C–H···π and Cl···π interactions. The adjacent molecules of [CuII2(Tolf)4(MeOH)2] interact via C11B–H11B···Cg1ii and C10B–Cl2···Cg2ii forming chains (ii = 1 −x, −y, 2 −z; the centers of the aromatic rings are denoted as Cg1 and Cg2; Cg1 = [C2A/C3A/C4A/C5A/C6A/C7A], Cg2 = [C2B/C3B/C4B/C5B/C6B/C7B]) (Figure 3, Tables S1 and S2). Interactions of [CuII2(Tolf)4(MeOH)2] molecules with uncoordinated methanol molecules through C–H···π (C2M–H2MC···Cg3, Cg3 = [C8A/C9A/C10A/C11A/C12A/C13A]) are also observed (Table S1).
Table 2.
Selected bond lengths in [CuII2(Tolf)4(MeOH)2]∙2MeOH.
Table 3.
Selected angles in [CuII2(Tolf)4(MeOH)2]∙2MeOH.
Table 4.
Geometry of hydrogen bonds in [CuII2(Tolf)4(MeOH)2]∙2MeOH.
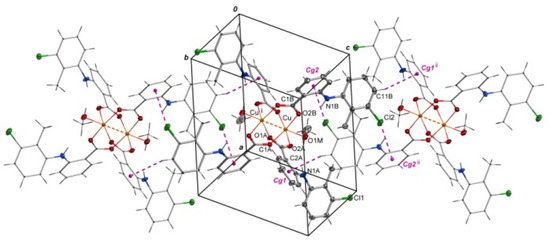
Figure 3.
Arrangement of molecules of [CuII2(Tolf)4(MeOH)2] in a chain where adjacent molecules contact via Cl···π and C–H···π interactions. For clarity, uncoordinated methanol molecules were removed. Symmetry codes: i = −x + 1, −y + 1, −z + 1; ii = 1 −x, −y, 2 −z.
2.2. QTAIM Analysis of [CuII2(Tolf)4(MeOH)2]∙2MeOH, QEBQIX and ACURCU01
The nature of the interaction between metal ions has long been studied by theoretical methods [30,31,32,33,34,35,36]. The use of the QTAIM analysis in the study of the interaction of Cu+ ions in model compounds [36] allowed for the conclusion that, contrary to expectations, this interaction is not repulsive but is a weak closed-shell interaction. In order to analyze the structure of the investigated compound, in particular the bond between two copper ions, the results of the QTAIM analysis for the investigated compound have been compared with the results for the selected previously determined structures of ACURCU01 and QEBQIX, for which the Cu2+∙∙∙Cu2+ distances are 2.5844(4), 2.624(1), and 2.6897(7) Å, respectively. QTAIM diagrams for the tolfenamic acid Cu(II) complex, QEBQIX, and ACURCU01 are presented in Figure 4 and Figure S1.
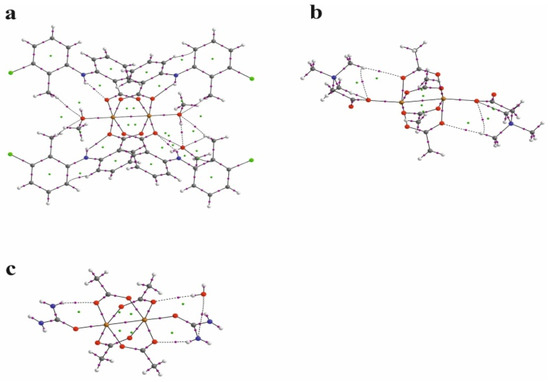
Figure 4.
QTAIM diagrams for [CuII2(Tolf)4(MeOH)2]∙2MeOH (a), QEBQIX (b), and ACURCU01 (c). The lines connecting the nuclei are the bond paths. Red and yellow dots represent the BCPs and RCPs, respectively.
QTAIM parameters describing the selected covalent chemical bond and weak interaction for Cu2+∙∙∙Cu2+ and Cu2+∙∙∙O for all the investigated compounds are collected in Table 5. In Table 6, the atom basin parameters are gathered. The parameter which allows differentiation of the open-shell and closed-shell interaction is the sign of Laplacian of the electron density. Depletion of the electron density between the atoms gives a positive sign of the Laplacian, which is typical for hydrogen bonds, van der Waals, and ionic interactions. According to the values in Table 5, the Cu2+∙∙∙Cu2+ and Cu2+∙∙∙O bonds are the closed-shell interactions when the C–O and C–C are the open-shell interactions typical for the covalent bonds. Taking into account only the sign of Laplacian, it is difficult to distinguish whether the interaction between the Cu2+ centers and between Cu2+ and carboxylate oxygen atoms can be classified as a weak interaction or an interaction between ions. The main parameter reflecting the chemical bond and the weak interaction strength is the electron density ρ(r) at the BCP located between the interacting atoms. Comparison of electron density at the Cu2+∙∙∙Cu2+ BCP for the investigated coordination compounds, QEBQIX and ACURCUO01, shows that for a greater distance of Cu2+∙∙∙Cu2+, the interaction between the cations is weaker, and the changes in electron density are sensitive to the changes in the distance between the Cu2+ ions. Comparison of the electron density at the critical point between Cu2+∙∙∙Cu2+ ions and at the critical point of a typical covalent bond shows how weak this interaction is.
Table 5.
QTAIM parameters for the chemical bonds and intermolecular interactions for [CuII2(Tolf)4(MeOH)2]∙2MeOH, QEBQIX, and ACURCU01.
Table 6.
QTAIM parameters for the selected atomic basins for [CuII2(Tolf)4(MeOH)2]∙2MeOH, QEBQIX, and ACURCU01.
Among the bonds listed in Table 5, the C–O bonds are characterized by the highest electron density at the BCP, which confirms the strength of the bond. Partially double-bond character of the C–O bonds is confirmed by the average number of the electrons delocalized between C and O atoms, which is higher than 1. For the single C–C bond, it is about 0.9. In contrast to the carboxylic group with single and double C–O bond, the geometrical and QTAIM parameters for both C–O bonds in every carboxylate group are similar, so both C–O bonds are identical with partially double character. The covalent C–C bond in tolfenamic acid is significantly weaker than the C–O bond. The closed-shell Cu2+∙∙∙O interaction between the Cu2+ cation and the tolfenamate carboxylate group is stronger than the interaction with the oxygen of methanol.
In line with the electron density is the delocalization index δ(A,B). The average number of electrons participating in the C–C bond is about 0.8. For the C–O bond, it is above 1, for Cu2+∙∙∙O about 0.4, and for the Cu2+∙∙∙O other than the carboxylate group, it is about 0.2. The delocalization index shows that the average number of electrons delocalized between Cu2+ cations is higher than between Cu2+∙∙∙O, but the electron density at the BCP indicates that the Cu2+∙∙∙O interaction is stronger. The other parameters in Table 5 confirm stability of every closed-shell interaction for the investigated compounds. Low ellipticity and linearity of the bonds and weak interactions suggest high stability of the complex.
Atomic properties are obtained by integration of three-dimensional electron density over the basin of the atom defined as the electron density local zero flux surfaces. The average electron population of atom N(A) can be obtained by numerical integration of the charge density over the basin of the atom. The net charge of atom q(A) is obtained by subtracting N(A) from the nuclear charge, q(A) = Zn − N(A) [55]. The results of the integration over the atomic basin are presented in Table 6. The open-shell interactions are connected with a higher percentage of the electrons for atoms that are engaged in a covalent bond. δBond(A,A’)/2 is the number of electrons in atom A that are delocalized (shared) between atom A and atoms to which atom A is bonded. It is characteristic that in the case of the Cu2+ cations, only about 5% of electrons participate in the weak interaction linking the cations together, when for the atoms participating in a covalent bond, the amount of electrons is significantly higher.
2.3. NCI Analysis of the Weak Interaction for the [CuII2(Tolf)4(MeOH)2]∙2MeOH and for QEBQIX
The second theoretical method, complementary to the QTAIM, which is very useful for characterizing the weak interaction, is the Noncovalent Interaction (NCI) approach [43]. This method allows differentiation of the repulsive and attractive interactions by using the reduced electron density gradient of the electron density, which describes the deviation from a homologous electron density distribution s = 1/(2(3π2)1/3)|∇ρ|/ρ4/3). The reduced gradient of electron density is very high for weak interactions but approaches zero for covalent bonds. The plot of the reduced electron density gradient versus the electron density multiplied by the sign of the second Hessian eigenvalue (λ2) of electron density makes it possible to differentiate a repulsive and attractive interaction. The spikes on the NCI diagram on the negative side of the vertical axis represent the attractive interactions, while those on the positive represent the repulsive interactions. Two symmetric peaks around zero represent the dispersive interaction. In addition to the NCI diagram, this method also offers a visualization of the interaction in the plots of the reduced density gradient in the real space for the molecule in colors traditionally used in the NCI approach: blue for attractive, red for repulsive, and green for intermediate-strength interactions (Figure 5, Figure S2).
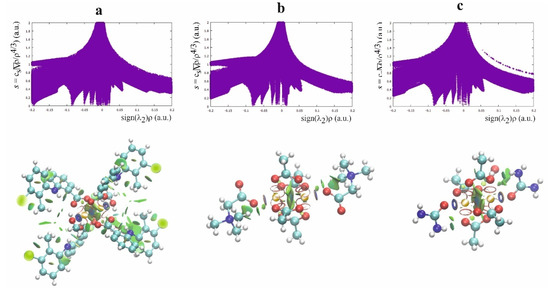
Figure 5.
NCI diagram and NCI plot for [CuII2(Tolf)4(MeOH)2]∙2MeOH (a), QEBQIX (b), and ACURCU01 (c).
According to the NCI method, the interaction between Cu2+ ions is dispersive rather than repulsive, which is consistent with the low electron density at the BCP between the cations. The strongest attractive interaction is for the Cu2+ cation and the methanol oxygen. The comparison of the Cu(II)∙∙∙O and Cu(II)∙∙∙Cu(II) interactions indicates a similar strength of these attractive interactions. A repulsive interaction is connected with the oxygen atoms of the carboxylate groups.
2.4. NBO, HOMO—LUMO and Fukui Parameters
Formation of the covalent bonds and the weak interaction are connected to the reorganization of the electron cloud which, other than the QTAIM analysis, can be described in a traditional way represented by the NBO analysis, which refers to a traditional Löwdin structure [56]. While QTAIM is based on the electron density, the NBO analysis refers to a traditional description of the molecular orbitals. The QTAIM and NBO approaches are complementary and strongly related to each other [57,58].
According to the NLMO analysis, the interaction between Cu2+ cations is formed by the lone pairs of both cations (Figure 6). Each Cu2+ cation has four lone pairs, and only one of them is directed to the lone pair of the other cation. For the Cu2+ cation, the weak interaction is formed by the lone pair which consists of 98.97% of the d orbital, 0.13% of p, and 0.9% of s. Participation of the orbital of the second Cu2+ is 0.303%, and the orbital consists of 83.98% of s, 11.25% of p, and 4.31% of d. Interaction of the Cu2+ cation with carboxylate oxygen atoms is also connected with overlapping of the lone pairs directed to the cation. In the interaction of oxygen, it participates with two Cu2+ cations in similar percentages (4.09 and 5.593%). The oxygen orbital that forms the interaction is built of the s orbital (13.88%) and p orbital (86.02%). As the interaction covers two cations, the orbital is localized only in 86.07%, which confirms delocalization of the dispersive interaction between the Cu2+ cations.
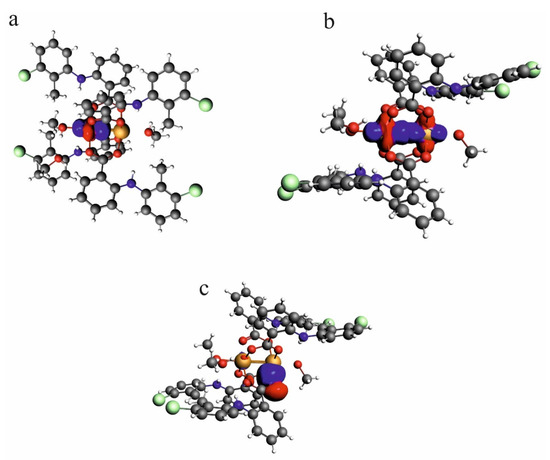
Figure 6.
NLMO orbitals for lone pair on one of the Cu2+ cations in the investigated compound—[CuII2(Tolf)4(MeOH)2]∙2MeOH (a), the weak interaction formed by lone pairs of two Cu2+ cations (b), lone pairs on the oxygen atom which is engaged in the interaction with Cu2+ cation (c).
HOMO and LUMO orbitals for CuII2(Tolf)4(MeOH)2]∙2MeOH, QEBQIX, and ACURCU01 are presented in Figure 7. It is characteristic that for every investigated compound, the frontier orbitals are located in the center of the molecule and spread over the Cu2+ cations and the carboxylic groups. The HOMO-LUMO energy gap is similar for the investigated compounds and equals 33.13, 32.94, and 31.69 kcal/mol for CuII2(Tolf)4(MeOH)2]∙2MeOH, QEBQIX, and ACURCU01, respectively.
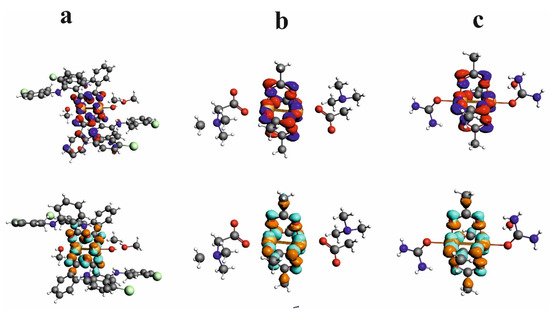
Figure 7.
HOMO (upper row) and LUMO (bottom row) orbitals for [CuII2(Tolf)4(MeOH)2]∙2MeOH (a), QEBQIX (b), and ACURCU01 (c).
The 3D model of molecular electrostatic potential is presented in Figure 8. The red color indicates the negatively charged oxygen atoms, while the blue represents positively charged Cu2+ cations. For QEBQIX, the positive and negative charge represents the betaine ligand.
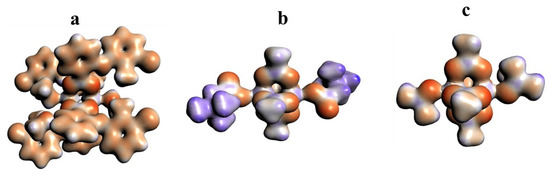
Figure 8.
Electrostatic potential surfaces for [CuII2(Tolf)4(MeOH)2]∙2MeOH (a), QEBQIX (b), and ACURCU01 (c). Field isosurfaces have been generated at the value of 0.03. Red—negative, blue—positive.
To investigate reactivity of the paddle-wheel compounds for QEBQIX and ACURCU01, Fukui indices have been calculated and presented in Figure 9. Fukui indices provide information regarding which part of the molecule can lose or accept an electron, that is, whether it may undergo nucleophilic or electrophilic attack [59]. Because the added electrons go into the HOMO orbital and the removed electrons come from LUMO, Fukui indices describe the change of electron density in a frontier orbital, as a result of adding or removing an electron from the frontier orbitals.
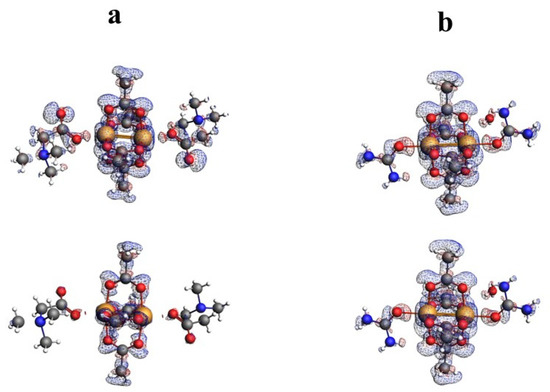
Figure 9.
Fukui surfaces for QEBQIX (a) and ACURCU01 (b). Field isosurfaces have been generated at the value of 0.001. Upper row—Fukui minus, bottom row—Fukui plus.
The Fukui minus and Fukui plus shown in Figure 9 indicate that the place of nucleophilic and electrophilic attack is located in the central part of the molecule containing Cu2+ cations and oxygens from carboxyl groups. On the other hand, the isosurface value of 0.001 indicates that this part of the molecule is not highly reactive.
2.5. Delocalization of Electrons
Delocalization of the electron density, except for the distribution of electron density in the molecule, can be visualized in the space of the molecule and indicates the bonds with mobile electrons. Electron delocalization can be visualized with the ACID (Anisotropy of the Current-Induced Density) method [60], which should be used to complement electron density analysis (Figure 10).
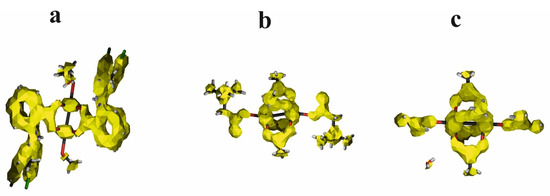
Figure 10.
ACID surfaces for [CuII2(Tolf)4(MeOH)2]∙2MeOH (a), QEBQIX (b), and ACURCU01 (c). ACID field isosurfaces have been generated at the value of 0.05.
According to the geometry and electron density parameters, the electrons for CO bonds with partially double character are characterized by high mobility identical to the aromatic rings. The C–C bond linking the carboxylate group with the aromatic ring is also characterized by high mobility. High mobility of the electrons is also characteristic of Cu2+ ions, but not for the weak interactions involving cations.
2.6. Decomposition of the Bonding Energy
The QTAIM and NCI methods illustrate the strength of the interactions. The NBO method explains the mechanism of orbital interaction. The energy decomposition according to Morokuma—Ziegler has been performed [61,62] and is an additional source of the interpretation of the bonding force linking the Cu2+∙∙∙Cu2+ cations with the surrounding atoms. The bonding energy is decomposed into electrostatic (Eelect), Pauli (EPauli), orbital (Eorb), and dispersive (Edysp) components.
where the Eelect is the Coulomb interaction between the unperturbed charge of the two interacting fragments, EPauli expresses the destabilizing Pauli repulsion between the occupied orbitals, Eorb illustrates the interaction energy between the orbitals of both complex components, and Edysp is the dispersion energy for the intermolecular interaction. The sum of Eelect and EPauli represents the steric interaction. The interaction energy has been calculated for two interacting fragments: the Cu2+∙∙∙Cu2+ core and the surrounding molecules. The calculated energy components in kcal/mol are as follows: Eelect −1679.20, EPauli 292.94, ESteric −1386.26, Edysp −13.58, Eorb −894.20. Total bonding energy is equal −2294.04. Energy decomposition shows that, despite the fact that the formation of the interactions with Cu2+ cations is connected with overlapping of the lone pairs, the complex is stabilized by the electrostatic interaction of the Cu2+∙∙∙ Cu2+ core and the surrounding molecules, although the orbital interaction is also significant. Dispersive interaction is important for the Cu2+∙∙∙Cu2+ cation interaction.
Ebonding = Eelect + EPauli + Eorb +Edysp
3. Materials and Methods
3.1. Single-Crystal X-ray Diffraction
Crystals of [CuII2(Tolf)4(MeOH)2]∙2MeOH coordination compound were measured with Xcalibur Ruby four-circle diffractometer equipped with a CCD area detector and a graphite monochromator using Mo Kα (λ = 0.71073 Å) radiation. The collected diffraction data were processed with the CrysAlis PRO program [63]. The structure was solved by the Patterson method and refined by the full-matrix least-squares method using SHELXT software [64,65]. The crystal data and structure refinement for the investigated compound are presented in Table 1. The program DIAMOND [66] was used for molecular graphics.
3.2. DTA, DTG
DTA (Differential Thermal Analysis) and DTG (Differential Thermal Gravimetry) were carried out by means of a Seteram SETSYS 16/18 instrument in the temperature range 330–870 K on a heating run at the rate of 5 K/min under N2.
3.3. IR, Raman
IR studies in the range of 400–4000 cm−1 were carried out using Thermo Scientific USA model Nicolet iS50 Fourier Transform Infrared Spectrometer FTIR using ATR. Raman spectrum was collected using Bruker Bravo Raman spectrometer.
3.4. Theoretical Analysis
The wave function for the experimental structure was obtained with Gaussian16 software [67]. The QTAIM analysis was carried with the AIMALL program [68]. Weak noncovalent interactions were investigated with the NCI program [43]. To describe the delocalization of the electrons, the ACID program [60] was used. NBO, molecular orbital analysis, and partition energy were performed without the molecule optimization using the ADF program [69].
3.5. Materials and Synthetic Procedures
Crystals of [CuII2(Tolf)4(MeOH)2]∙2MeOH coordination compound were obtained according to the analogous procedure for the synthesis of a copper coordination compound with meclofenamate ligand [17]. Copper(II) acetate monohydrate ([Cu(CH3COO)2(H2O)]2) (0.0250 g, 0.125 mmol) was dissolved in methanol (small amount), and the solution was added to a solution of tolfenamic acid (0.0654 g, 0.250 mmol) in methanol (about 5 mL). According to the preparative methods for the coordination compound of Cu(II) with meclofenamic acid, a few drops of triethylamine were added to the reaction mixture until the pH value was about 7. The reaction mixture was stirred for 2 h at room temperature, and then it was cooled in the refrigerator overnight. Slow evaporation of the solvent resulted in needle-like green crystals. The yield was fair (above 50%). M.p. 132–134 °C. IR (cm−1): 3330 w, ν(NH); 3078 vw; 2952 vw; 2833 vw; 1622 m, νasym(COO-); 1586 s; 1565 m; 1512 s; 1459 s; 1447 s; 1392 vs, νsym(COO-); 1323 vw; 1286 s; 1230 vw; 1208 vw; 1181 vw; 1164 m; 1152 m; 1122 vw; 1098 vw; 1079 vw; 1054 vw; 1044 vw; 1014 s; 957 vw; 912 w; 850 w; 805 w; 777 s; 751 vs; 736 w; 708 m; 680 s; 617 w. Raman (cm−1): 3160 w, 3130 w, 3078 w, 1980 w, 1951 w, 1915 w, 1858 w, 1611 vs, 1580 s, 1461 w, 1384 s, 1319 w, 1280 s, 1226 w, 1204 w, 1158 s, 1075 m, 1047 m, 849 s, 685 vw, 616 m, 520 m. Anal. calc. for C58H56N4O10Cl4Cu2 (coordination compound without uncoordinated methanol molecules) (1238 g mol−1): C, 56.3; H, 4.5; N, 4.5; O, 12.9; Cl, 11.5; Cu, 10.3; found: C, 56.8; H, 4.3; N, 4.4%.
The IR spectrum (Figures S3 and S4) exhibits the weak bands at 3330 (w) cm−1 derived from ν(NH) and at 3078 (vw) cm−1 attributed to ν(CH) from the aromatic ring. Bands at 2952 (vw) and 2833 (vw) cm−1 can be assigned to νasym(CH3) and νsym(CH3), respectively. The infrared spectrum for the studied crystal exhibits characteristic bands observed at 1622 (m) and 1392 (vs) cm−1 that can be assigned to νasym(COO-) and νsym(COO-) according to Hurtado et al. [70] (the band at 1392 (vs) cm−1 is broad; therefore, a composition of vibrations of νsym(COO-) and δas(CH3) [71] can occur). Bands at 1586 (s), 1565 (m), and 1512 (s) cm−1 can be attributed to a composition of ν(CN), bending vibrations of δ(NH) [71], and stretching vibrations of aromatic rings of ν(CC) [72], which correspond to analogous vibrations observed in the tolfenamic acid. The bands at 1459 (s) and 1447 (s) cm−1 could be assigned to δ(CH) as well as ν(CC) from aromatic rings. In the region of 1000–1300 cm−1, the δ(CH) from the aromatic ring appear, but the 1286 (s) cm−1 can also be assigned to ν(CN) vibration. According to Jabeen et al. [71], the strong band observed at 1014 (s) cm−1 can be attributed to ν(CCl) as well as ρ(CH3). In the region of 1000–675, bands observed can be assigned to δ(CH). Bands observed between 400–540 cm−1 may be interpreted as derived from ν(CCl) and δ(CCl) [71,72].
In the Raman spectrum (Figure S5), bands at about 3160, 3130, and 3078 cm−1 could be assigned to ν(CH) from aromatic rings. Broad weak bands with a maximum at about 2900 cm−1 can be attributed to νasym(CH3) and νsym(CH3) vibrations. Bands at about 1611 (vs) and 1580 (s) cm−1 can be assigned to δ(NH) and ν(CN), respectively. At 1461 (w) cm−1, the deformation modes δ(CH) can be expected. The strong band at 1384 (s) cm−1 can be interpreted as derived from δ(CH3). The bands observed at 1319 (w), 1280 (s) cm−1 can be attributed to ν(CN), and those at 1226 (w), 1204 (w), 1158 (s), 1075 (m), 1047 (m), 849 (s) cm−1 can be associated with ring deformations δ(CH) and δ(CC). At 685 (vw) cm−1, the deformation modes δ(NH) can be assigned. Bands observed at about 616 (m), 520 (m) cm−1 may be interpreted as derived from δ(CCl) [71].
Results of DTA (Differential Thermal Analysis) and DTG (Differential Thermal Gravimetry) studies are presented in Figure S6. DTA and DTG curves prove chemical and thermal stability of the studied compound up to about 330 K. Between 330 and 460 K, a distinct weight loss is observed, which is connected to the loss of methanol molecules present in the crystal as well as coordinated to copper(II) ions. Above 465 K, rapid weight loss attributed to further decomposition of the studied compound is observed.
4. Conclusions
- From the point of view of potential applications of double-core copper complexes with pharmaceutical ligands as new, promising therapeutic substances, it seems important to study the structure of compounds, the nature of chemical bonds, and weak interactions in the solid. Studying the nature of chemical bonds and interactions in the crystal may in the future be crucial in interpreting the mechanisms of drug delivery to cells and binding to proteins.
- The interactions linking the Cu2+∙∙∙Cu2+ cations as well as the core of the compound and the surrounding molecules are a weak, closed shell but very stable. Taking into account the value of the electron density at the critical point and the stability of the attracting Cu2+∙∙∙Cu2+ interaction, this interaction can be considered as one of the noncovalent interactions, affecting the overall geometry of the compound, the spatial arrangement of molecules in the crystal, and thus physicochemical properties, similar to the hydrogen bond, van der Waals, or pnictogen interaction.
- The interactions are formed by the orbital overlapping. The stabilizing force for the compounds is the electrostatic interaction of the Cu2+∙∙∙Cu2+ core with the rest of the molecule, especially with the carboxylate oxygen atoms.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24021745/s1.
Author Contributions
Conceptualization, I.M. and M.S.K.; methodology, I.M. and M.S.K.; software, I.M. and M.S.K.; validation, I.M. and M.S.K.; formal analysis, I.M. and M.S.K.; investigation, I.M. and M.S.K.; data curation, I.M. and M.S.K.; writing—original draft preparation, I.M. and M.S.K.; writing—review and editing, I.M. and M.S.K.; visualization, I.M. and M.S.K.; funding acquisition, I.M. and M.S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Structural data are available in the Cambridge Structural Database with deposition number: 2202870. Calculation data can be obtained from the authors upon request.
Acknowledgments
The authors would like to thank Tadeusz Lis for the X-ray measurement. The Wroclaw Center for Networking and Supercomputing is acknowledged for generous allocation of computer time. The Medical University is gratefully acknowledged for support of the research SUBZ.D050.22.025.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Herendeen, J.M.; Lindley, C. Use of NSAIDs for the chemoprevention of colorectal cancer. Ann. Pharmacother 2003, 37, 1664–1674. [Google Scholar] [CrossRef]
- Rao, C.V.; Reddy, B.S. NSAIDs and chemoprevention. Curr. Cancer Drug Targets 2004, 4, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Suh, N. Chemoprevention of cancer. Carcinogenesis 2000, 21, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Grossman, E.M.; Longo, W.E.; Panesar, N.; Mazuski, J.E.; Kaminski, D.L. The role of cyclooxygenase enzymes in the growth of human gall bladder cancer cells. Carcinogenesis 2000, 21, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Ritland, S.R.; Gendler, S.J. Chemoprevention of intestinal adenomas in the ApcMin mouse by piroxicam: Kinetics, strain effects and resistance to chemosuppression. Carcinogenesis 1999, 20, 51–58. [Google Scholar] [CrossRef] [PubMed]
- de Groot, D.J.A.; de Vries, E.G.E.; Groen, H.J.M.; de Jong, S. Non-steroidal anti-inflammatory drugs to potentiate chemotherapy effects: From lab to clinic. Crit. Rev. Oncol. Hematol. 2007, 61, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Orido, T.; Fujino, H.; Hasegawa, Y.; Toyomura, K.; Kawashima, T.; Murayama, T. Indomethacin Decreases Arachidonic Acid Uptake in HCA-7 Human Colon Cancer Cells. J. Pharmacol. Sci. 2008, 108, 389–392. [Google Scholar] [CrossRef]
- Glazko, A.J. Experimental observations on flufenamic, mefenamic and meclofenamic acids. 3. Metabolic disposition. Ann. Phys. Med. 1966, 23–36. [Google Scholar]
- Kauppila, A.; Ylikorkala, O. Indomethacin and tolfenamic acid in primary dysmenorrhea. Eur. J. Obstet. Gynecol. Reprod. Biol. 1977, 7, 59–64. [Google Scholar] [CrossRef]
- Kajander, A.; Martio, J.; Mutru, O.; Gothoni, G. Prolonged treatment with tolfenamic acid in inflammatory rheumatic diseases. Scand. J. Rheum. 1976, 4, 158–160. [Google Scholar] [CrossRef]
- Rejholec, V.; Vapaatalo, H.; Tokola, O.; Gothoni, G. A comparative, double-blind study on tolfenamic acid in the treatment of rheumatoid arthritis. Scand. J. Rheum. 1979, 24, 13–16. [Google Scholar] [CrossRef]
- Thorsteinn, L. Drug solubilization by complexation. Int. J. Pharm. 2017, 531, 276–280. [Google Scholar]
- Rentrew, A.K. Transition metal complexes with bioactive ligands: Mechanisms for selective ligand release and applications for drug delivery. Metallomics 2014, 6, 1324–1335. [Google Scholar]
- Jurca, T.; Marian, E.; Vicaş, L.G.; Mureşan, M.; Fritea, L. Metal Complexes of Pharmaceutical Substances. In Spectroscopic Analyses-Developments and Applications; Sharmin, E., Zafar, F., Eds.; IntechOpen: London, UK, 2017; Available online: https://www.intechopen.com/chapters/54928 (accessed on 14 November 2022). [CrossRef]
- Zhang, X.C.; Lippard, S.J. New metal complexes as potential therapeutics. Curr. Opin. Chem. Biol. 2003, 7, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Drewry, J.A.; Gunning, P.T. Recent advances in biosensory and medicinal therapeutic applications of zinc(II) and copper(II) coordination complexes. Coord. Chem. Rev. 2011, 255, 459–472. [Google Scholar] [CrossRef]
- Kovala-Demertzi, D.; Staninska, M.; Garcia-Santos, I.; Castineiras, A.; Demertzis, M.A. Synthesis, crystal structures and spectroscopy of meclofenamic acid and its metal complexes with manganese(II), copper(II), zinc(II) and cadmium(II). Antiproliferative and superoxide dismutase activity. J. Inorg. Biochem. 2011, 105, 1187–1195. [Google Scholar] [CrossRef]
- Roy, S.; Banerjee, R.; Sarkar, M.J. Direct binding of Cu(II)-complexes of oxicam NSAIDs with DNA backbone. Inorg. Biochem. 2006, 100, 1320–1331. [Google Scholar] [CrossRef]
- Weder, J.E.; Dillon, C.T.; Hambley, T.W.; Kennedy, B.J.; Lay, P.A.; Biffin, J.T.; Regtop, H.L.; Davies, N.M. Copper Complexes of Non-Steroidal Anti-Inflammatory Drugs: An Opportunity Yet to Be Realized. Coord. Chem. Rev. 2002, 232, 95–126. [Google Scholar] [CrossRef]
- Marzano, C.; Pellei, M.; Tisato, F.; Santini, C. Copper complexes as anticancer agents. Anticancer Agents Med. Chem. 2009, 9, 185–211. [Google Scholar] [CrossRef]
- Tisato, F.; Marzano, C.; Porchia, M.; Pellei, M.; Santini, C. Copper in diseases and treatments, and copper-based anticancer strategies. Med. Res. Rev. 2010, 30, 708–749. [Google Scholar] [CrossRef]
- Tardito, S.; Marchiò, L. Copper compounds in anticancer strategies. Curr. Med. Chem. 2009, 16, 1325–1348. [Google Scholar] [CrossRef] [PubMed]
- Hobza, P.; Zahradník, R.; Müller-Dethlefs, K. The world of non-covalent interactions. Collect. Czech. Chem. Commun. 2006, 71, 443–531. [Google Scholar] [CrossRef]
- de Azevedo Santos, L.; Hamlin, T.A.; Ramalho, T.C.; Bickelhaupt, F.M. The pnictogen bond: A quantitative molecular orbital picture. Phys. Chem. Chem. Phys. 2021, 23, 13842–13852. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; Resnati, G.; Pombeiro, A.J.L. Pnictogen bonding in coordination chemistry. Coord. Chem. Rev. 2020, 418, 213381. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Definition of the Pnictogen Bond: A Perspective. Inorganics 2022, 10, 149. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Inomata, Y.; Howell, F.S. Characterization and crystal structure of copper(II) complex with betaine having a water-wheel-like structure. Inorg. Chim. Acta 1999, 295, 121–124. [Google Scholar] [CrossRef]
- Uekusa, H.; Ohba, S.; Saito, Y.; Kato, M.; Tokii, T.; Muto, Y. Structural comparison between dimeric copper(II) formate and acetate in pyridine and urea adducts. Acta Cryst. 1989, C45, 377–388. [Google Scholar] [CrossRef]
- Mehrotra, P.K.; Hoffmann, R. Copper(I)-copper(I) interactions. Bonding relationships in d10-d10 systems. Inorg. Chem. 1978, 17, 2187–2189. [Google Scholar] [CrossRef]
- Jiang, Y.; Alvarez, S.; Hoffmann, R. Binuclear and polymeric gold(I) complexes. Inorg. Chem. 1985, 24, 749–757. [Google Scholar] [CrossRef]
- Cotton, F.A.; Feng, X.; Matusz, M.; Poli, R. Experimental and theoretical studies of the copper(I) and silver(I) dinuclear N,N′-di-p-tolylformamidinato complexes. J. Am. Chem. Soc. 1988, 110, 7077–7083. [Google Scholar] [CrossRef]
- Pyykkӧ, P. Strong Closed-Shell Interactions in Inorganic Chemistry. Chem. Rev. 1997, 97, 597–636. [Google Scholar] [CrossRef] [PubMed]
- Pyykkӧ, P. Theoretical chemistry of gold. III. Chem. Soc. Rev. 2008, 37, 1967–1997. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, M.A.; Alvarez, S.; Novoa, J.J. The Nature of Intermolecular CuI⋅CuI Interactions: A Combined Theoretical and Structural Database Analysis. Chem. Eur. J. 2004, 10, 2117–2132. [Google Scholar] [CrossRef] [PubMed]
- Dinda, A.G. Samuelson, The Nature of Bond Critical Points in Dinuclear Copper(I) Complexes. Chem. Eur. J. 2012, 18, 3032–3042. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum Theory. In Atoms in Molecules; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Bader, R.F.W. Bond Paths Are Not Chemical Bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef]
- Bader, R.F.W. Definition of Molecular Structure: By Choice or by Appeal to Observation? J. Phys. Chem. A 2010, 114, 7431–7444. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essen, H. The Characterization of Atomic Interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Bader, R.F.W. Effect of Twisting a Polypeptide on Its Geometry and Electron Distribution. J. Phys. Chem. 1994, 98, 4473–4481. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Characterization of a Dihydrogen Bond on the Basis of the Electron Density. J. Phys. Chem. 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Kovala-Demertzi, D.; Galani, A.; Demertzis, M.A.; Skoulika, S.; Kotoglou, C. Binuclear copper(II) complexes of tolfenamic: Synthesis, crystal structure, spectroscopy and superoxide dismutase activity. J. Inorg. Biochem. 2004, 98, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, S.; Andre, V.; Fernandes, A.; Duarte, M.T. Mechanochemistry–A green synthetic methodology leading to metallodrugs, metallopharmaceuticals and bio-inspired metal-organic frameworks. Inorg. Chim. Acta 2017, 455, 309–318. [Google Scholar] [CrossRef]
- Facchin, G.; Torre, M.H.; Kremer, E.; Piro, O.E.; Baran, E.J. Crystal Structure and Spectroscopic Behaviour of a Binuclear Copper(II) Complex of Mefenamic Acid and Dimethylsulfoxide. Z. Naturforsch. B Chem.Sci. 1998, 53, 871–874. [Google Scholar] [CrossRef]
- Mys’kiv, M.G.; Olijnik, V.V.; Kriss, E.E.; Konakhovich, N.F.; Grigor’eva, A.S. STRUCTURE CRISTALLINE DE COMPLEXES DE CUIVRE (II) AVEC DES DERIVES DE L’ACIDE N-PHENYLANTHRANILIQUE. Koord. Khim. Russ. Coord. Chem. 1982, 8, 1415. [Google Scholar]
- Xin, C.-W.; Liu, F.-C. Tetrakis(μ-2-anilinobenzoato)bis[methanolcopper(II)](Cu-Cu). Acta Cryst. 2008, E64, m1589. [Google Scholar] [CrossRef] [PubMed]
- Tas, M.; Yesilel, O.Z.; Buyukgungor, O. Novel Copper(II) Complexes of N-Phenylanthranilic Acid Containing Ethanol and Hydroxo Ligands. J. Inorg. Organomet. Polym. Mater. 2010, 20, 298–305. [Google Scholar] [CrossRef]
- Batsanov, A.S.; Struchkov, Y.T.; Grigor’eva, A.S.; Kriss, E.E.; Konakhovich, N.F.; Fialkov, Y.A. Structure of Copper(II) Complex with N-3,4-dimethyl Phenyl Anthranilic Acid, Isomefenamic Acid. Koord. Khim. Coord. Chem. 1981, 7, 784. [Google Scholar]
- Tolia, C.; Papadopoulos, A.N.; Raptopoulou, C.P.; Psycharis, V.; Garino, C.; Salassa, L.; Psomas, G. Copper(II) interacting with the non-steroidal antiinflammatory drug flufenamic acid: Structure, antioxidant activity and binding to DNA and albumins. J. Inorg. Biochem. 2013, 123, 53–65. [Google Scholar] [CrossRef]
- Sabirov, V.K.; Batsanov, A.S.; Struchkov, Y.T.; Grigor’eva, A.S.; Kriss, E.E.; Konakhovich, N.F.; Fialkov, Y.A. Synthesis and Crystal-Structure of Dimeric Cooper(II) Complexes with N-3-Difluoromethylthiophenylanthranilic Acid. Koord. Khim. Russ. Coord. Chem. 1984, 10, 1474. [Google Scholar]
- Tas, M.; Karabag, E.; Titiz, A.; Kaya, M.; Ataseven, M.; Dal, H. Synthesis and characterization of Tetrakis-μ-[N-phenylanthranilato](O,O′)-bis[(4-vinylpyridine copper(II)] complex. Z. Krist. Cryst. Mater. 2013, 228, 92–99. [Google Scholar] [CrossRef]
- Tas, M.; Titiz, A.; Karabag, E.; Kaya, M.; Ataseven, M.; Dal, H. The Tetra–μ–[N-phenylanthranilato](O,O’)–bis [(2-amino-4-methyl)pyridine Copper(II)] Complex Crystal. Synth. React. Inorg., Met.-Org. Nano-Met. Chem. 2013, 43, 1212–1223. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Bader, R.F.W.; Lau, C.D.H. A Theoretical Analysis of Hydrocarbon Properties: II. Additivity of Group Properties and the Origin of Strain Energy. J. Am. Chem. Soc. 1987, 109, 1001–1012. [Google Scholar] [CrossRef]
- Löwdin, P.-O. Quantum Theory of Many-Particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the Method of Configurational Interaction. Phys. Rev. 1955, 97, 1474–1489. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Natural Bond Orbitals and Extensions of Localized Bonding Concepts. Chem. Educ. Res. Pract. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Herges, R.; Geuenich, D. Delocalization of Electrons in Molecules. J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar] [CrossRef]
- Morokuma, K. Molecular Orbital Studies of Hydrogen Bonds. III. C=O... H–O Hydrogen Bond in H2CO. H2O and H2CO 2H2O. J. Chem. Phys. 1971, 55, 1236–1244. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Rigaku, O.D. CrysAlis PRO; Rigaku Oxford Diffraction: Yarnton, UK, 2015. [Google Scholar]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond; Crystal Impact GbR: Bonn, Germany, 2014. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian Inc 16, Revision, A. 03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Keith, T.A. AIMALL (Version 19.10.12); TK Gristmill Software: Overland Park, KS, USA, 2014. [Google Scholar]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Hurtado, M.; Sankpal, U.T.; Chhabra, J.; Brown, D.T.; Maram, R.; Patel, R.; Gurung, R.K.; Simecka, J.; Holder, A.A.; Basha, R. Copper-tolfenamic acid: Evaluation of stability and anti-cancer activity. Investig. New Drugs 2019, 37, 27–34. [Google Scholar] [CrossRef]
- Jabeen, S.; Dines, T.J.; Leharne, S.A.; Babur, Z.C. Raman and IR spectroscopic studies of fenamates–Conformational differences in polymorphs of flufenamic acid, mefenamic acid and tolfenamic acid. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 96, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Sheraz, M.A.; Yorucu, C.; ur Rehman, I. Quantitative determination of tolfenamic acid and its pharmaceutical formulation using FTIR and UV spectrometry. Cent. Eur. J. Chem. 2013, 11, 1533–1541. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).