
Top Selling (2026) Small Molecule Orphan Drugs: A Journey into Their Chemistry

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Top Selling Orphan Drugs (Forecast 2026)
2.1. Ibrutinib (Brand Name Imbruvica®)
2.2. Elexacaftor/Tezacaftor/Ivacaftor (Brand Name Trikafta®)
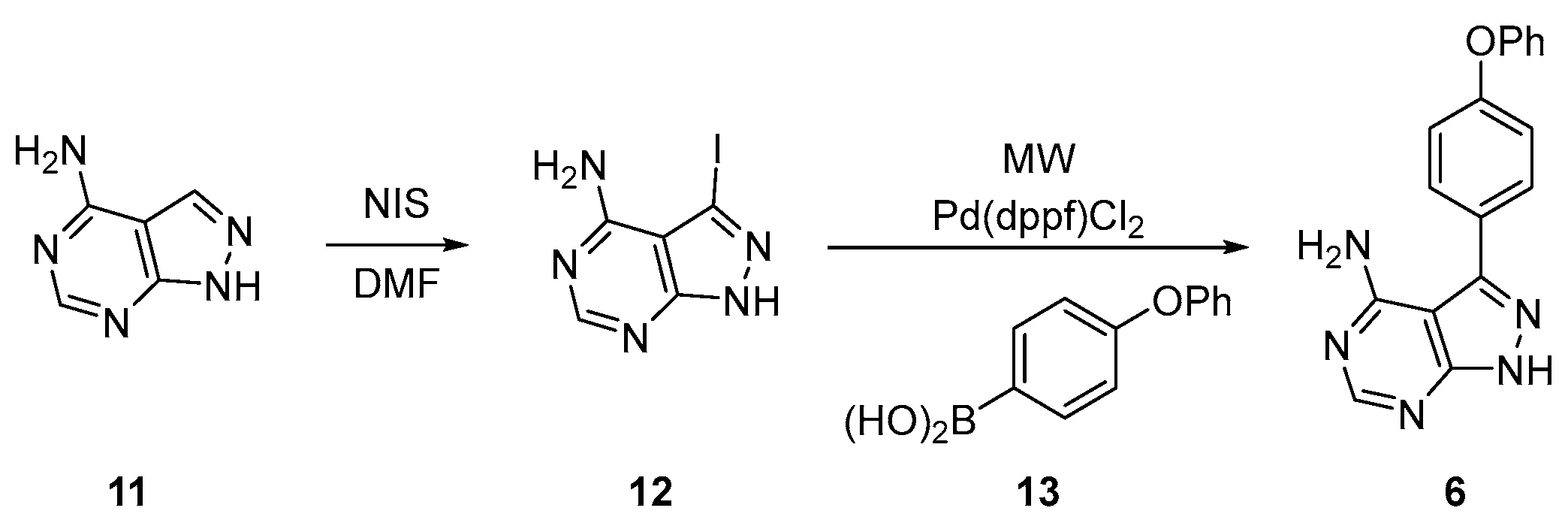
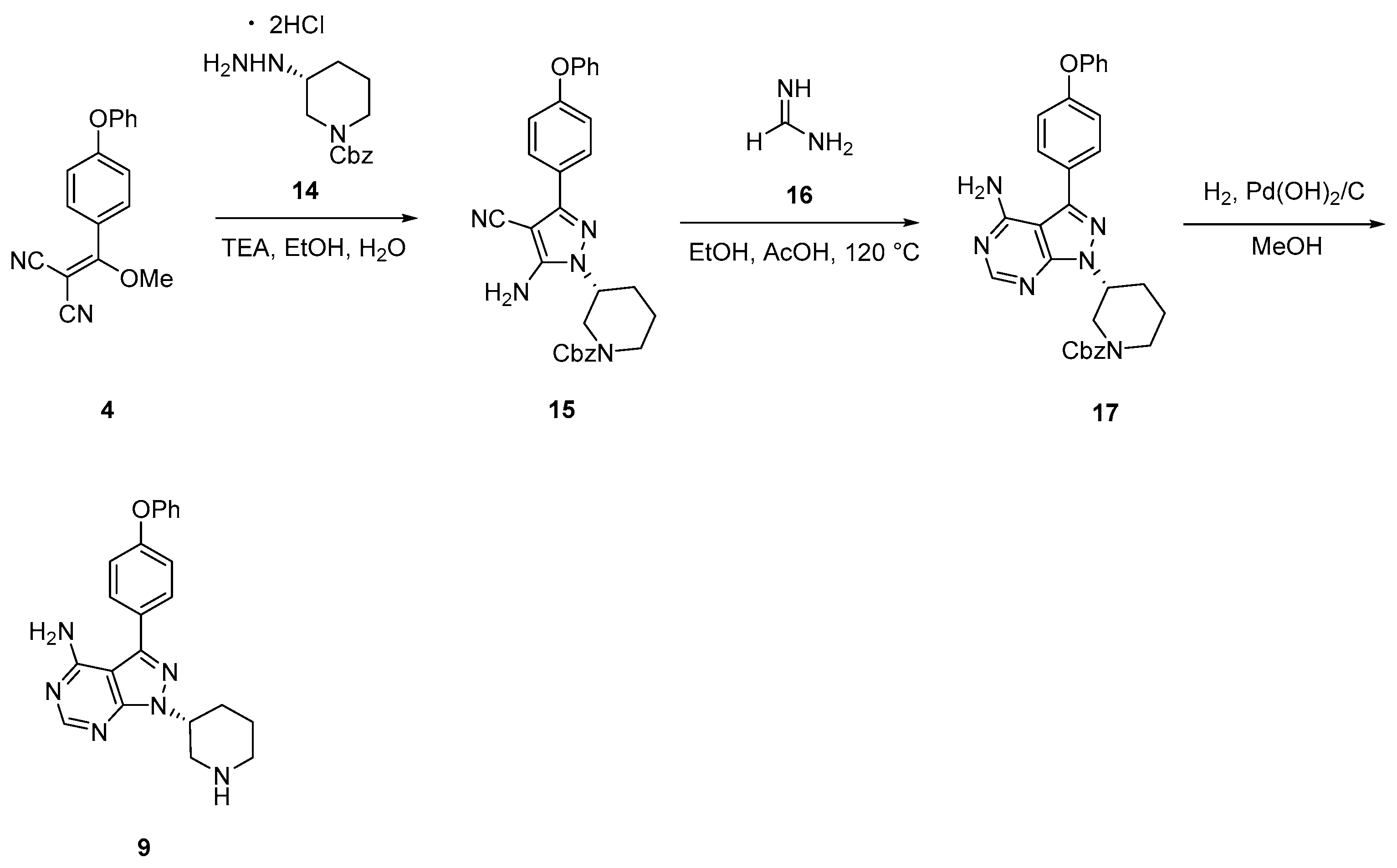
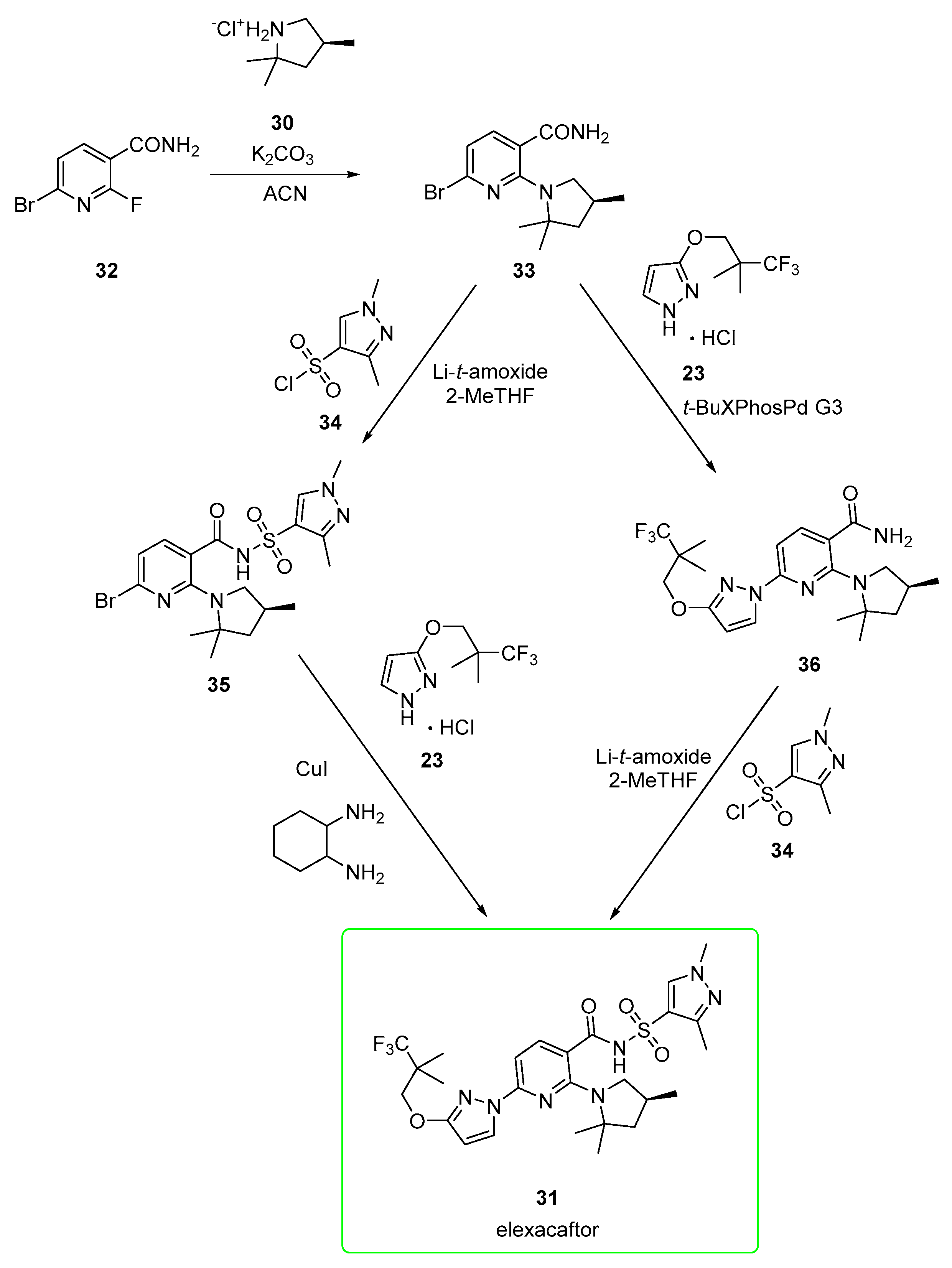
2.2.1. Elexacaftor
2.2.2. Tezacaftor
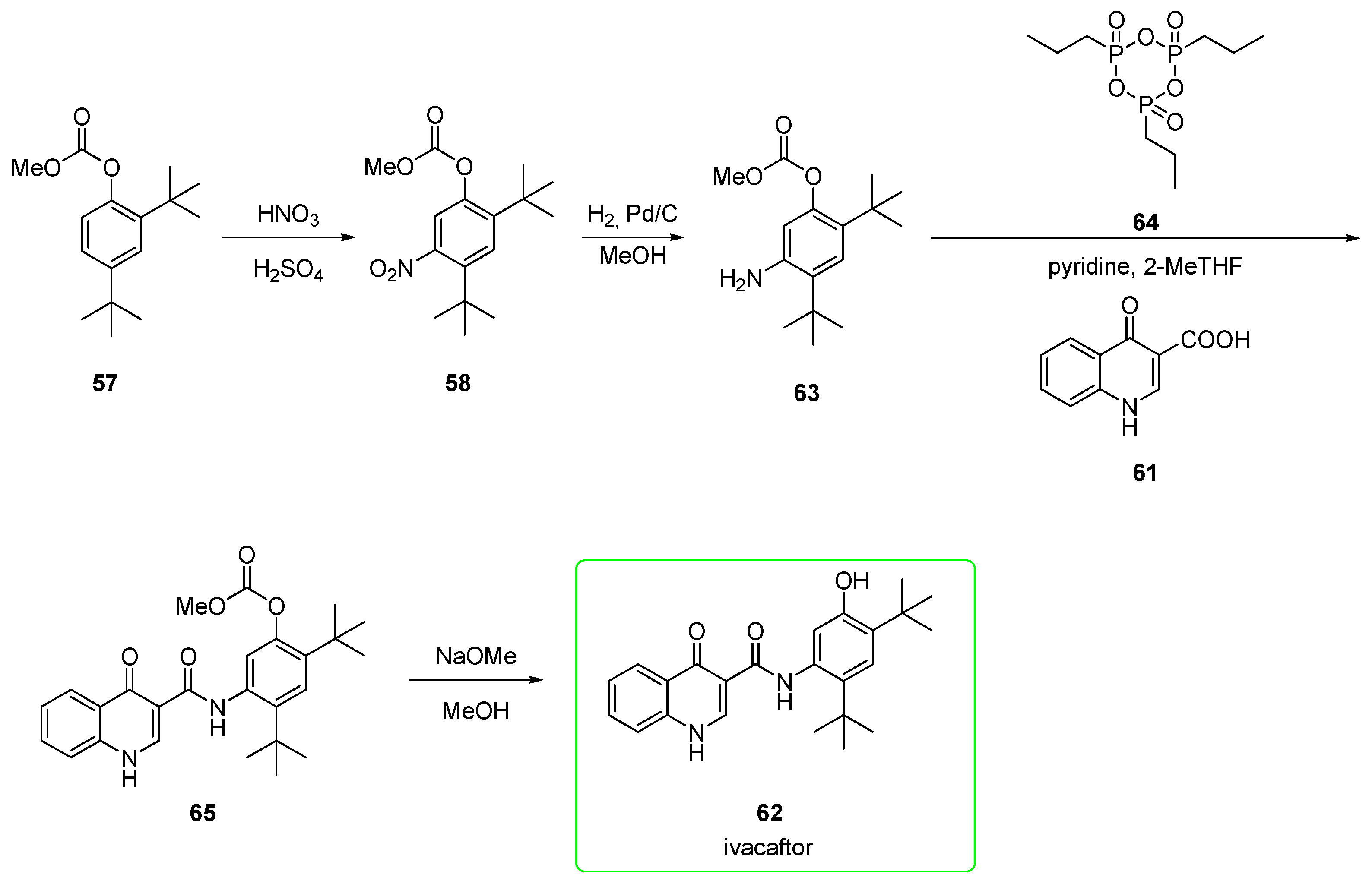
2.2.3. Ivacaftor
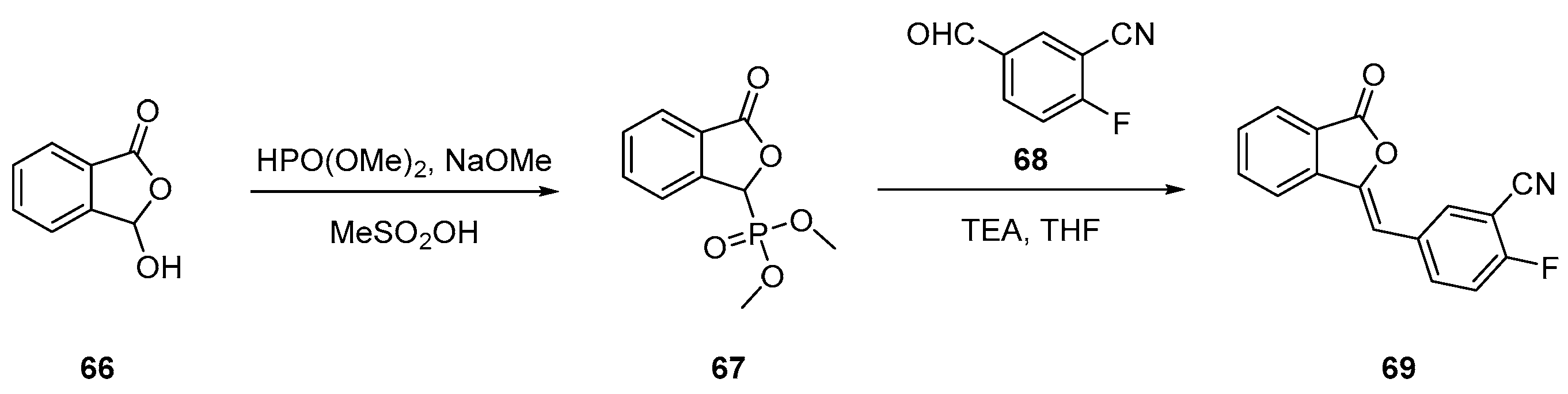
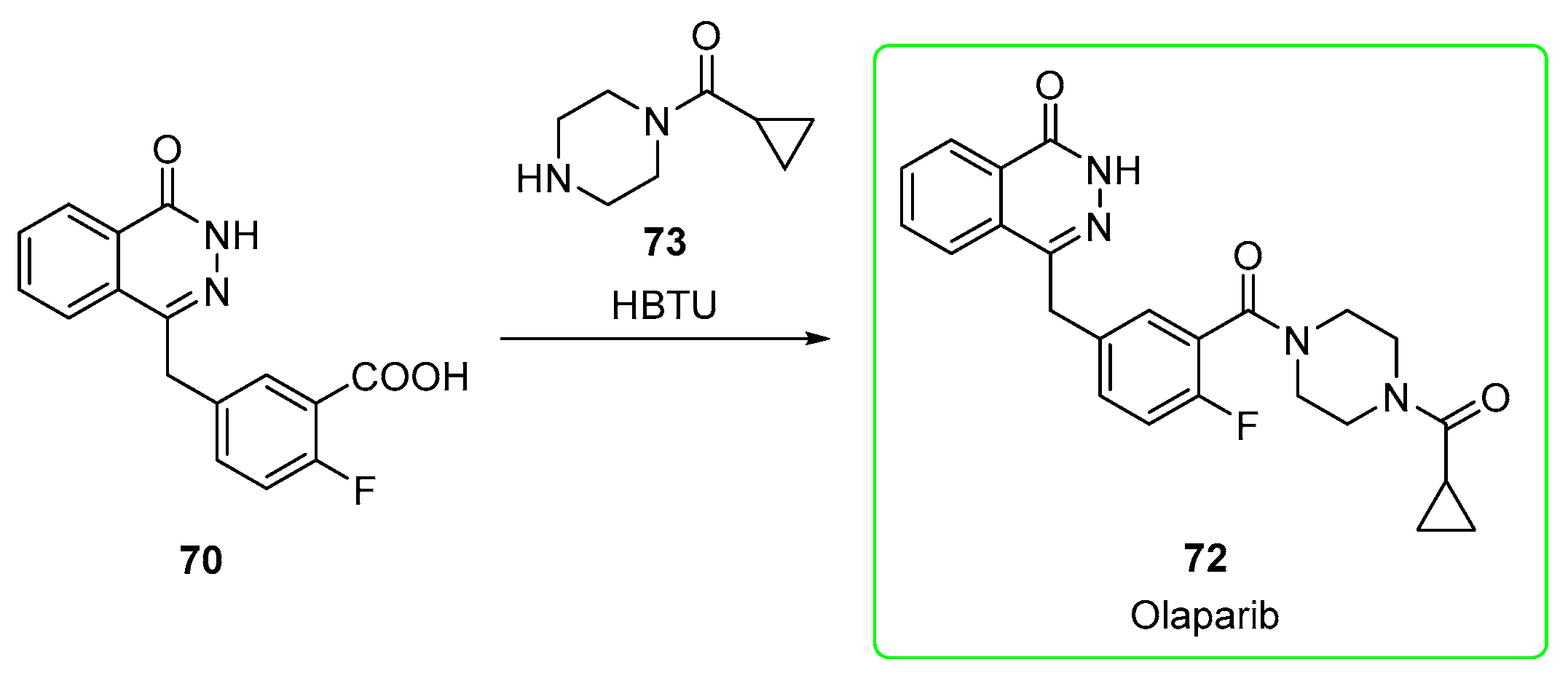
2.3. Olaparib
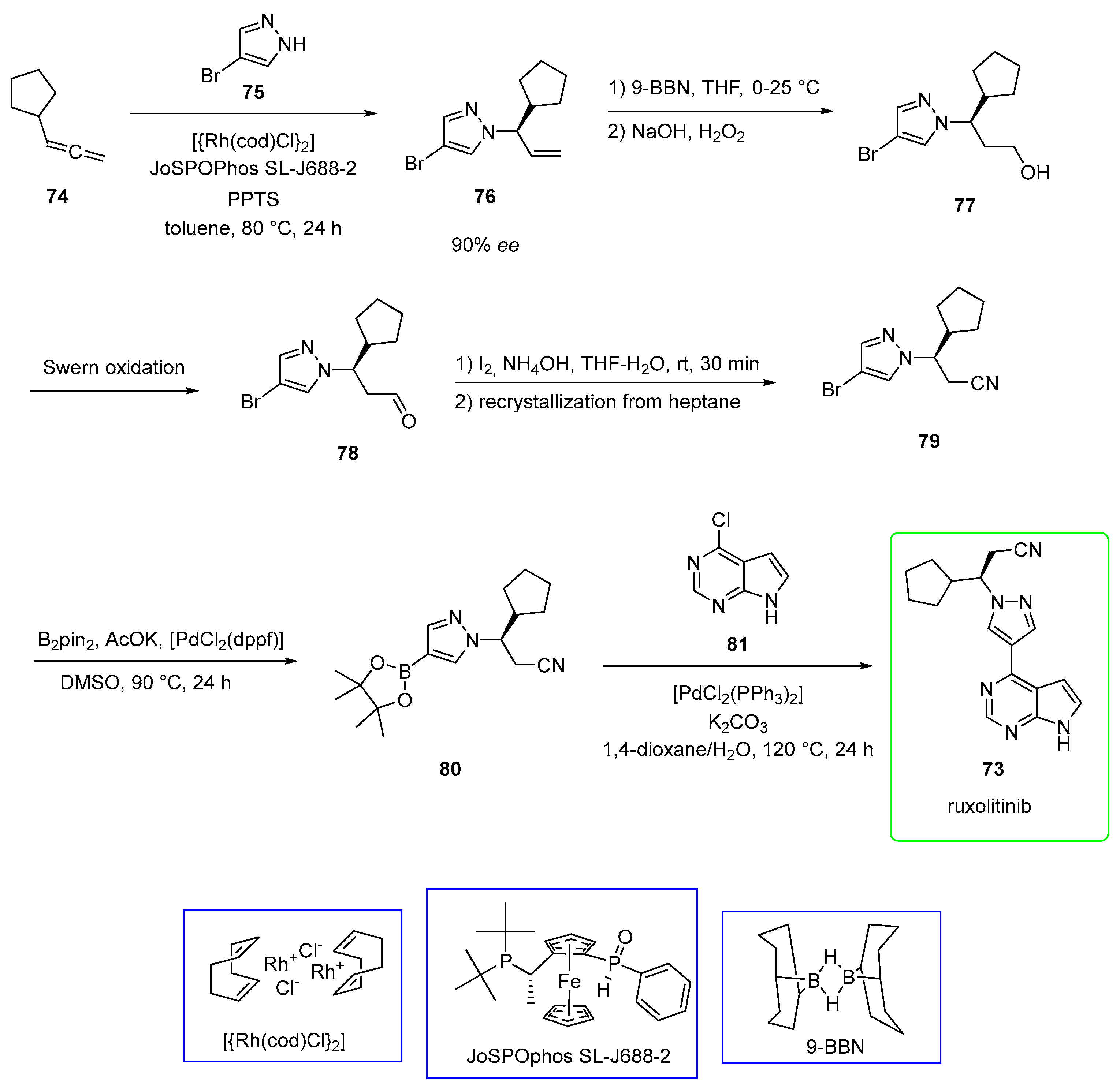
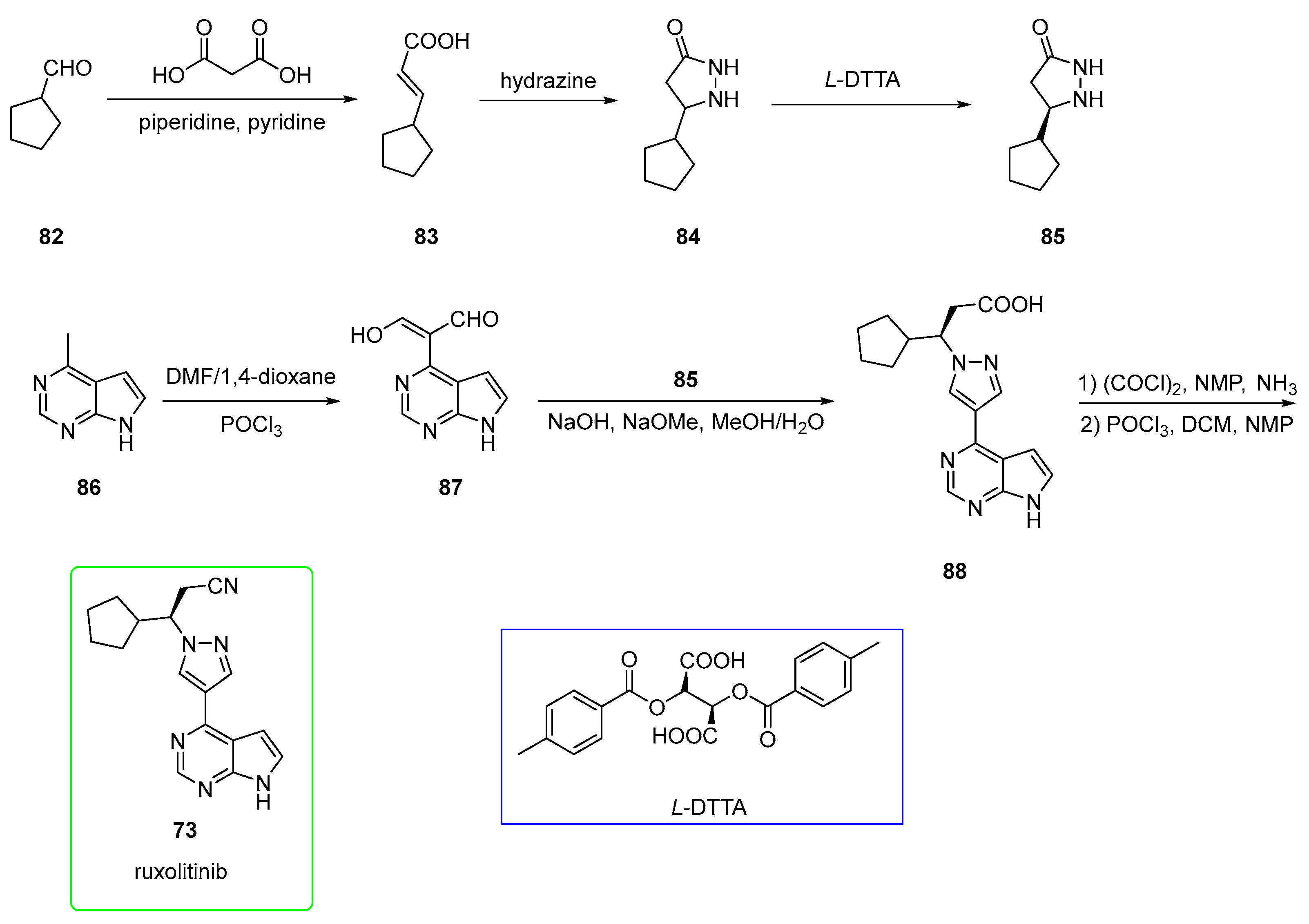
2.4. Ruxolitinib (Brand Name Jakafi®)
2.5. Venetoclax (Brand Name Venclexta®)
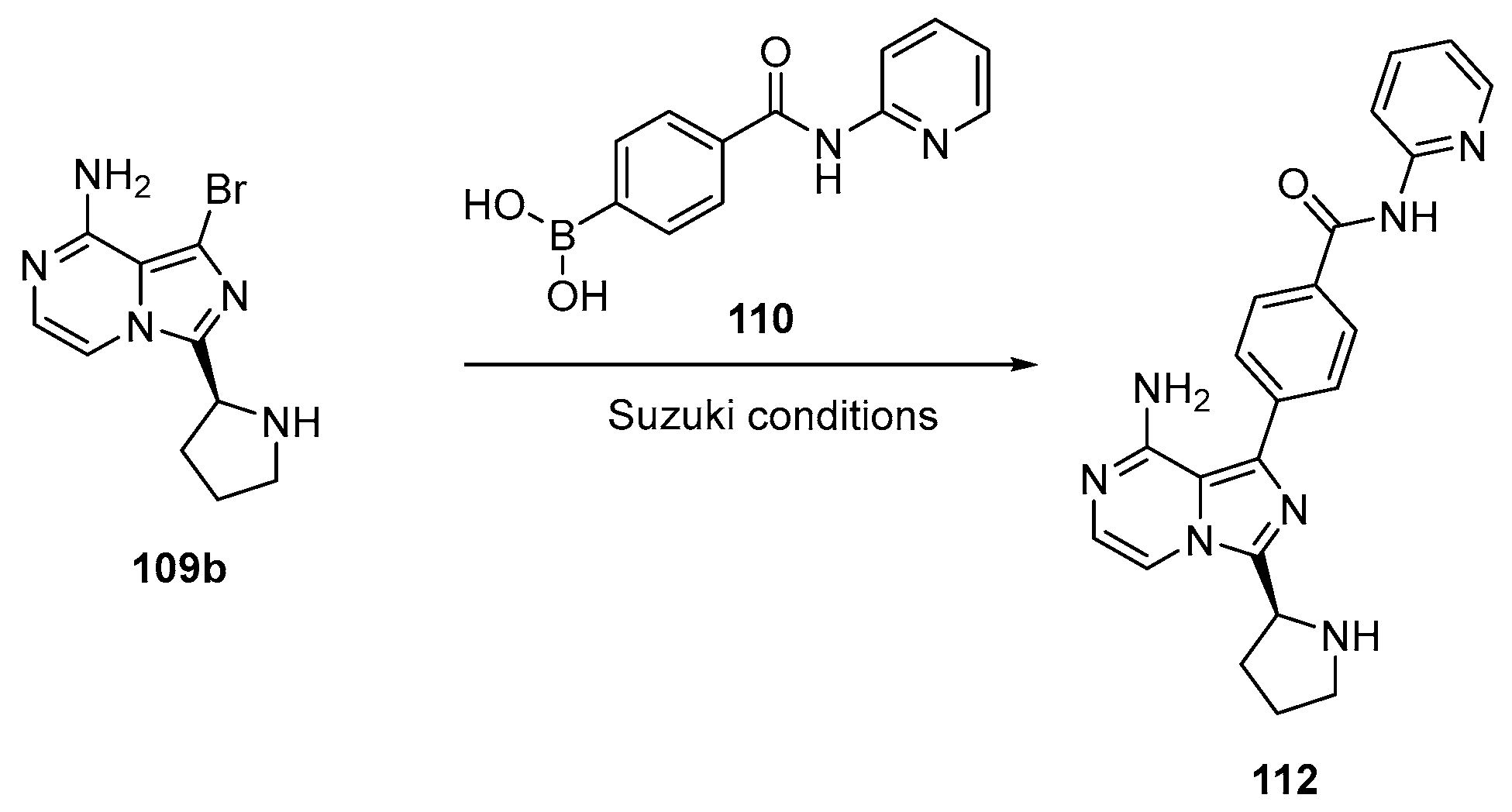
2.6. Acalabrutinib (Brand Name Calquence®)
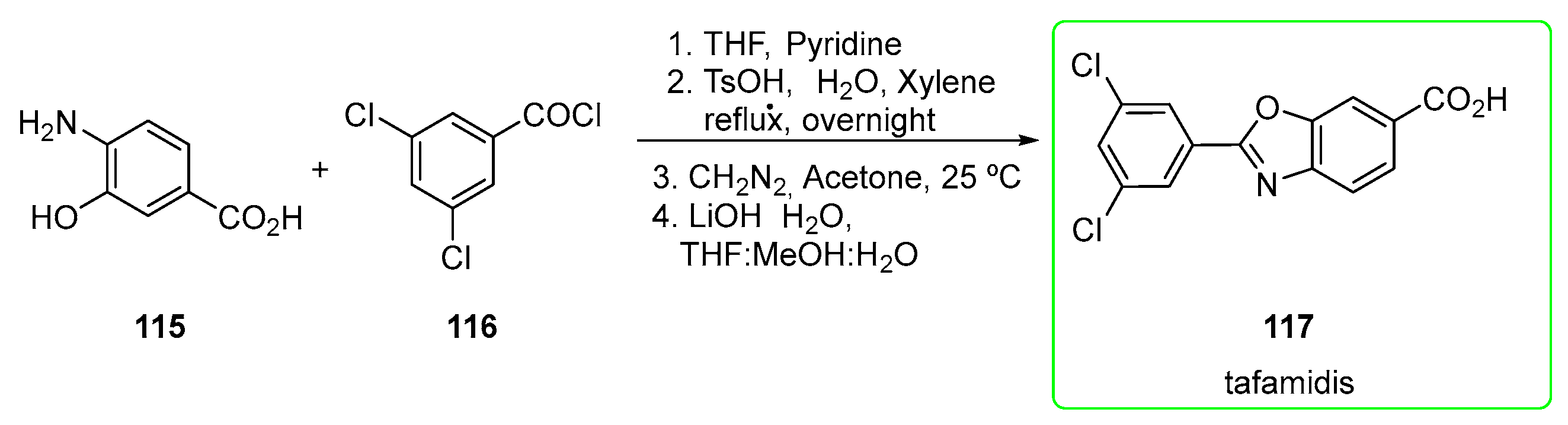
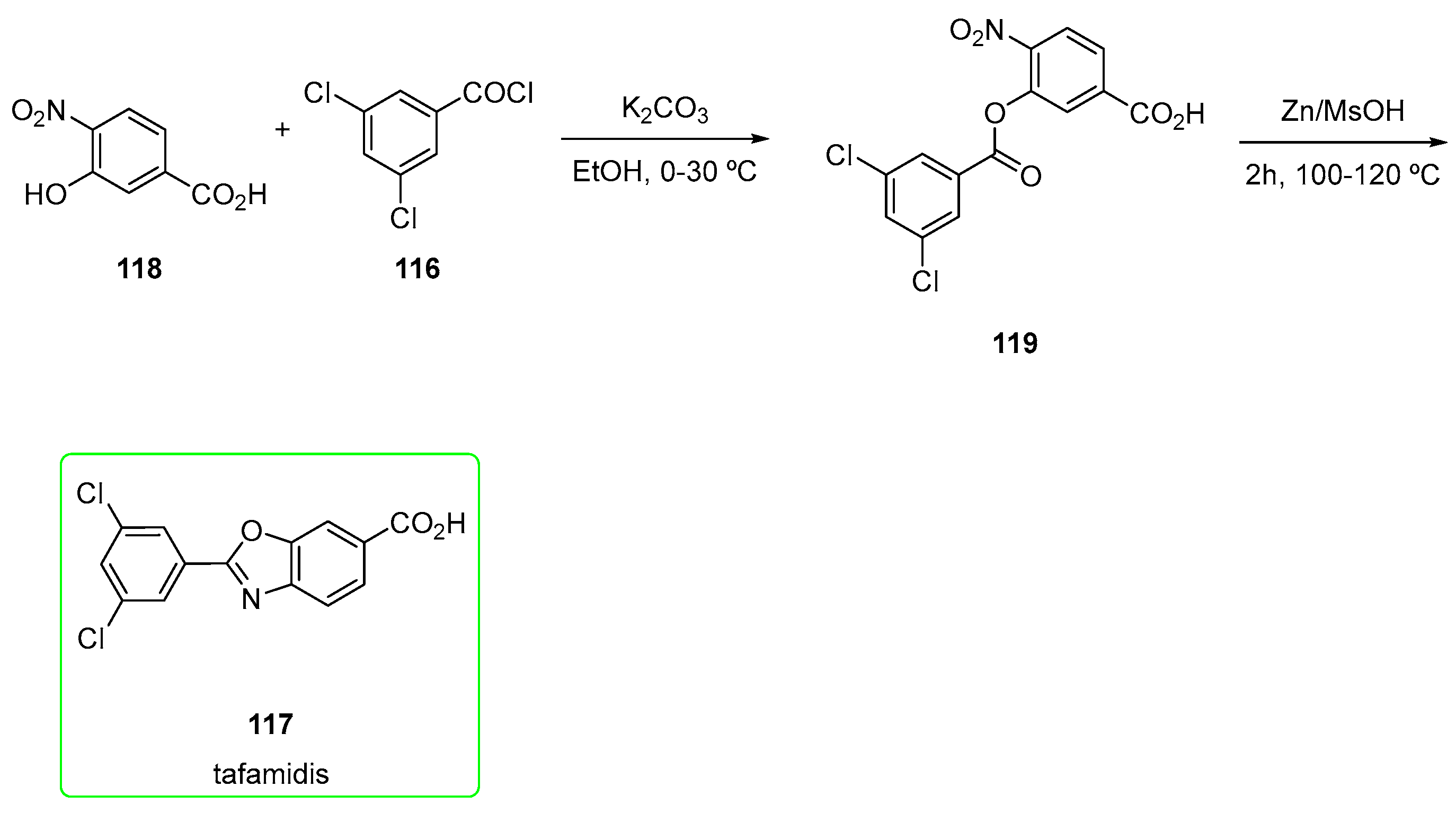
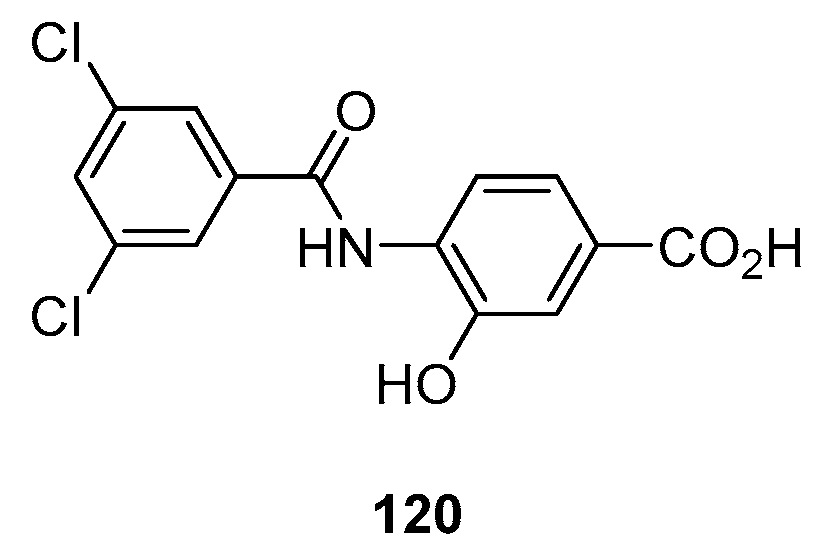
2.7. Tafamidis (Brand Name Vyndaqel®)
3. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Milne, C.-P.; Cabanilla, L.A. Orphan Drugs and Generics. In Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherlands, 2007; pp. 655–680. ISBN 978-0-08-045044-5. [Google Scholar]
- Designating an Orphan Product: Drugs and Biological Products. Available online: https://www.fda.gov/industry/medical-products-rare-diseases-and-conditions/designating-orphan-product-drugs-and-biological-products (accessed on 26 October 2022).
- EMA Orphan Designation: Overview. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview (accessed on 26 October 2022).
- The Story behind the Orphan Drug Act. Available online: https://www.fda.gov/industry/fdas-rare-disease-day/story-behind-orphan-drug-act (accessed on 26 October 2022).
- Aronson, J.K. Rare Diseases and Orphan Drugs. Br. J. Clin. Pharmacol. 2006, 61, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Lavandeira, A. Orphan Drugs: Legal Aspects, Current Situation. Haemophilia 2002, 8, 194–198. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on Orphan Medicinal Products; European Commission: Brussels, Belgium, 1999; Volume 018. [Google Scholar]
- Asbury, C.H. The Orphan Drug Act: The First 7 Years. JAMA 1991, 265, 893–897. [Google Scholar] [CrossRef]
- Mariz, S.; Reese, J.H.; Westermark, K.; Greene, L.; Goto, T.; Hoshino, T.; Llinares-Garcia, J.; Sepodes, B. Worldwide Collaboration for Orphan Drug Designation. Nat. Rev. Drug Discov. 2016, 15, 440–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rare Diseases at FDA. Available online: https://www.fda.gov/patients/rare-diseases-fda (accessed on 26 October 2022).
- U.S. Congress. H.R. 1—An Act to Provide for Reconciliation Pursuant to Titles II and V of the Concurrent Resolution on the Budget for Fiscal Year 2018. Available online: https://www.congress.gov/115/plaws/publ97/PLAW-115publ97.pdf (accessed on 22 November 2022).
- Zhang, M.; Zhu, C.; Jacomy, A.; Lu, L.J.; Jegga, A.G. The Orphan Disease Networks. Am. J. Hum. Genet. 2011, 88, 755–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontoghiorghe, C.N. World Health Dilemmas: Orphan and Rare Diseases, Orphan Drugs and Orphan Patients. WJM 2014, 4, 163. [Google Scholar] [CrossRef]
- Orphan Drugs: From Niche to Mainstream. Available online: https://www.pharmexec.com/view/orphan-drugs-from-niche-to-mainstream (accessed on 9 November 2022).
- Ponader, S.; Chen, S.-S.; Buggy, J.J.; Balakrishnan, K.; Gandhi, V.; Wierda, W.G.; Keating, M.J.; O’Brien, S.; Chiorazzi, N.; Burger, J.A. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Thwarts Chronic Lymphocytic Leukemia Cell Survival and Tissue Homing in Vitro and in Vivo. Blood 2012, 119, 1182–1189. [Google Scholar] [CrossRef]
- Paydas, S. Management of Adverse Effects/Toxicity of Ibrutinib. Crit. Rev. Oncol./Hematol. 2019, 136, 56–63. [Google Scholar] [CrossRef]
- Nocco, S.; Andriano, T.M.; Bose, A.; Chilov, M.; Godwin, K.; Dranitsaris, G.; Wu, S.; Lacouture, M.E.; Roeker, L.E.; Mato, A.R.; et al. Ibrutinib-Associated Dermatologic Toxicities: A Systematic Review and Meta-Analysis. Crit. Rev. Oncol./Hematol. 2022, 174, 103696. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef]
- Hughes, D.L. Patent Review of Manufacturing Routes to Recently Approved Oncology Drugs: Ibrutinib, Cobimetinib, and Alectinib. Org. Process Res. Dev. 2016, 20, 1855–1869. [Google Scholar] [CrossRef]
- Honigberg, L.; Verner, E.; Pan, Z. Inhibitors of Bruton’s Tyrosine Kinase. U.S. Patent US2010022561(A1), 28 January 2010. [Google Scholar]
- Pan, Z.; Li, S.J.; Schereens, H.; Honigberg, L.; Verner, E. Bruton’s Tyrosine Kinase Activity Probe and Method of Using. European Patent. Office Patent No. EP2089391A4, 17 March 2010. [Google Scholar]
- Chen, W.; Loury, D.J.; Mody, T.D. Pyrazolo-Pyrimidine Inhibitors of Bruton’s Tyrosine Kinase. U.S. Patent US7718662B1, 18 May 2010. [Google Scholar]
- Pan, Z.; Scheerens, H.; Li, S.-J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef]
- Liu, H.; Pan, Z. Ibrutinib (Imbruvica): The First-in-Class Btk Inhibitor for Mantle Cell Lymphoma, Chronic Lymphocytic Leukemia, and Waldenstrom’s Macroglobulinemia. In Innovative Drug Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 157–166. ISBN 978-1-118-81995-1. [Google Scholar]
- Owens, T.D. Ibrutinib, a Carboxylic Acid Amide Inhibitor of Bruton’s Tyrosine Kinase. In Bioactive Carboxylic Compound Classes; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016; pp. 197–208. ISBN 978-3-527-69393-1. [Google Scholar]
- Pye, P.; Haim, C.B.; Conza, M.; Houpis, I.N. Processes and Intermediates for Preparing a Medicament. U.S. Patent US20140275126A1, 18 September 2014. [Google Scholar]
- Pye, P.; Haim, C.B.; Conza, M.; Houpis, I.N. Processes and Intermediates for Preparing a Medicament. U.S. Patent US9156847B2, 13 October 2015. [Google Scholar]
- Fang, T.; Chen, Y.; Jiang, M.; Zhang, W.; Sun, K.; Chen, X. Synthesis Method of Ibrutinib. CN Patent CN107674079B, 13 December 2019. [Google Scholar]
- Jaychandra, S.; Sebastian, S.; Rao, J.; Naidu, H.; Adla, M.; Mannava, S.R.; Sabbella, S.R.; Dandala, R. Process for the Preparation of Ibrutinib. WO2016132383A1, 25 August 2016. [Google Scholar]
- Sharma, K.; Thanki, B.P.; Khanna, M.S.; Prasad, M. A Process for the Preparation of Ibrutinib. WO Patent WO2016079693A1, 25 May 2016. [Google Scholar]
- Ridley, K.; Condren, M. Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy. J. Pediatr. Pharmacol. Ther. 2020, 25, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Sala, M.A.; Jain, M. Tezacaftor for the Treatment of Cystic Fibrosis. Expert Rev. Respir. Med. 2018, 12, 725–732. [Google Scholar] [CrossRef]
- Hughes, D.L. Patent Review of Synthetic Routes and Crystalline Forms of the CFTR-Modulator Drugs Ivacaftor, Lumacaftor, Tezacaftor, and Elexacaftor. Org. Process Res. Dev. 2019, 23, 2302–2322. [Google Scholar] [CrossRef]
- Shaughnessy, C.A.; Zeitlin, P.L.; Bratcher, P.E. Elexacaftor Is a CFTR Potentiator and Acts Synergistically with Ivacaftor during Acute and Chronic Treatment. Sci. Rep. 2021, 11, 19810. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Molecular Structures Reveal Synergistic Rescue of Δ508 CFTR by Trikafta Modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of Multiple Class II CFTR Mutations by Elexacaftor+tezacaftor+ivacaftor Mediated in Part by the Dual Activities of Elexacaftor as Both Corrector and Potentiator. Eur. Respir. J. 2021, 57, 2002774. [Google Scholar] [CrossRef]
- Abela, A.R.; Alcacio, T.; Anderson, C.; Angell, P.T.; Baek, M.; Clemens, J.J.; Cleveland, T.; Ferris, L.A.; Grootenhuis, P.D.J.; Gross, R.S.; et al. Modulator of Cystic Fibrosis Transmembrane Conductance Regulator, Pharmaceutical Compositions, Methods of Treatment, and Process for Making the Modulator. WO Patent WO2018107100A1, 14 June 2018. [Google Scholar]
- Tezacaftor (VX-661) for Cystic Fibrosis. Available online: https://cysticfibrosisnewstoday.com/tezacaftor-vx-661-for-cystic-fibrosis (accessed on 9 November 2022).
- Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/210491s007lbl.pdf (accessed on 30 December 2022).
- Looker, A.; Littler, B.J.; Choudhury, A.; Harrison, C.; Veluri, R.; Ryan, M.P.; Jiang, L.; Luss-Lusis, E. Modulators of Atp-Binding Cassette Transporters. U.S. Patent US20130116238A1, 9 May 2013. [Google Scholar]
- Modulators of ATP-Binding Cassette Transporters Patent Application. Available online: https://uspto.report/patent/app/20050148648 (accessed on 3 November 2022).
- Tanoury, G.J.; Harrison, C.; Littler, B.J.; Rose, P.J.; Huges, R.M.; Jung, Y.C.; Siesel, D.A.; Lee, E.C.; Belmont, D.T. Process of Producing Cycloalkylcarboxamido-Indole Compounds. WO Patent WO2011US33396, 21 April 2011. [Google Scholar]
- Deeks, E.D. Ivacaftor: A Review of Its Use in Patients with Cystic Fibrosis. Drugs 2013, 73, 1595–1604. [Google Scholar] [CrossRef]
- Fohner, A.E.; McDonagh, E.M.; Clancy, J.P.; Whirl Carrillo, M.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Ivacaftor Pathway, Pharmacokinetics/Pharmacodynamics. Pharm. Genom. 2017, 27, 39–42. [Google Scholar] [CrossRef]
- Lai, C.K.; Song, T.V.; Li, J.J. Ivacaftor (Kalydeco): A CFTR Potentiator for the Treatment of Cystic Fibrosis. In Innovative Drug Synthesis; Li, J.J., Johnson, D.S., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 303–316. ISBN 978-1-118-81995-1. [Google Scholar]
- Arekar, S.G.; Johnston, S.C.; Krawiec, M.; Medek, A.; Mudunuri, P.; Sullivan, M.J. Solid Forms of N-[2,4-Bis(1,1-Dimethylethyl)-5-Hydroxyphenyl]-1,4-Dihydro-4-Oxoquinoline-3-Carboxamide. WO Patent WO2011116397A1, 22 September 2011. [Google Scholar]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2021, 26, e164–e172. [Google Scholar] [CrossRef] [PubMed]
- Rohit Deepak Nalawade; Bhagwan Dilip Devkar Olaparib an Anticancer Drug: A Review. World J. Adv. Res. Rev. 2021, 11, 329–336. [CrossRef]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)— unction in Genome Maintenance and Relevance of Inhibitors for Anti-Cancer Therapy. Front. Mol. Biosci. 2020, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Menear, K.A.; Adcock, C.; Boulter, R.; Cockcroft, X.L.; Copsey, L.; Cranston, A.; Dillon, K.J.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2Hphthalazin-1-one: A novel bioavailable inhibitor of poly (ADP-ribose) polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, T.Y.; Chang, Y.H. Processes for Preparing Olaparib. WO Patent WO2018038680A1, 1 March 2018. [Google Scholar]
- Martin, N.M.B.; Smith, G.C.; Jackson, S.P.; Loh, V.J.M.; Cockcroft, X.-L.F.; Matthews, I.T.W.; Menear, K.A.; Kerrigan, F.; Ashworth, A. Phthalazinone Derivatives. U.S. Patent US7449464B2, 11 November 2008. [Google Scholar]
- Menear, K.A.; Ottridge, A.P.; Londesbrough, D.J.; Hallett, M.R.; Mulholland, K.R.; Pittam, J.D.; Laffan, D.D.P.; Ashworth, I.W.; Jones, M.F.; Cherryman, J.H. Phthalazinone Derivative. U.S. Patent US8247416B2, 21 August 2012. [Google Scholar]
- McKeage, K. Ruxolitinib: A Review in Polycythaemia Vera. Drugs 2015, 75, 1773–1781. [Google Scholar] [CrossRef]
- Purandare, A.V.; Lorenzi, M.V.; Lombardo, L.J. Janus Kinase 2 (JAK2) Inhibitors for the Treatment of Myeloproliferative Neoplasm (MPN). In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2010; Volume 45, pp. 210–227. ISBN 978-0-12-380902-5. [Google Scholar]
- Przepiorka, D.; Luo, L.; Subramaniam, S.; Qiu, J.; Gudi, R.; Cunningham, L.C.; Nie, L.; Leong, R.; Ma, L.; Sheth, C.; et al. FDA Approval Summary: Ruxolitinib for Treatment of Steroid-Refractory Acute Graft-Versus-Host Disease. Oncologist 2020, 25, e328–e334. [Google Scholar] [CrossRef] [Green Version]
- Coricello, A.; Mesiti, F.; Lupia, A.; Maruca, A.; Alcaro, S. Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates. Molecules 2020, 25, 3321. [Google Scholar] [CrossRef]
- Haydl, A.M.; Xu, K.; Breit, B. Regio- and Enantioselective Synthesis of N-Substituted Pyrazoles by Rhodium-Catalyzed Asymmetric Addition to Allenes. Angew. Chem. Int. Ed. 2015, 54, 7149–7153. [Google Scholar] [CrossRef]
- Lin, Q.; Meloni, D.; Pan, Y.; Xia, M.; Rodgers, J.; Shepard, S.; Li, M.; Galya, L.; Metcalf, B.; Yue, T.-Y.; et al. Enantioselective Synthesis of Janus Kinase Inhibitor INCB018424 via an Organocatalytic Aza-Michael Reaction. Org. Lett. 2009, 11, 1999–2002. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, A.; Zhou, Z.; YANG, L.; Yao, H.; Zhu, X.; Wang, H. Synthesis Process of Ruxolitinib. U.S. Patent US10562904B2, 18 February 2020. [Google Scholar]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef]
- Salem, A.H.; Agarwal, S.K.; Dunbar, M.; Nuthalapati, S.; Chien, D.; Freise, K.J.; Wong, S.L. Effect of low and high-fat meals on the pharmacokinetics of venetoclax, a selective first-in-class BCL-2 inhibitor. J. Clin. Pharmacol. 2016, 56, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Ku, Y.-Y.; Chan, V.S.; Christesen, A.; Grieme, T.; Mulhern, M.; Pu, Y.-M.; Wendt, M.D. Development of a Convergent Large-Scale Synthesis for Venetoclax, a First-in-Class BCL-2 Selective Inhibitor. J. Org. Chem. 2019, 84, 4814–4829. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.S.; Christesen, A.C.; Grieme, T.A.; Ku, Y.-Y.; Mulhern, M.M.; Pu, Y.-M.M. Processes For The Preparation Of An Apoptosis-Inducing Agent. U.S. Patent US20140275540A1, 18 September 2014. [Google Scholar]
- Markham, A.; Dhillon, S. Acalabrutinib: First Global Approval. Drugs 2018, 78, 139–145. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Acalabrutinib for CLL and SLL. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/project-orbis-fda-approves-acalabrutinib-cll-and-sll (accessed on 28 October 2022).
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, V.; Balakrishnan, K.; Bibikova, E.; Ayres, M.; Keating, M.J.; Wierda, W.G.; Gandhi, V. Comparison of Acalabrutinib, A Selective Bruton Tyrosine Kinase Inhibitor, with Ibrutinib in Chronic Lymphocytic Leukemia Cells. Clin. Cancer Res. 2017, 23, 3734–3743. [Google Scholar] [CrossRef] [Green Version]
- Budideti, S.R.; Konduri, S.K.M.; Sanapureddy, J.M.R.; Remella, R.K.; Thoota, S.K.; Kothamunireddygari, S.; Muddasani, P.R.; Nannapaneni, V.C. Novel Process for the Preparation of Acalabrutinib and Its Intermediates. WO Patent WO2021111465A1, 10 June 2021. [Google Scholar]
- Barf, T.A.; Jans, C.; de Man, A.; Oubrie, A.A.; Raaijmakers, H.C.A.; Rewinkel, J.; Sterrenburg, J.-G.; Wijkmans, J.C.H.M. 4-Imidazopyridazin-1-Yl-Benzamides and 4-Imidazotriazin-1-Yl-Benzamides as Btk-Inhibitors. WO Patent WO2013010868, 24 January 2013. [Google Scholar]
- Karumanchi, K.; Natarajan, S.K.; Gadde, S.; Vanchanagiri, K.; Moturu, K.V.R. A New Synthesis of Tafamidis via Zinc-MsOH Mediated Reductive Cyclisation Strategy. J. Chem. Sci. 2021, 133, 48. [Google Scholar] [CrossRef]
- Simões, C.J.V.; Almeida, Z.L.; Costa, D.; Jesus, C.S.H.; Cardoso, A.L.; Almeida, M.R.; Saraiva, M.J.; Pinho e Melo, T.M.V.D.; Brito, R.M.M. A Novel Bis-Furan Scaffold for Transthyretin Stabilization and Amyloid Inhibition. Eur. J. Med. Chem. 2016, 121, 823–840. [Google Scholar] [CrossRef]
- Burge, J.A. BTK Inhibitors: Present and future. Cancer J. 2019, 25, 386–393. [Google Scholar] [CrossRef]
- Sellers, W.R.; Fisher, D.E. Apoptosis and cancer drug targeting. J. Clin. Investig. 1999, 104, 1655–1661. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Ma, X.; Ouyang, D.; Williams III, R.O. Emerging Artificial Intelligence (AI) Technologies Used in the Development of Solid Dosage Forms. Pharmaceutics 2022, 14, 2257. [Google Scholar] [CrossRef]
- Liu, Q. Artificial intelligence and drug discovery. Acad. J. Second Mil. Med. Univ. 2018, 39, 869–872. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedetto Tiz, D.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. Top Selling (2026) Small Molecule Orphan Drugs: A Journey into Their Chemistry. Int. J. Mol. Sci. 2023, 24, 930. https://doi.org/10.3390/ijms24020930
Benedetto Tiz D, Bagnoli L, Rosati O, Marini F, Sancineto L, Santi C. Top Selling (2026) Small Molecule Orphan Drugs: A Journey into Their Chemistry. International Journal of Molecular Sciences. 2023; 24(2):930. https://doi.org/10.3390/ijms24020930
Chicago/Turabian StyleBenedetto Tiz, Davide, Luana Bagnoli, Ornelio Rosati, Francesca Marini, Luca Sancineto, and Claudio Santi. 2023. "Top Selling (2026) Small Molecule Orphan Drugs: A Journey into Their Chemistry" International Journal of Molecular Sciences 24, no. 2: 930. https://doi.org/10.3390/ijms24020930
APA StyleBenedetto Tiz, D., Bagnoli, L., Rosati, O., Marini, F., Sancineto, L., & Santi, C. (2023). Top Selling (2026) Small Molecule Orphan Drugs: A Journey into Their Chemistry. International Journal of Molecular Sciences, 24(2), 930. https://doi.org/10.3390/ijms24020930