
Biglycan Involvement in Heart Fibrosis: Modulation of Adenosine 2A Receptor Improves Damage in Immortalized Cardiac Fibroblasts

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Treatment with Istradefylline and ZM241385, Two Different A2A Receptor Antagonists, Does Not Affect IM-HCF Cell Viability
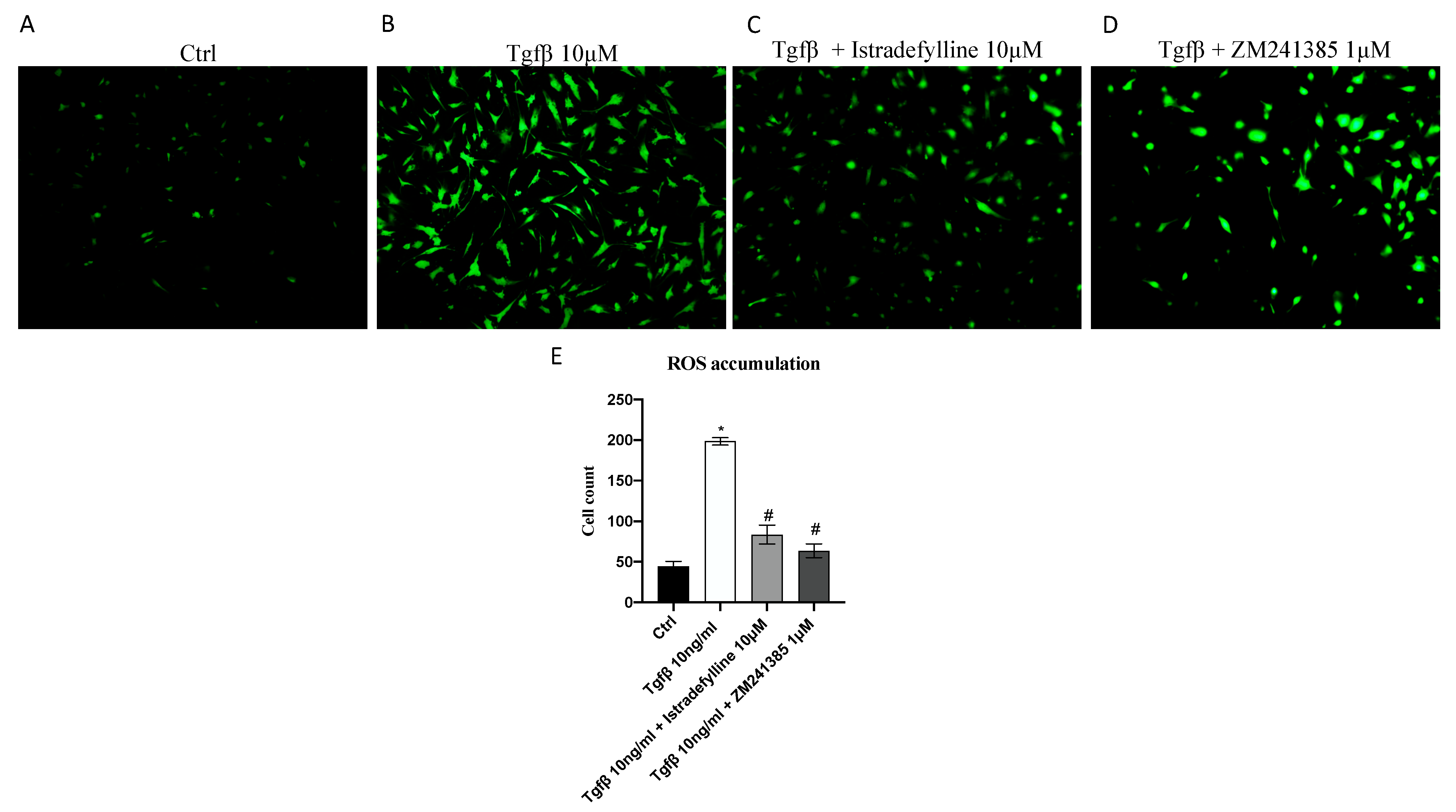
2.2. Antagonism of A2AR Reduces Intracellular ROS Production
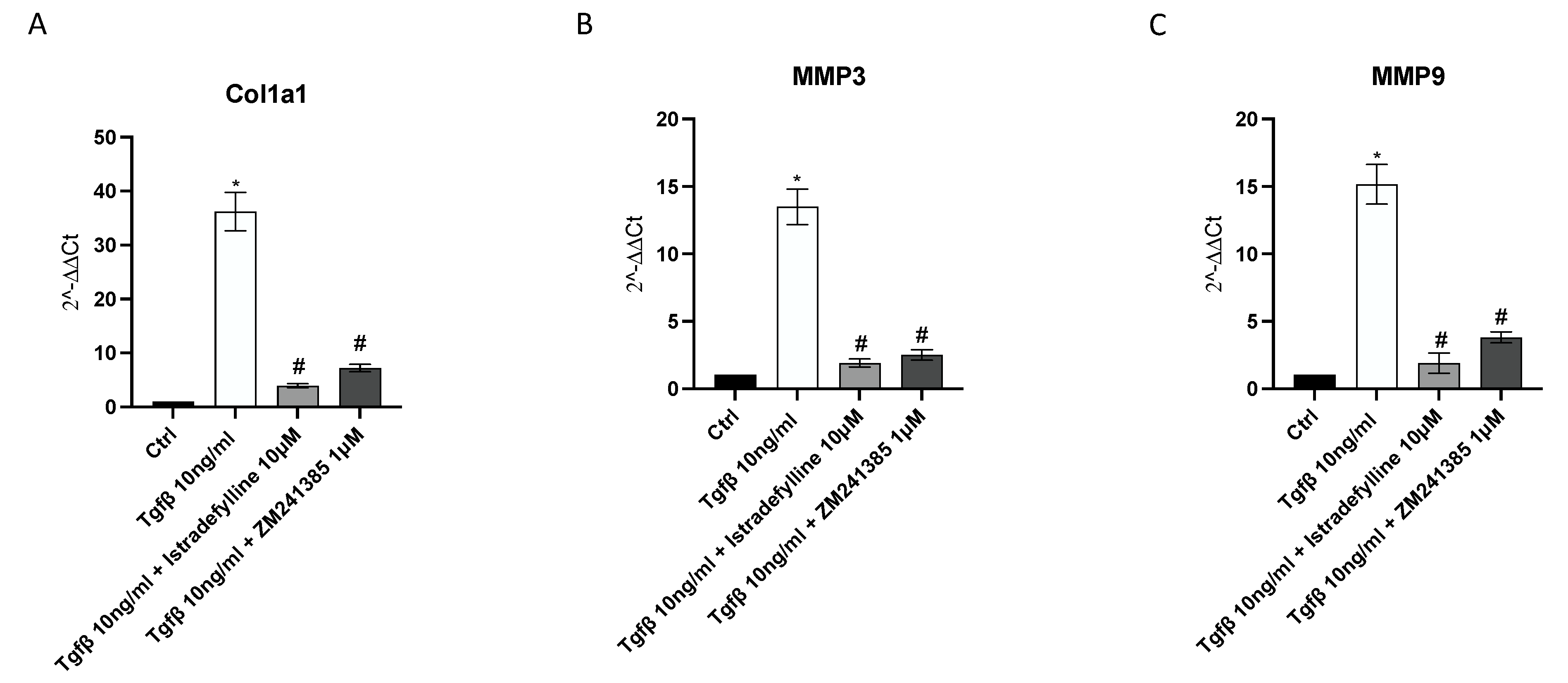
2.3. A2AR Inhibition Produces an Anti-Fibrotic Effect in Human Cardiac Fibroblasts
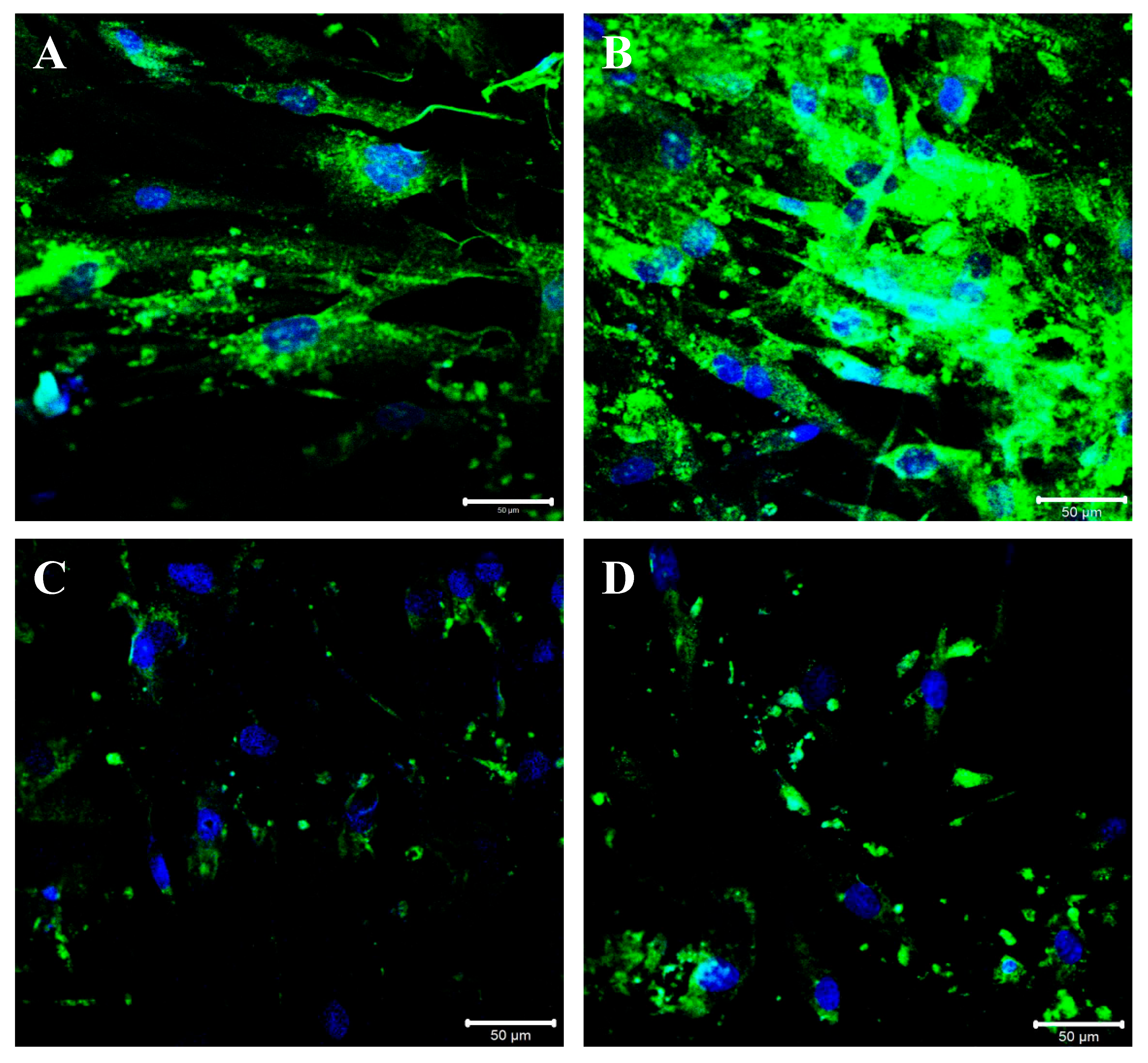
2.4. A2AR Inhibition Modulates BGN, IL-1β and Caspase-1 Expression following TGF-β Stimulation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Treatments
4.3. MTT Assay
4.4. Measurement of ROS Production
4.5. Real-Time PCR Assay
4.6. Western Blot
4.7. Immunofluorescence
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, W.; Xiong, Y.; Li, X.; Yang, Y. Cardiac Fibrosis: Cellular Effectors, Molecular Pathways, and Exosomal Roles. Front. Cardiovasc. Med. 2021, 8, 715258. [Google Scholar] [CrossRef] [PubMed]
- van Putten, S.; Shafieyan, Y.; Hinz, B. Mechanical control of cardiac myofibroblasts. J. Mol. Cell. Cardiol. 2016, 93, 133–142. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.W.; Chung, C.C.; Lee, T.I.; Lin, Y.K.; Kao, Y.H.; Chen, Y.J. Fibroblast Growth Factor 23 Stimulates Cardiac Fibroblast Activity through Phospholipase C-Mediated Calcium Signaling. Int. J. Mol. Sci. 2021, 23, 166. [Google Scholar] [CrossRef] [PubMed]
- Scalise, R.F.M.; De Sarro, R.; Caracciolo, A.; Lauro, R.; Squadrito, F.; Carerj, S.; Bitto, A.; Micari, A.; Bella, G.D.; Costa, F.; et al. Fibrosis after Myocardial Infarction: An Overview on Cellular Processes, Molecular Pathways, Clinical Evaluation and Prognostic Value. Med. Sci. 2021, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Lu, Z.; Ye, W. TGF-β1 Induces Interlukin-11 Expression and Pro-Fibrotic Effect by DNA Demethylation in Subconjunctival Fibroblasts. Evid.-Based Complement. Altern. Med. Ecam 2022, 2022, 7729827. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.J.; Tsai, F.C.; Chang, G.J.; Chang, S.H.; Huang, C.C.; Chen, W.J.; Yeh, Y.H. miR-181b targets semaphorin 3A to mediate TGF-β-induced endothelial-mesenchymal transition related to atrial fibrillation. J. Clin. Investig. 2022, 132, 142548. [Google Scholar] [CrossRef]
- Li, N.; Hang, W.; Shu, H.; Zhou, N. Pirfenidone alleviates cardiac fibrosis induced by pressure overload via inhibiting TGF-β1/Smad3 signalling pathway. J. Cell. Mol. Med. 2022, 26, 4548–4555. [Google Scholar] [CrossRef]
- Saljic, A.; Grandi, E.; Dobrev, D. TGF-β1-induced endothelial-mesenchymal transition: A potential contributor to fibrotic remodeling in atrial fibrillation? J. Clin. Investig. 2022, 132, 161070. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Seo, E.; Oh, Y.S.; Jun, H.S. TGF-β activates NLRP3 inflammasome by an autocrine production of TGF-β in LX-2 human hepatic stellate cells. Mol. Cell. Biochem. 2022, 477, 1329–1338. [Google Scholar] [CrossRef]
- Kang, L.L.; Zhang, D.M.; Ma, C.H.; Zhang, J.H.; Jia, K.K.; Liu, J.H.; Wang, R.; Kong, L.D. Cinnamaldehyde and allopurinol reduce fructose-induced cardiac inflammation and fibrosis by attenuating CD36-mediated TLR4/6-IRAK4/1 signaling to suppress NLRP3 inflammasome activation. Sci. Rep. 2016, 6, 27460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R., Jr.; Fedak, P.W.; Beck, P.L.; Muruve, D.A.; Duff, H.J. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014, 289, 19571–19584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis via Activating the NLRP3 Inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Chen, S.J.; Zhou, S.C.; Wu, S.Z.; Wang, H. Inflammasomes and Fibrosis. Front. Immunol. 2021, 12, 643149. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Lv, X.; Wang, Q.; Zhao, H.; Yang, F.; Yang, Y.; Li, J. Involvement of cAMP-PKA pathway in adenosine A1 and A2A receptor-mediated regulation of acetaldehyde-induced activation of HSCs. Biochimie 2015, 115, 59–70. [Google Scholar] [CrossRef]
- Vasiukov, G.; Menshikh, A.; Owens, P.; Novitskaya, T.; Hurley, P.; Blackwell, T.; Feoktistov, I.; Novitskiy, S.V. Adenosine/TGFβ axis in regulation of mammary fibroblast functions. PLoS ONE 2021, 16, e0252424. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, G.; Cronstein, B. Signaling pathways involving adenosine A2A and A2B receptors in wound healing and fibrosis. Purinergic Signal. 2016, 12, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.S.; Øie, E.; Vinge, L.E.; Yndestad, A.; Andersen, G.G.; Andersson, Y.; Attramadal, T.; Attramadal, H. Induction of myocardial biglycan in heart failure in rats--an extracellular matrix component targeted by AT (1) receptor antagonism. Cardiovasc. Res. 2003, 60, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Bereczki, E.; Gonda, S.; Csont, T.; Korpos, E.; Zvara, A.; Ferdinandy, P.; Santha, M. Overexpression of biglycan in the heart of transgenic mice: An antibody microarray study. J. Proteome Res. 2007, 6, 854–861. [Google Scholar] [CrossRef]
- Bereczki, E.; Sántha, M. The role of biglycan in the heart. Connect Tissue Res 2008, 49, 129–132. [Google Scholar] [CrossRef]
- Schönherr, E.; Järveläinen, H.T.; Kinsella, M.G.; Sandell, L.J.; Wight, T.N. Platelet-derived growth factor and transforming growth factor-beta 1 differentially affect the synthesis of biglycan and decorin by monkey arterial smooth muscle cells. Arterioscler. Thromb. J. Vasc. Biol. 1993, 13, 1026–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Krull, N.B. Transcriptional regulation of the human biglycan gene. J. Biol. Chem. 1996, 271, 15787–15795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, L.; Beck, K.F.; Raslik, I.; Walpen, S.; Mihalik, D.; Micegova, M.; Macakova, K.; Schonherr, E.; Seidler, D.G.; Varga, G.; et al. Biglycan, a nitric oxide-regulated gene, affects adhesion, growth, and survival of mesangial cells. J. Biol. Chem. 2003, 278, 26227–26237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.F.; Yin, X.J.; Zhao, W.J.; Liu, L.C.; Wang, Z.P. Biglycan as a potential diagnostic and prognostic biomarker in multiple human cancers. Oncol. Lett. 2020, 19, 1673–1682. [Google Scholar] [CrossRef] [Green Version]
- Schulz, M.; Diehl, V.; Trebicka, J.; Wygrecka, M.; Schaefer, L. Biglycan: A regulator of hepatorenal inflammation and autophagy. Matrix Biol. J. Int. Soc. Matrix Biol. 2021, 100, 150–161. [Google Scholar] [CrossRef]
- Melchior-Becker, A.; Dai, G.; Ding, Z.; Schäfer, L.; Schrader, J.; Young, M.F.; Fischer, J.W. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J. Biol. Chem. 2011, 286, 17365–17375. [Google Scholar] [CrossRef] [Green Version]
- Beetz, N.; Rommel, C.; Schnick, T.; Neumann, E.; Lother, A.; Monroy-Ordonez, E.B.; Zeeb, M.; Preissl, S.; Gilsbach, R.; Melchior-Becker, A.; et al. Ablation of biglycan attenuates cardiac hypertrophy and fibrosis after left ventricular pressure overload. J. Mol. Cell. Cardiol. 2016, 101, 145–155. [Google Scholar] [CrossRef]
- Roberts, V.S.; Cowan, P.J.; Alexander, S.I.; Robson, S.C.; Dwyer, K.M. The role of adenosine receptors A2A and A2B signaling in renal fibrosis. Kidney Int. 2014, 86, 685–692. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, M.R.; Vance, C.O.; Morschl, E.; Wilson, C.N. Adenosine receptors and inflammation. Handb. Exp. Pharmacol. 2009, 193, 215–269. [Google Scholar] [CrossRef]
- Cronstein, B.N. Adenosine receptors and fibrosis: A translational review. F1000 Biol. Rep. 2011, 3, 21. [Google Scholar] [CrossRef]
- Guo, F.; Wang, X.; Guo, Y.; Wan, W.; Cui, Y.; Wang, J.; Liu, W. Shenfu Administration Improves Cardiac Fibrosis in Rats With Myocardial Ischemia-Reperfusion Through Adenosine A(2a) Receptor Activation. Hum. Exp. Toxicol. 2022, 41, 9603271221077684. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, E.A.; White, P.J.; May, L.T. Targeting Adenosine Receptors for the Treatment of Cardiac Fibrosis. Front. Pharmacol. 2017, 8, 243. [Google Scholar] [CrossRef] [Green Version]
- Guieu, R.; Deharo, J.C.; Maille, B.; Crotti, L.; Torresani, E.; Brignole, M.; Parati, G. Adenosine and the Cardiovascular System: The Good and the Bad. J. Clin. Med. 2020, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef]
- Perez-Aso, M.; Fernandez, P.; Mediero, A.; Chan, E.S.; Cronstein, B.N. Adenosine 2A receptor promotes collagen production by human fibroblasts via pathways involving cyclic AMP and AKT but independent of Smad2/3. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Rønnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenes. Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Siwik, D.A.; Pagano, P.J.; Colucci, W.S. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 280, C53–C60. [Google Scholar] [CrossRef]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [Green Version]
- Dobaczewski, M.; Bujak, M.; Li, N.; Gonzalez-Quesada, C.; Mendoza, L.H.; Wang, X.F.; Frangogiannis, N.G. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010, 107, 418–428. [Google Scholar] [CrossRef]
- Saadat, S.; Noureddini, M.; Mahjoubin-Tehran, M.; Nazemi, S.; Shojaie, L.; Aschner, M.; Maleki, B.; Abbasi-Kolli, M.; Rajabi Moghadam, H.; Alani, B.; et al. Pivotal Role of TGF-β/Smad Signaling in Cardiac Fibrosis: Non-coding RNAs as Effectual Players. Front. Cardiovasc. Med. 2020, 7, 588347. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, S.; Sharma, M.V.R.; Brownson, B.; Gallicano, V.E.; Gallicano, G.I. Cardiac inducing colonies halt fibroblast activation and induce cardiac/endothelial cells to move and expand via paracrine signaling. Mol. Biol. Cell 2022, 33, ar96. [Google Scholar] [CrossRef] [PubMed]
- Medzikovic, L.; Heese, H.; van Loenen, P.B.; van Roomen, C.; Hooijkaas, I.B.; Christoffels, V.M.; Creemers, E.E.; de Vries, C.J.M.; de Waard, V. Nuclear Receptor Nur77 Controls Cardiac Fibrosis through Distinct Actions on Fibroblasts and Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 1600. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Cibi, D.M.; Ghani, S.; Singh, A.; Tee, N.; Sivakumar, V.; Bogireddi, H.; Cook, S.A.; Mao, J.; Singh, M.K. Loss of Yap/Taz in cardiac fibroblasts attenuates adverse remodelling and improves cardiac function. Cardiovasc. Res. 2022, 118, 1785–1804. [Google Scholar] [CrossRef]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef]
- Westermann, D.; Mersmann, J.; Melchior, A.; Freudenberger, T.; Petrik, C.; Schaefer, L.; Lüllmann-Rauch, R.; Lettau, O.; Jacoby, C.; Schrader, J.; et al. Biglycan is required for adaptive remodeling after myocardial infarction. Circulation 2008, 117, 1269–1276. [Google Scholar] [CrossRef] [Green Version]
- Perez-Aso, M.; Chiriboga, L.; Cronstein, B.N. Pharmacological blockade of adenosine A2A receptors diminishes scarring. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 4254–4263. [Google Scholar] [CrossRef] [Green Version]
- Tachampa, K.; Wongtawan, T. Unique patterns of cardiogenic and fibrotic gene expression in rat cardiac fibroblasts. Vet. World 2020, 13, 1697–1708. [Google Scholar] [CrossRef]
- Babelova, A.; Moreth, K.; Tsalastra-Greul, W.; Zeng-Brouwers, J.; Eickelberg, O.; Young, M.F.; Bruckner, P.; Pfeilschifter, J.; Schaefer, R.M.; Grone, H.J.; et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J. Biol. Chem. 2009, 284, 24035–24048. [Google Scholar] [CrossRef] [Green Version]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef]
- Ouyang, X.; Ghani, A.; Malik, A.; Wilder, T.; Colegio, O.R.; Flavell, R.A.; Cronstein, B.N.; Mehal, W.Z. Adenosine is required for sustained inflammasome activation via the A₂A receptor and the HIF-1α pathway. Nat. Commun. 2013, 4, 2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonuccio, P.; Micali, A.G.; Romeo, C.; Freni, J.; Vermiglio, G.; Puzzolo, D.; Squadrito, F.; Irrera, N.; Marini, H.R.; Rana, R.A.; et al. NLRP3 Inflammasome: A New Pharmacological Target for Reducing Testicular Damage Associated with Varicocele. Int. J. Mol. Sci. 2021, 22, 1319. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, C.M.; Song, Y.S.; Zaitoun, I.S.; Wang, S.; Hanna, B.A.; Darjatmoko, S.R.; Gurel, Z.; Fisk, D.L.; McDowell, C.M.; McAdams, R.M.; et al. Caffeine Inhibits Choroidal Neovascularization Through Mitigation of Inflammatory and Angiogenesis Activities. Front. Cell Dev. Biol. 2021, 9, 737426. [Google Scholar] [CrossRef]
- Mediero, A.; Wilder, T.; Reddy, V.S.; Cheng, Q.; Tovar, N.; Coelho, P.G.; Witek, L.; Whatling, C.; Cronstein, B.N. Ticagrelor regulates osteoblast and osteoclast function and promotes bone formation in vivo via an adenosine-dependent mechanism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 3887–3900. [Google Scholar] [CrossRef] [PubMed]
- Picciolo, G.; Pallio, G.; Altavilla, D.; Vaccaro, M.; Oteri, G.; Irrera, N.; Squadrito, F. β-Caryophyllene Reduces the Inflammatory Phenotype of Periodontal Cells by Targeting CB2 Receptors. Biomedicines 2020, 8, 164. [Google Scholar] [CrossRef]
- Micali, A.; Pallio, G.; Irrera, N.; Marini, H.; Trichilo, V.; Puzzolo, D.; Pisani, A.; Malta, C.; Santoro, G.; Laurà, R.; et al. Flavocoxid, a Natural Antioxidant, Protects Mouse Kidney from Cadmium-Induced Toxicity. Oxid. Med. Cell Longev. 2018, 2018, 9162946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenga, S.; Marini, H.R.; Micali, A.; Freni, J.; Pallio, G.; Irrera, N.; Squadrito, F.; Altavilla, D.; Antonelli, A.; Ferrari, S.M.; et al. Protective Effects of Myo-Inositol and Selenium on Cadmium-Induced Thyroid Toxicity in Mice. Nutrients 2020, 12, 1222. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scuruchi, M.; Mannino, F.; Imbesi, C.; Pallio, G.; Vermiglio, G.; Bagnato, G.; Minutoli, L.; Bitto, A.; Squadrito, F.; Irrera, N. Biglycan Involvement in Heart Fibrosis: Modulation of Adenosine 2A Receptor Improves Damage in Immortalized Cardiac Fibroblasts. Int. J. Mol. Sci. 2023, 24, 1784. https://doi.org/10.3390/ijms24021784
Scuruchi M, Mannino F, Imbesi C, Pallio G, Vermiglio G, Bagnato G, Minutoli L, Bitto A, Squadrito F, Irrera N. Biglycan Involvement in Heart Fibrosis: Modulation of Adenosine 2A Receptor Improves Damage in Immortalized Cardiac Fibroblasts. International Journal of Molecular Sciences. 2023; 24(2):1784. https://doi.org/10.3390/ijms24021784
Chicago/Turabian StyleScuruchi, Michele, Federica Mannino, Chiara Imbesi, Giovanni Pallio, Giovanna Vermiglio, Gianluca Bagnato, Letteria Minutoli, Alessandra Bitto, Francesco Squadrito, and Natasha Irrera. 2023. "Biglycan Involvement in Heart Fibrosis: Modulation of Adenosine 2A Receptor Improves Damage in Immortalized Cardiac Fibroblasts" International Journal of Molecular Sciences 24, no. 2: 1784. https://doi.org/10.3390/ijms24021784