
MAFLD and Celiac Disease in Children



{kind=link}
Abstract
:1. Introduction
2. Research Strategy
3. CD and MAFLD
4. The Gut–Liver Axis
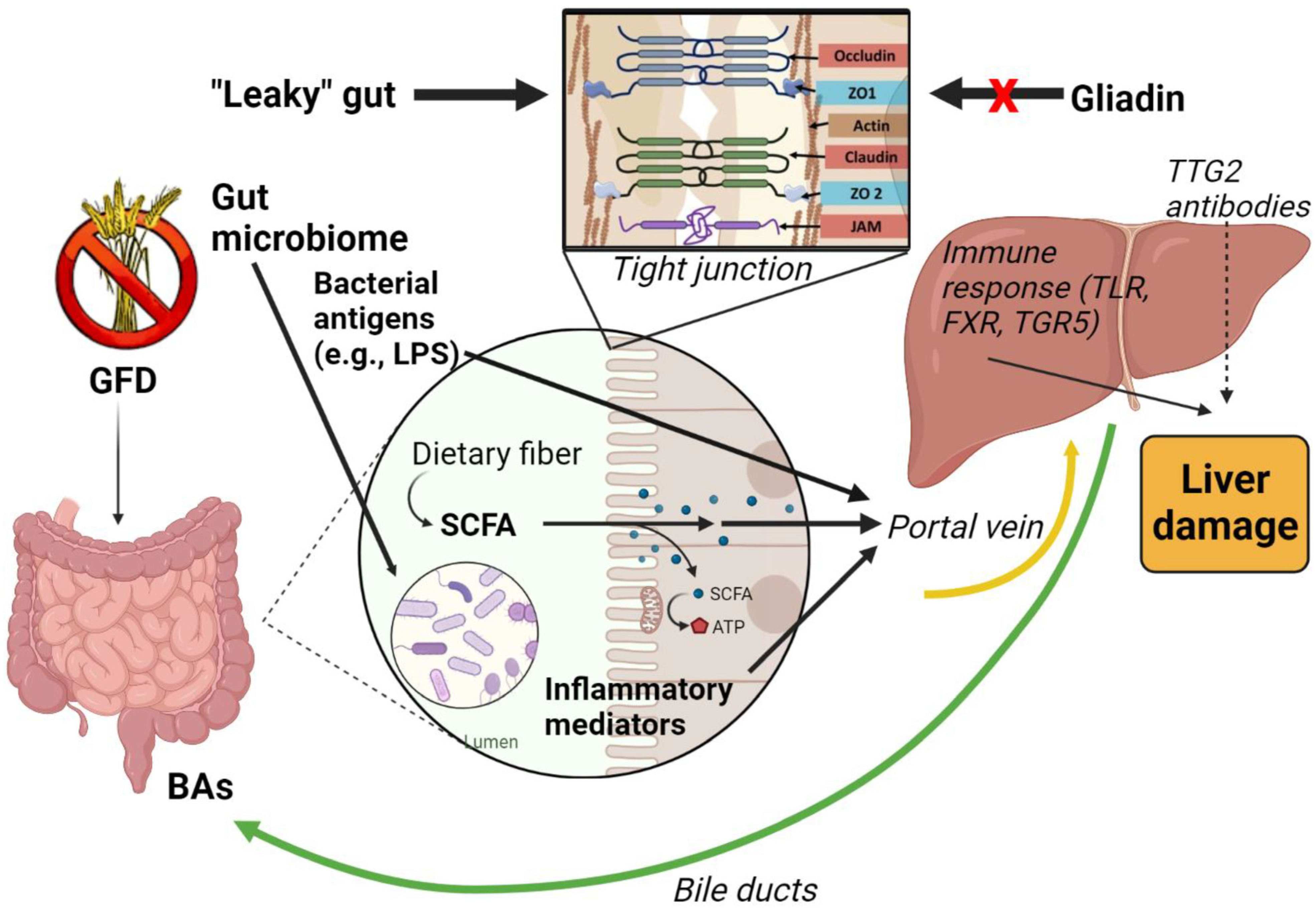
5. Intestinal Barrier Dysfunction and the “Leaky Gut”
6. Gut Microbiome
7. Bacterial Products and Metabolites
8. Malnutrition and Compensatory Theory
9. Bile Acids (BAs) as Mediator of Hepatic Damage in CD Patients
10. The Role of Tissue Transglutaminase (TTG2) Antibodies and Vitamin D
11. Genetic Contribution
12. GFD and MAFLD
12.1. Obesity
12.2. IR and Glucose Metabolism Alterations
12.3. Serum Lipids
12.4. Metabolic Syndrome (MetS)
13. Final Considerations and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Husby, S.; Koletzko, S.; Korponay-Szabó, I.R.; Mearin, M.L.; Phillips, A.; Shamir, R.; Troncone, R.; Giersiepen, K.; Branski, D.; Catassi, C.; et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 136–160. [Google Scholar] [CrossRef] [PubMed]
- Gujral, N.; Freeman, H.J.; Thomson, A.B. Celiac disease: Prevalence, diagnosis, pathogenesis and treatment. World J. Gastroenterol. 2012, 18, 6036–6059. [Google Scholar] [CrossRef]
- Riznik, P.; De Leo, L.; Dolinsek, J.; Gyimesi, J.; Klemenak, M.; Koletzko, B.; Koletzko, S.; Korponay-Szabó, I.R.; Krencnik, T.; Not, T.; et al. Clinical Presentation in Children With Coeliac Disease in Central Europe. J. Pediatr. Gastroenterol. Nutr. 2021, 72, 546–551. [Google Scholar] [CrossRef]
- Al-Toma, A.; Volta, U.; Auricchio, R.; Castillejo, G.; Sanders, D.S.; Cellier, C.; Mulder, C.J.; Lundin, K.E.A. European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United Eur. Gastroenterol. J. 2019, 7, 583–613. [Google Scholar] [CrossRef] [PubMed]
- Ben Houmich, T.; Admou, B. Celiac disease: Understandings in diagnostic, nutritional, and medicinal aspects. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211008709. [Google Scholar] [CrossRef] [PubMed]
- Valletta, E.; Fornaro, M.; Cipolli, M.; Conte, S.; Bissolo, F.; Danchielli, C. Celiac disease and obesity: Need for nutritional follow-up after diagnosis. Eur. J. Clin. Nutr. 2010, 64, 1371–1372. [Google Scholar] [CrossRef] [Green Version]
- Norsa, L.; Shamir, R.; Zevit, N.; Verduci, E.; Hartman, C.; Ghisleni, D.; Riva, E.; Giovannini, M. Cardiovascular disease risk factor profiles in children with celiac disease on gluten-free diets. World J. Gastroenterol. 2013, 19, 5658–5664. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Eslam, M.; Alkhouri, N.; Vajro, P.; Baumann, U.; Weiss, R.; Socha, P.; Marcus, C.; Lee, W.S.; Kelly, D.; Porta, G.; et al. Defining paediatric metabolic (dysfunction)-associated fatty liver disease: An international expert consensus statement. Lancet Gastroenterol. Hepatol. 2021, 6, 864–873. [Google Scholar] [CrossRef]
- Hoffmanová, I.; Sánchez, D.; Tučková, L.; Tlaskalová-Hogenová, H. Celiac Disease and Liver Disorders: From Putative Pathogenesis to Clinical Implications. Nutrients 2018, 10, 892. [Google Scholar] [CrossRef]
- Vos, M.B.; Abrams, S.H.; Barlow, S.E.; Caprio, S.; Daniels, S.R.; Kohli, R.; Mouzaki, M.; Sathya, P.; Schwimmer, J.B.; Sundaram, S.S.; et al. NASPGHAN Clinical Practice Guideline for the Diagnosis and Treatment of Nonalcoholic Fatty Liver Disease in Children: Recommendations from the Expert Committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN). J. Pediatr. Gastroenterol. Nutr. 2017, 64, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Le Garf, S.; Nègre, V.; Anty, R.; Gual, P. Metabolic Fatty Liver Disease in Children: A Growing Public Health Problem. Biomedicines 2021, 9, 1915. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Wieckowska, A.; Feldstein, A.E. Diagnosis of nonalcoholic fatty liver disease: Invasive versus noninvasive. Semin. Liver Dis. 2008, 28, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Alisi, A.; Newton, K.P.; Schwimmer, J.B. Comparison of the Phenotype and Approach to Pediatric vs Adult Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1798–1810. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Mu, C.; Li, K.; Luo, H.; Liu, Y.; Li, Z. Estimating Global Prevalence of Metabolic Dysfunction-Associated Fatty Liver Disease in Overweight or Obese Children and Adolescents: Systematic Review and Meta-Analysis. Int. J. Public Health 2021, 66, 1604371. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Milic, N.; De Lorenzo, A.; Luzza, F. A pathogenetic link between non-alcoholic fatty liver disease and celiac disease. Endocrine 2013, 43, 65–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludvigsson, J.F.; Elfström, P.; Broomé, U.; Ekbom, A.; Montgomery, S.M. Celiac disease and risk of liver disease: A general population-based study. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2007, 5, 63–69.e1. [Google Scholar] [CrossRef]
- Reilly, N.R.; Lebwohl, B.; Hultcrantz, R.; Green, P.H.; Ludvigsson, J.F. Increased risk of non-alcoholic fatty liver disease after diagnosis of celiac disease. J. Hepatol. 2015, 62, 1405–1411. [Google Scholar] [CrossRef] [Green Version]
- Hill, I.D.; Fasano, A.; Guandalini, S.; Hoffenberg, E.; Levy, J.; Reilly, N.; Verma, R. NASPGHAN Clinical Report on the Diagnosis and Treatment of Gluten-related Disorders. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 156–165. [Google Scholar] [CrossRef]
- Yodoshi, T.; Orkin, S.; Arce-Clachar, A.C.; Bramlage, K.; Xanthakos, S.A.; Valentino, P.L.; Mouzaki, M. Alternative Etiologies of Liver Disease in Children With Suspected NAFLD. Pediatrics 2021, 147, 2274. [Google Scholar] [CrossRef]
- Zimmet, P.; Alberti, K.G.; Kaufman, F.; Tajima, N.; Silink, M.; Arslanian, S.; Wong, G.; Bennett, P.; Shaw, J.; Caprio, S. The metabolic syndrome in children and adolescents—An IDF consensus report. Pediatr. Diabetes 2007, 8, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Freeman, H.J. Hepatic manifestations of celiac disease. Clin. Exp. Gastroenterol. 2010, 3, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Wiest, R.; Albillos, A.; Trauner, M.; Bajaj, J.S.; Jalan, R. Targeting the gut-liver axis in liver disease. J. Hepatol. 2017, 67, 1084–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturgeon, C.; Fasano, A. Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers 2016, 4, e1251384. [Google Scholar] [CrossRef] [Green Version]
- Pekáriková, A.; Sánchez, D.; Palová-Jelínková, L.; Simsová, M.; Benes, Z.; Hoffmanová, I.; Drastich, P.; Janatková, I.; Mothes, T.; Tlaskalová-Hogenová, H.; et al. Calreticulin is a B cell molecular target in some gastrointestinal malignancies. Clin. Exp. Immunol. 2010, 160, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, D.; Tucková, L.; Mothes, T.; Kreisel, W.; Benes, Z.; Tlaskalová-Hogenová, H. Epitopes of calreticulin recognised by IgA autoantibodies from patients with hepatic and coeliac disease. J. Autoimmun. 2003, 21, 383–392. [Google Scholar] [CrossRef]
- Milosevic, I.; Vujovic, A.; Barac, A.; Djelic, M.; Korac, M.; Radovanovic Spurnic, A.; Gmizic, I.; Stevanovic, O.; Djordjevic, V.; Lekic, N.; et al. Gut-Liver Axis, Gut Microbiota, and Its Modulation in the Management of Liver Diseases: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 395. [Google Scholar] [CrossRef] [Green Version]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Michaudel, C.; Sokol, H. The Gut Microbiota at the Service of Immunometabolism. Cell Metab. 2020, 32, 514–523. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, Y.; Yamamoto, K. Role of gut microbiota in liver diseases. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2013, 43, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Delzenne, N.M. The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- De Re, V.; Magris, R.; Cannizzaro, R. New Insights into the Pathogenesis of Celiac Disease. Front. Med. 2017, 4, 137. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Hoffmanová, I.; Sánchez, D.; Hábová, V.; Anděl, M.; Tučková, L.; Tlaskalová-Hogenová, H. Serological markers of enterocyte damage and apoptosis in patients with celiac disease, autoimmune diabetes mellitus and diabetes mellitus type 2. Physiol. Res. 2015, 64, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Novacek, G.; Miehsler, W.; Wrba, F.; Ferenci, P.; Penner, E.; Vogelsang, H. Prevalence and clinical importance of hypertransaminasaemia in coeliac disease. Eur. J. Gastroenterol. Hepatol. 1999, 11, 283–288. [Google Scholar] [CrossRef]
- Fasano, A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann. N. Y. Acad. Sci. USA 2012, 1258, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiman, Y.; Friedman, S.L. The role of chemokines in acute liver injury. Front. Physiol. 2012, 3, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.L.K.; Edington, C.; Suter, E.; Yu, J.; Velazquez, J.J.; Velazquez, J.G.; Shockley, M.; Large, E.M.; Venkataramanan, R.; Hughes, D.J.; et al. Integrated gut/liver microphysiological systems elucidates inflammatory inter-tissue crosstalk. Biotechnol. Bioeng. 2017, 114, 2648–2659. [Google Scholar] [CrossRef] [Green Version]
- Barilli, A.; Rotoli, B.M.; Visigalli, R.; Ingoglia, F.; Cirlini, M.; Prandi, B.; Dall’Asta, V. Gliadin-mediated production of polyamines by RAW264.7 macrophages modulates intestinal epithelial permeability in vitro. Biochim. Biophys. Acta 2015, 1852, 1779–1786. [Google Scholar] [CrossRef] [Green Version]
- Cinova, J.; Palová-Jelínková, L.; Smythies, L.E.; Cerná, M.; Pecharová, B.; Dvorák, M.; Fruhauf, P.; Tlaskalová-Hogenová, H.; Smith, P.D.; Tucková, L. Gliadin peptides activate blood monocytes from patients with celiac disease. J. Clin. Immunol. 2007, 27, 201–209. [Google Scholar] [CrossRef]
- Barilli, A.; Gaiani, F.; Prandi, B.; Cirlini, M.; Ingoglia, F.; Visigalli, R.; Rotoli, B.M.; de’Angelis, N.; Sforza, S.; de’Angelis, G.L.; et al. Gluten peptides drive healthy and celiac monocytes toward an M2-like polarization. J. Nutr. Biochem. 2018, 54, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Scaldaferri, F.; Bruno, G.; Petito, V.; Franceschi, F.; Gasbarrini, A. The therapeutic management of gut barrier leaking: The emerging role for mucosal barrier protectors. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1068–1076. [Google Scholar]
- Assimakopoulos, S.F.; Tsamandas, A.C.; Tsiaoussis, G.I.; Karatza, E.; Triantos, C.; Vagianos, C.E.; Spiliopoulou, I.; Kaltezioti, V.; Charonis, A.; Nikolopoulou, V.N.; et al. Altered intestinal tight junctions’ expression in patients with liver cirrhosis: A pathogenetic mechanism of intestinal hyperpermeability. Eur. J. Clin. Investig. 2012, 42, 439–446. [Google Scholar] [CrossRef]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Surewaard, B.G.; Deniset, J.F.; Zemp, F.J.; Amrein, M.; Otto, M.; Conly, J.; Omri, A.; Yates, R.M.; Kubes, P. Identification and treatment of the Staphylococcus aureus reservoir in vivo. J. Exp. Med. 2016, 213, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef]
- Miura, K.; Ishioka, M.; Iijima, K. The Roles of the Gut Microbiota and Toll-like Receptors in Obesity and Nonalcoholic Fatty Liver Disease. J. Obes. Metab. Syndr. 2017, 26, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Su, G.L.; Klein, R.D.; Aminlari, A.; Zhang, H.Y.; Steinstraesser, L.; Alarcon, W.H.; Remick, D.G.; Wang, S.C. Kupffer cell activation by lipopolysaccharide in rats: Role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 2000, 31, 932–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E.; et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G433–G441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganami, T.; Mieda, T.; Itoh, M.; Shimoda, Y.; Kamei, Y.; Ogawa, Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochem. Biophys. Res. Commun. 2007, 354, 45–49. [Google Scholar] [CrossRef]
- Fujita, T.; Narumiya, S. Roles of hepatic stellate cells in liver inflammation: A new perspective. Inflamm. Regen. 2016, 36, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdu, E.F.; Galipeau, H.J.; Jabri, B. Novel players in coeliac disease pathogenesis: Role of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 497–506. [Google Scholar] [CrossRef]
- Grander, C.; Adolph, T.E.; Wieser, V.; Lowe, P.; Wrzosek, L.; Gyongyosi, B.; Ward, D.V.; Grabherr, F.; Gerner, R.R.; Pfister, A.; et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 2018, 67, 891–901. [Google Scholar] [CrossRef]
- Woodhouse, C.A.; Patel, V.C.; Singanayagam, A.; Shawcross, D.L. Review article: The gut microbiome as a therapeutic target in the pathogenesis and treatment of chronic liver disease. Aliment. Pharmacol. Ther. 2018, 47, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef] [Green Version]
- Luck, H.; Tsai, S.; Chung, J.; Clemente-Casares, X.; Ghazarian, M.; Revelo, X.S.; Lei, H.; Luk, C.T.; Shi, S.Y.; Surendra, A.; et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab. 2015, 21, 527–542. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Birchenough, G.M.H.; Ståhlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Bäckhed, F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 2018, 23, 27–40.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Tsilingiri, K.; Rescigno, M. Postbiotics: What else? Benef. Microbes 2013, 4, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Mosca, F.; Gianni, M.L.; Rescigno, M. Can Postbiotics Represent a New Strategy for NEC? Adv. Exp. Med. Biol. 2019, 1125, 37–45. [Google Scholar] [CrossRef]
- Huda-Faujan, N.; Abdulamir, A.S.; Fatimah, A.B.; Anas, O.M.; Shuhaimi, M.; Yazid, A.M.; Loong, Y.Y. The impact of the level of the intestinal short chain Fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem. J. 2010, 4, 53–58. [Google Scholar] [CrossRef]
- Gonçalves, P.; Martel, F. Regulation of colonic epithelial butyrate transport: Focus on colorectal cancer. Porto. Biomed. J. 2016, 1, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, R.; Koebnick, C.; Schildt, J.; Mueller, M.; Radke, M.; Blaut, M. Effects of Bifidobacterium lactis Bb12 supplementation on body weight, fecal pH, acetate, lactate, calprotectin, and IgA in preterm infants. Pediatr. Res. 2008, 64, 418–422. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, B.S.; Mathan, V.I. Colonic dysfunction in acute diarrhoea: The role of luminal short chain fatty acids. Gut 1993, 34, 1215–1218. [Google Scholar] [CrossRef] [Green Version]
- Scheppach, W.; Bartram, H.P.; Richter, F. Role of short-chain fatty acids in the prevention of colorectal cancer. Eur. J. Cancer 1995, 31, 1077–1080. [Google Scholar] [CrossRef]
- Farup, P.G.; Rudi, K.; Hestad, K. Faecal short-chain fatty acids—A diagnostic biomarker for irritable bowel syndrome? BMC Gastroenterol. 2016, 16, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niccolai, E.; Baldi, S.; Ricci, F.; Russo, E.; Nannini, G.; Menicatti, M.; Poli, G.; Taddei, A.; Bartolucci, G.; Calabrò, A.S.; et al. Evaluation and comparison of short chain fatty acids composition in gut diseases. World J. Gastroenterol. 2019, 25, 5543–5558. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.M.; Vanhoutvin, S.A.; Troost, F.J.; Rijkers, G.; de Bruïne, A.; Bast, A.; Venema, K.; Brummer, R.J. Effect of butyrate enemas on inflammation and antioxidant status in the colonic mucosa of patients with ulcerative colitis in remission. Clin. Nutr. 2010, 29, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; O’Riordan, M.X. Regulation of bacterial pathogenesis by intestinal short-chain Fatty acids. Adv. Appl. Microbiol. 2013, 85, 93–118. [Google Scholar] [CrossRef] [Green Version]
- Bhaskaran, N.; Quigley, C.; Paw, C.; Butala, S.; Schneider, E.; Pandiyan, P. Role of Short Chain Fatty Acids in Controlling T(regs) and Immunopathology During Mucosal Infection. Front. Microbiol. 2018, 9, 1995. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Eberl, G. Type 3 regulatory T cells at the interface of symbiosis. J. Microbiol. 2018, 56, 163–171. [Google Scholar] [CrossRef]
- Belkaid, Y.; Harrison, O.J. Homeostatic Immunity and the Microbiota. Immunity 2017, 46, 562–576. [Google Scholar] [CrossRef] [Green Version]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.W.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 2019, 50, 432–445.e7. [Google Scholar] [CrossRef] [PubMed]
- Kellow, N.J.; Coughlan, M.T.; Reid, C.M. Metabolic benefits of dietary prebiotics in human subjects: A systematic review of randomised controlled trials. Br. J. Nutr. 2014, 111, 1147–1161. [Google Scholar] [CrossRef]
- Gasaly, N.; de Vos, P.; Hermoso, M.A. Impact of Bacterial Metabolites on Gut Barrier Function and Host Immunity: A Focus on Bacterial Metabolism and Its Relevance for Intestinal Inflammation. Front. Immunol. 2021, 12, 658354. [Google Scholar] [CrossRef]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of butyrate on intestinal barrier function in a Caco-2 cell monolayer model of intestinal barrier. Pediatr. Res. 2007, 61, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappenden, K.A.; Thomson, A.B.; Wild, G.E.; McBurney, M.I. Short-chain fatty acid-supplemented total parenteral nutrition enhances functional adaptation to intestinal resection in rats. Gastroenterology 1997, 112, 792–802. [Google Scholar] [CrossRef]
- Tappenden, K.A.; McBurney, M.I. Systemic short-chain fatty acids rapidly alter gastrointestinal structure, function, and expression of early response genes. Dig. Dis. Sci. 1998, 43, 1526–1536. [Google Scholar] [CrossRef]
- Bartholome, A.L.; Albin, D.M.; Baker, D.H.; Holst, J.J.; Tappenden, K.A. Supplementation of total parenteral nutrition with butyrate acutely increases structural aspects of intestinal adaptation after an 80% jejunoileal resection in neonatal piglets. JPEN J. Parenter. Enter. Nutr. 2004, 28, 210–222, discussion 222–223. [Google Scholar] [CrossRef]
- Tappenden, K.A.; Drozdowski, L.A.; Thomson, A.B.; McBurney, M.I. Short-chain fatty acid-supplemented total parenteral nutrition alters intestinal structure, glucose transporter 2 (GLUT2) mRNA and protein, and proglucagon mRNA abundance in normal rats. Am. J. Clin. Nutr. 1998, 68, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [Green Version]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjellström, B.; Stenhammar, L.; Högberg, L.; Fälth-Magnusson, K.; Magnusson, K.E.; Midtvedt, T.; Sundqvist, T.; Norin, E. Gut microflora associated characteristics in children with celiac disease. Am. J. Gastroenterol. 2005, 100, 2784–2788. [Google Scholar] [CrossRef] [Green Version]
- Tjellström, B.; Högberg, L.; Stenhammar, L.; Fälth-Magnusson, K.; Magnusson, K.E.; Norin, E.; Sundqvist, T.; Midtvedt, T. Faecal short-chain fatty acid pattern in childhood coeliac disease is normalised after more than one year’s gluten-free diet. Microb. Ecol. Health Dis. 2013, 24, 20905. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Rau, M.; Rehman, A.; Dittrich, M.; Groen, A.K.; Hermanns, H.M.; Seyfried, F.; Beyersdorf, N.; Dandekar, T.; Rosenstiel, P.; Geier, A. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United Eur. Gastroenterol. J. 2018, 6, 1496–1507. [Google Scholar] [CrossRef]
- Semeraro, L.A.; Barwick, K.W.; Gryboski, J.D. Obesity in celiac sprue. J. Clin. Gastroenterol. 1986, 8, 177–180. [Google Scholar] [CrossRef]
- Czaja-Bulsa, G.; Garanty-Bogacka, B.; Syrenicz, M.; Gebala, A. Obesity in an 18-year-old boy with untreated celiac disease. J. Pediatr. Gastroenterol. Nutr. 2001, 32, 226. [Google Scholar] [CrossRef]
- Franzese, A.; Iannucci, M.P.; Valerio, G.; Ciccimarra, E.; Spaziano, M.; Mandato, C.; Vajro, P. Atypical celiac disease presenting as obesity-related liver dysfunction. J. Pediatr. Gastroenterol. Nutr. 2001, 33, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Oso, O.; Fraser, N.C. A boy with coeliac disease and obesity. Acta Paediatr. (Oslo Nor. 1992) 2006, 95, 618–619. [Google Scholar] [CrossRef] [PubMed]
- Arslan, N.; Esen, I.; Demircioglu, F.; Yilmaz, S.; Unuvar, T.; Bober, E. The changing face of celiac disease: A girl with obesity and celiac disease. J. Paediatr. Child Health 2009, 45, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Balamtekin, N.; Demir, H.; Baysoy, G.; Uslu, N.; Yüce, A. Obesity in adolescents with celiac disease: Two adolescents and two different presentations. Turk. J. Pediatr. 2011, 53, 314–316. [Google Scholar]
- Bjørndal, B.; Alterås, E.K.; Lindquist, C.; Svardal, A.; Skorve, J.; Berge, R.K. Associations between fatty acid oxidation, hepatic mitochondrial function, and plasma acylcarnitine levels in mice. Nutr. Metab. (Lond.) 2018, 15, 10. [Google Scholar] [CrossRef] [Green Version]
- Gaur, K.; Sakhuja, P.; Puri, A.S.; Majumdar, K. Gluten-Free hepatomiracle in “celiac hepatitis”: A case highlighting the rare occurrence of nutrition-induced near total reversal of advanced steatohepatitis and cirrhosis. Saudi J. Gastroenterol. Off. J. Saudi Gastroenterol. Assoc. 2016, 22, 461–464. [Google Scholar] [CrossRef]
- Newnham, E.D. Coeliac disease in the 21st century: Paradigm shifts in the modern age. J. Gastroenterol. Hepatol. 2017, 32 (Suppl. 1), 82–85. [Google Scholar] [CrossRef] [Green Version]
- Sydor, S.; Best, J.; Messerschmidt, I.; Manka, P.; Vilchez-Vargas, R.; Brodesser, S.; Lucas, C.; Wegehaupt, A.; Wenning, C.; Aßmuth, S.; et al. Altered Microbiota Diversity and Bile Acid Signaling in Cirrhotic and Noncirrhotic NASH-HCC. Clin. Transl. Gastroenterol. 2020, 11, e00131. [Google Scholar] [CrossRef]
- Manka, P.; Sydor, S.; Wase, N.; Best, J.; Brandenburg, M.; Hellbeck, A.; Schänzer, J.; Vilchez-Vargas, R.; Link, A.; Figge, A.; et al. Anti-TNFα treatment in Crohn’s disease: Impact on hepatic steatosis, gut-derived hormones and metabolic status. Liver Int. Off. J. Int. Assoc. Study Liver 2021, 41, 2646–2658. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [Green Version]
- Manka, P.; Sydor, S.; Schänzer-Ocklenburg, J.M.; Brandenburg, M.; Best, J.; Vilchez-Vargas, R.; Link, A.; Heider, D.; Brodesser, S.; Figge, A.; et al. A Potential Role for Bile Acid Signaling in Celiac Disease-Associated Fatty Liver. Metabolites 2022, 12, 130. [Google Scholar] [CrossRef]
- Ejderhamn, J.; Samuelson, K.; Strandvik, B. Serum primary bile acids in the course of celiac disease in children. J. Pediatr. Gastroenterol. Nutr. 1992, 14, 443–449. [Google Scholar] [CrossRef]
- DiMagno, E.P.; Go, W.L.; Summerskill, W.H. Impaired cholecystokinin-pancreozymin secretion, intraluminal dilution, and maldigestion of fat in sprue. Gastroenterology 1972, 63, 25–32. [Google Scholar] [CrossRef]
- Low-Beer, T.S.; Heaton, K.W.; Pomare, E.W.; Read, A.E. The effect of coeliac disease upon bile salts. Gut 1973, 14, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maton, P.N.; Selden, A.C.; Fitzpatrick, M.L.; Chadwick, V.S. Defective gallbladder emptying and cholecystokinin release in celiac disease. Reversal by gluten-free diet. Gastroenterology 1985, 88, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Behar, J.; Mawe, G.M.; Carey, M.C. Roles of cholesterol and bile salts in the pathogenesis of gallbladder hypomotility and inflammation: Cholecystitis is not caused by cystic duct obstruction. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2013, 25, 283–290. [Google Scholar] [CrossRef]
- Peláez-Luna, M.; Schmulson, M.; Robles-Díaz, G. Intestinal involvement is not sufficient to explain hypertransaminasemia in celiac disease? Med. Hypotheses 2005, 65, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elli, L.; Bergamini, C.M.; Bardella, M.T.; Schuppan, D. Transglutaminases in inflammation and fibrosis of the gastrointestinal tract and the liver. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2009, 41, 541–550. [Google Scholar] [CrossRef]
- Drastich, P.; Honsová, E.; Lodererová, A.; Jarešová, M.; Pekáriková, A.; Hoffmanová, I.; Tučková, L.; Tlaskalová-Hogenová, H.; Spičák, J.; Sánchez, D. Celiac disease markers in patients with liver diseases: A single center large scale screening study. World J. Gastroenterol. 2012, 18, 6255–6262. [Google Scholar] [CrossRef]
- Esposito, C.; Paparo, F.; Caputo, I.; Rossi, M.; Maglio, M.; Sblattero, D.; Not, T.; Porta, R.; Auricchio, S.; Marzari, R.; et al. Anti-tissue transglutaminase antibodies from coeliac patients inhibit transglutaminase activity both in vitro and in situ. Gut 2002, 51, 177–181. [Google Scholar] [CrossRef]
- Hoffmanová, I.; Sánchez, D.; Džupa, V. Bone and Joint Involvement in Celiac Disease. Acta Chir. Orthop. Et Traumatol. Cechoslov. 2015, 82, 308–312. [Google Scholar]
- Carambia, A.; Herkel, J. Dietary and metabolic modulators of hepatic immunity. Semin. Immunopathol. 2018, 40, 175–188. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.C.; Wu, C.C.; Ni, Y.H. New Perspectives on Genetic Prediction for Pediatric Metabolic Associated Fatty Liver Disease. Front. Pediatr. 2020, 8, 603654. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobili, V.; Alisi, A.; Valenti, L.; Miele, L.; Feldstein, A.E.; Alkhouri, N. NAFLD in children: New genes, new diagnostic modalities and new drugs. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Tortora, R.; Rispo, A.; Alisi, A.; Imperatore, N.; Crudele, A.; Ferretti, F.; Nobili, V.; Miele, L.; Gerbino, N.; Caporaso, N.; et al. PNPLA3 rs738409 Polymorphism Predicts Development and Severity of Hepatic Steatosis but Not Metabolic Syndrome in Celiac Disease. Nutrients 2018, 10, 1239. [Google Scholar] [CrossRef] [Green Version]
- Mangge, H.; Baumgartner, B.G.; Zelzer, S.; Prüller, F.; Schnedl, W.J.; Reininghaus, E.Z.; Haybaeck, J.; Lackner, C.; Stauber, R.; Aigner, E.; et al. Patatin-like phospholipase 3 (rs738409) gene polymorphism is associated with increased liver enzymes in obese adolescents and metabolic syndrome in all ages. Aliment. Pharmacol. Ther. 2015, 42, 99–105. [Google Scholar] [CrossRef]
- Shen, J.; Wong, G.L.; Chan, H.L.; Chan, H.Y.; Yeung, D.K.; Chan, R.S.; Chim, A.M.; Chan, A.W.; Choi, P.C.; Woo, J.; et al. PNPLA3 gene polymorphism accounts for fatty liver in community subjects without metabolic syndrome. Aliment. Pharmacol. Ther. 2014, 39, 532–539. [Google Scholar] [CrossRef]
- Del Ben, M.; Polimeni, L.; Brancorsini, M.; Di Costanzo, A.; D’Erasmo, L.; Baratta, F.; Loffredo, L.; Pastori, D.; Pignatelli, P.; Violi, F.; et al. Non-alcoholic fatty liver disease, metabolic syndrome and patatin-like phospholipase domain-containing protein3 gene variants. Eur. J. Intern. Med. 2014, 25, 566–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rispo, A.; Imperatore, N.; Guarino, M.; Tortora, R.; Alisi, A.; Cossiga, V.; Testa, A.; Ricciolino, S.; Fiorentino, A.; Morisco, F. Metabolic-associated fatty liver disease (MAFLD) in coeliac disease. Liver Int. Off. J. Int. Assoc. Study Liver 2021, 41, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Reilly, N.R.; Aguilar, K.; Hassid, B.G.; Cheng, J.; Defelice, A.R.; Kazlow, P.; Bhagat, G.; Green, P.H. Celiac disease in normal-weight and overweight children: Clinical features and growth outcomes following a gluten-free diet. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Anania, C.; Pacifico, L.; Olivero, F.; Perla, F.M.; Chiesa, C. Cardiometabolic risk factors in children with celiac disease on a gluten-free diet. World J. Clin. Pediatr. 2017, 6, 143–148. [Google Scholar] [CrossRef]
- Zanini, B.; Mazzoncini, E.; Lanzarotto, F.; Ricci, C.; Cesana, B.M.; Villanacci, V.; Lanzini, A. Impact of gluten-free diet on cardiovascular risk factors. A retrospective analysis in a large cohort of coeliac patients. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2013, 45, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Brar, P.; Kwon, G.Y.; Holleran, S.; Bai, D.; Tall, A.R.; Ramakrishnan, R.; Green, P.H. Change in lipid profile in celiac disease: Beneficial effect of gluten-free diet. Am. J. Med. 2006, 119, 786–790. [Google Scholar] [CrossRef]
- de Onis, M.; Blössner, M.; Borghi, E. Global prevalence and trends of overweight and obesity among preschool children. Am. J. Clin. Nutr. 2010, 92, 1257–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barera, G.; Mora, S.; Brambilla, P.; Ricotti, A.; Menni, L.; Beccio, S.; Bianchi, C. Body composition in children with celiac disease and the effects of a gluten-free diet: A prospective case-control study. Am. J. Clin. Nutr. 2000, 72, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Brar, P.S.; Lee, A.R.; Green, P.H. Body mass index in celiac disease: Beneficial effect of a gluten-free diet. J. Clin. Gastroenterol. 2010, 44, 267–271. [Google Scholar] [CrossRef]
- Radlović, N.; Mladenović, M.; Leković, Z.; Zivanović, D.; Brdar, R.; Radlović, V.; Ristić, D.; Pavlović, M.; Stojsić, Z.; Vuletić, B.; et al. Effect of gluten-free diet on the growth and nutritional status of children with coeliac disease. Srp. Arh. Za Celok. Lek. 2009, 137, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, P.; Picca, M.; Dilillo, D.; Meneghin, F.; Cravidi, C.; Tischer, M.C.; Vivaldo, T.; Bedogni, G.; Zuccotti, G.V. Changes of body mass index in celiac children on a gluten-free diet. Nutr. Metab. Cardiovasc. Dis. NMCD 2013, 23, 177–182. [Google Scholar] [CrossRef]
- Penagini, F.; Dilillo, D.; Meneghin, F.; Mameli, C.; Fabiano, V.; Zuccotti, G.V. Gluten-free diet in children: An approach to a nutritionally adequate and balanced diet. Nutrients 2013, 5, 4553–4565. [Google Scholar] [CrossRef] [PubMed]
- Mariani, P.; Viti, M.G.; Montuori, M.; La Vecchia, A.; Cipolletta, E.; Calvani, L.; Bonamico, M. The gluten-free diet: A nutritional risk factor for adolescents with celiac disease? J. Pediatr. Gastroenterol. Nutr. 1998, 27, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Patton, H.M.; Yates, K.; Unalp-Arida, A.; Behling, C.A.; Huang, T.T.; Rosenthal, P.; Sanyal, A.J.; Schwimmer, J.B.; Lavine, J.E. Association between metabolic syndrome and liver histology among children with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2010, 105, 2093–2102. [Google Scholar] [CrossRef] [Green Version]
- Tortora, R.; Capone, P.; De Stefano, G.; Imperatore, N.; Gerbino, N.; Donetto, S.; Monaco, V.; Caporaso, N.; Rispo, A. Metabolic syndrome in patients with coeliac disease on a gluten-free diet. Aliment. Pharmacol. Ther. 2015, 41, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Mishima, Y.; Kuyama, A.; Tada, A.; Takahashi, K.; Ishioka, T.; Kibata, M. Relationship between serum tumor necrosis factor-alpha and insulin resistance in obese men with Type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2001, 52, 119–123. [Google Scholar] [CrossRef]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Luciani, A.; Villella, V.R.; Vasaturo, A.; Giardino, I.; Pettoello-Mantovani, M.; Guido, S.; Cexus, O.N.; Peake, N.; Londei, M.; Quaratino, S.; et al. Lysosomal accumulation of gliadin p31–43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut 2010, 59, 311–319. [Google Scholar] [CrossRef]
- Wheeler, E.; Barroso, I. Genome-wide association studies and type 2 diabetes. Brief. Funct. Genom. 2011, 10, 52–60. [Google Scholar] [CrossRef]
- Rosenthal, E.; Hoffman, R.; Aviram, M.; Benderly, A.; Erde, P.; Brook, J.G. Serum lipoprotein profile in children with celiac disease. J. Pediatr. Gastroenterol. Nutr. 1990, 11, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Ciampolini, M.; Bini, S. Serum lipids in celiac children. J. Pediatr. Gastroenterol. Nutr. 1991, 12, 459–460. [Google Scholar] [CrossRef]
- Pillan, M.N.; Spandrio, S.; Sleiman, I.; Meini, A.; Scalvini, T.; Balestrieri, G.P. Effects of a gluten-free diet on serum lipids and lipoprotein (a) levels in a group of patients with celiac disease. J. Pediatr. Gastroenterol. Nutr. 1994, 18, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Forchielli, M.L.; Fernicola, P.; Diani, L.; Scrivo, B.; Salfi, N.C.; Pessina, A.C.; Lima, M.; Conti, V.; Pession, A. Gluten-Free Diet and Lipid Profile in Children With Celiac Disease: Comparison With General Population Standards. J. Pediatr. Gastroenterol. Nutr. 2015, 61, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scapaticci, S.; Venanzi, A.; Chiarelli, F.; Giannini, C. MAFLD and Celiac Disease in Children. Int. J. Mol. Sci. 2023, 24, 1764. https://doi.org/10.3390/ijms24021764
Scapaticci S, Venanzi A, Chiarelli F, Giannini C. MAFLD and Celiac Disease in Children. International Journal of Molecular Sciences. 2023; 24(2):1764. https://doi.org/10.3390/ijms24021764
Chicago/Turabian StyleScapaticci, Serena, Annamaria Venanzi, Francesco Chiarelli, and Cosimo Giannini. 2023. "MAFLD and Celiac Disease in Children" International Journal of Molecular Sciences 24, no. 2: 1764. https://doi.org/10.3390/ijms24021764
APA StyleScapaticci, S., Venanzi, A., Chiarelli, F., & Giannini, C. (2023). MAFLD and Celiac Disease in Children. International Journal of Molecular Sciences, 24(2), 1764. https://doi.org/10.3390/ijms24021764