
The Autophagy Marker LC3 Is Processed during the Sperm Capacitation and the Acrosome Reaction and Translocates to the Acrosome Where It Colocalizes with the Acrosomal Membranes in Horse Spermatozoa
 , , ,
, , ,  and
and 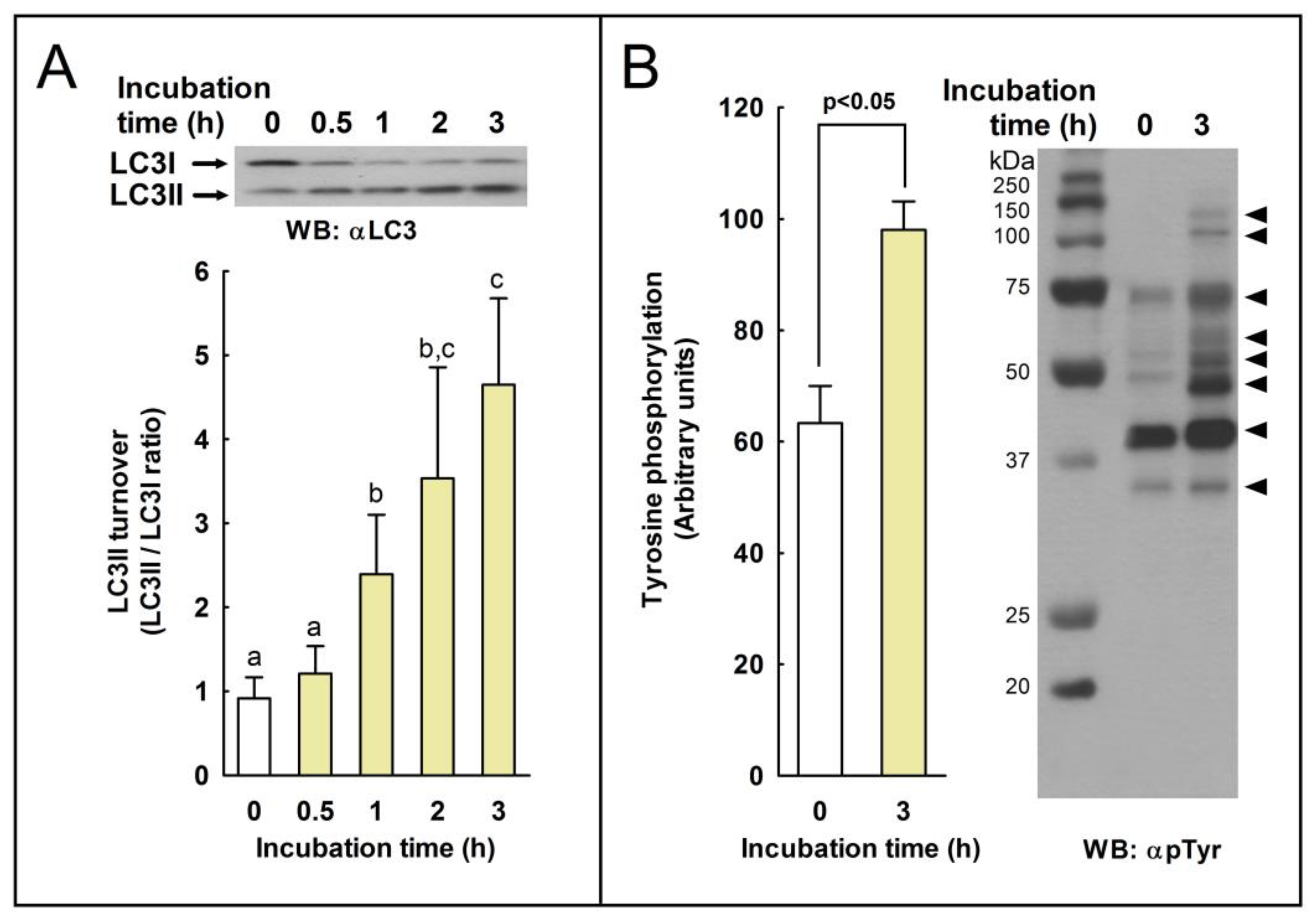{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
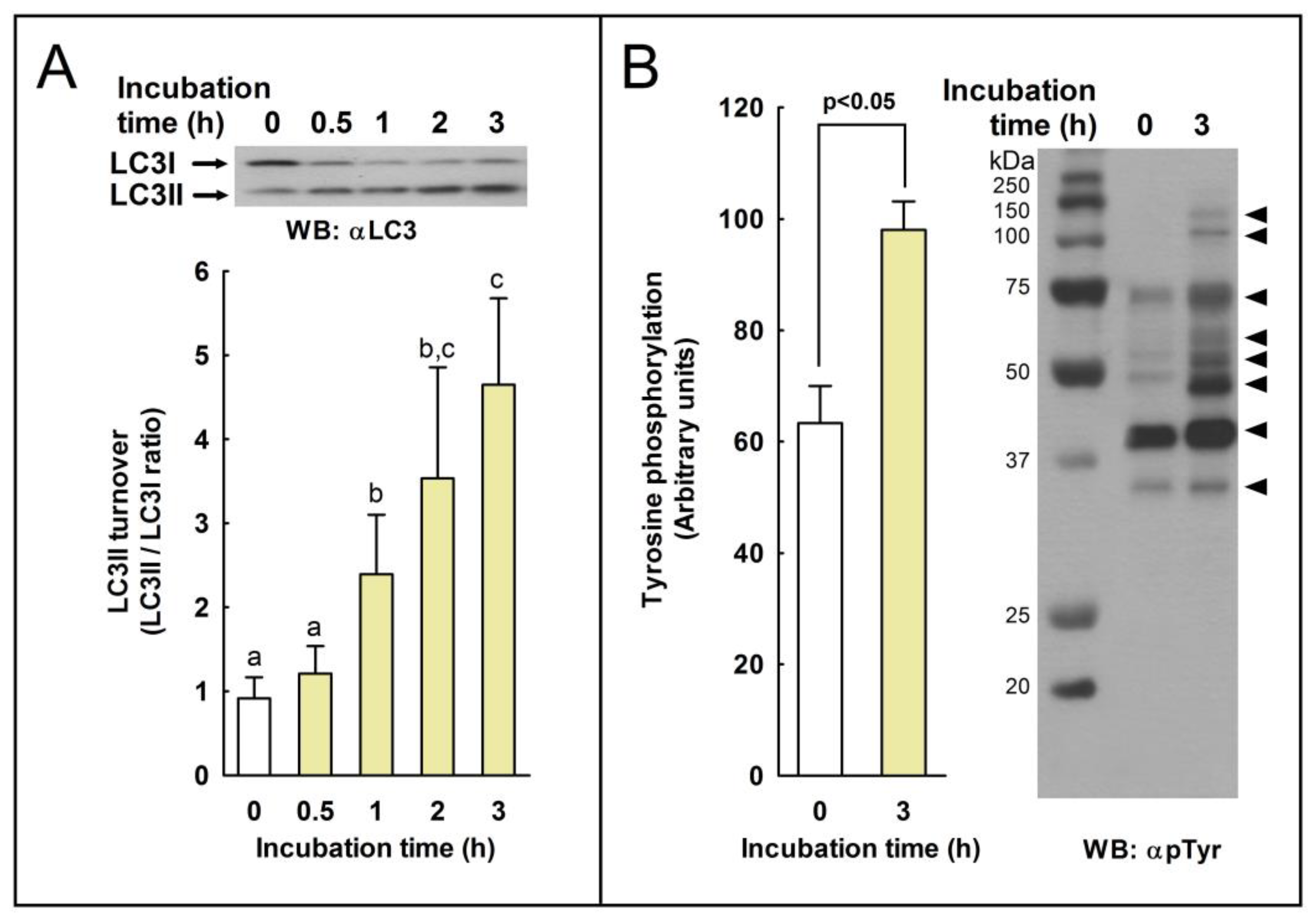
2.1. LC3 Processing (LC3II/LC3I Ratio) during Sperm Capacitation
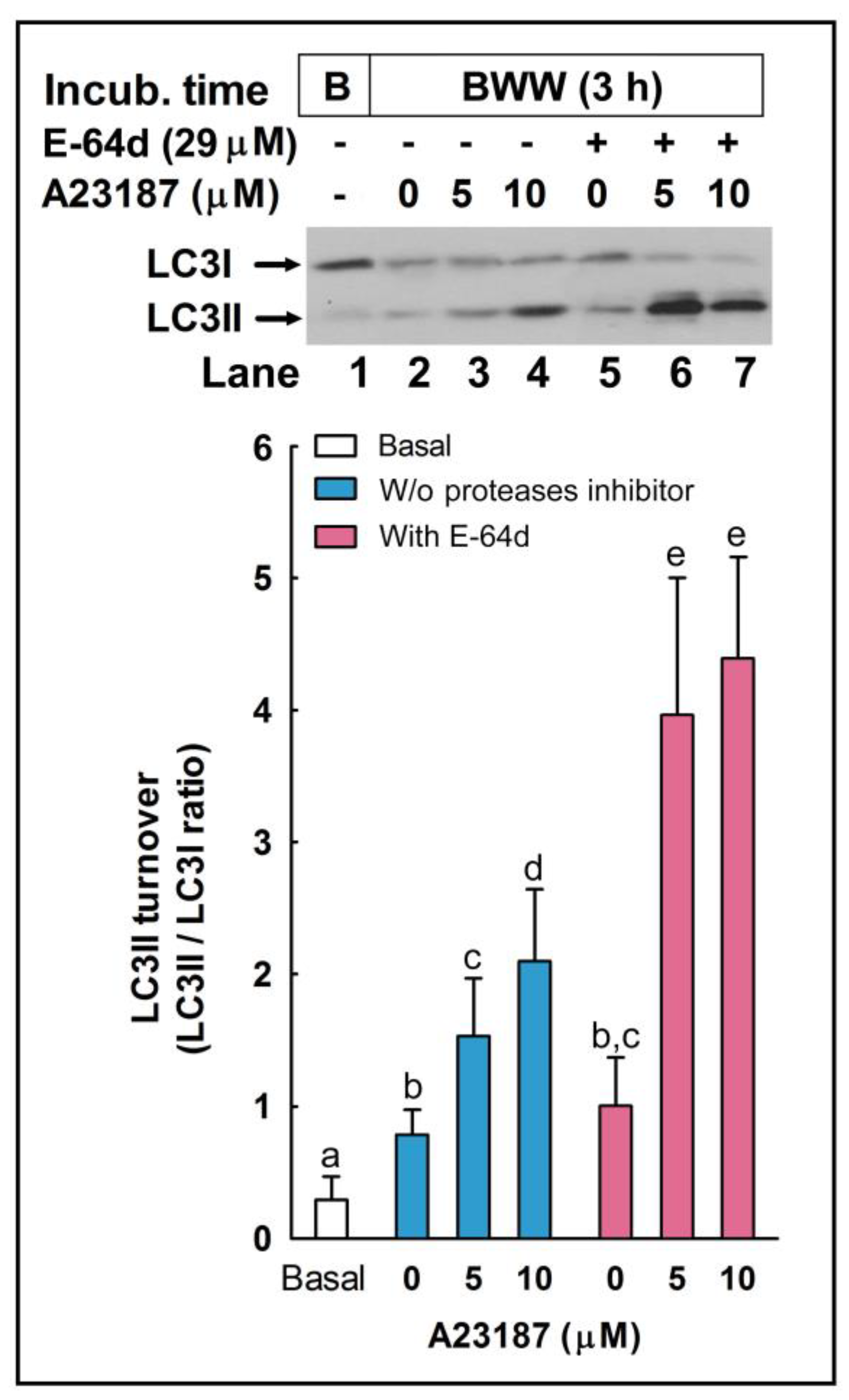
2.2. LC3 Processing during the Acrosome Reaction
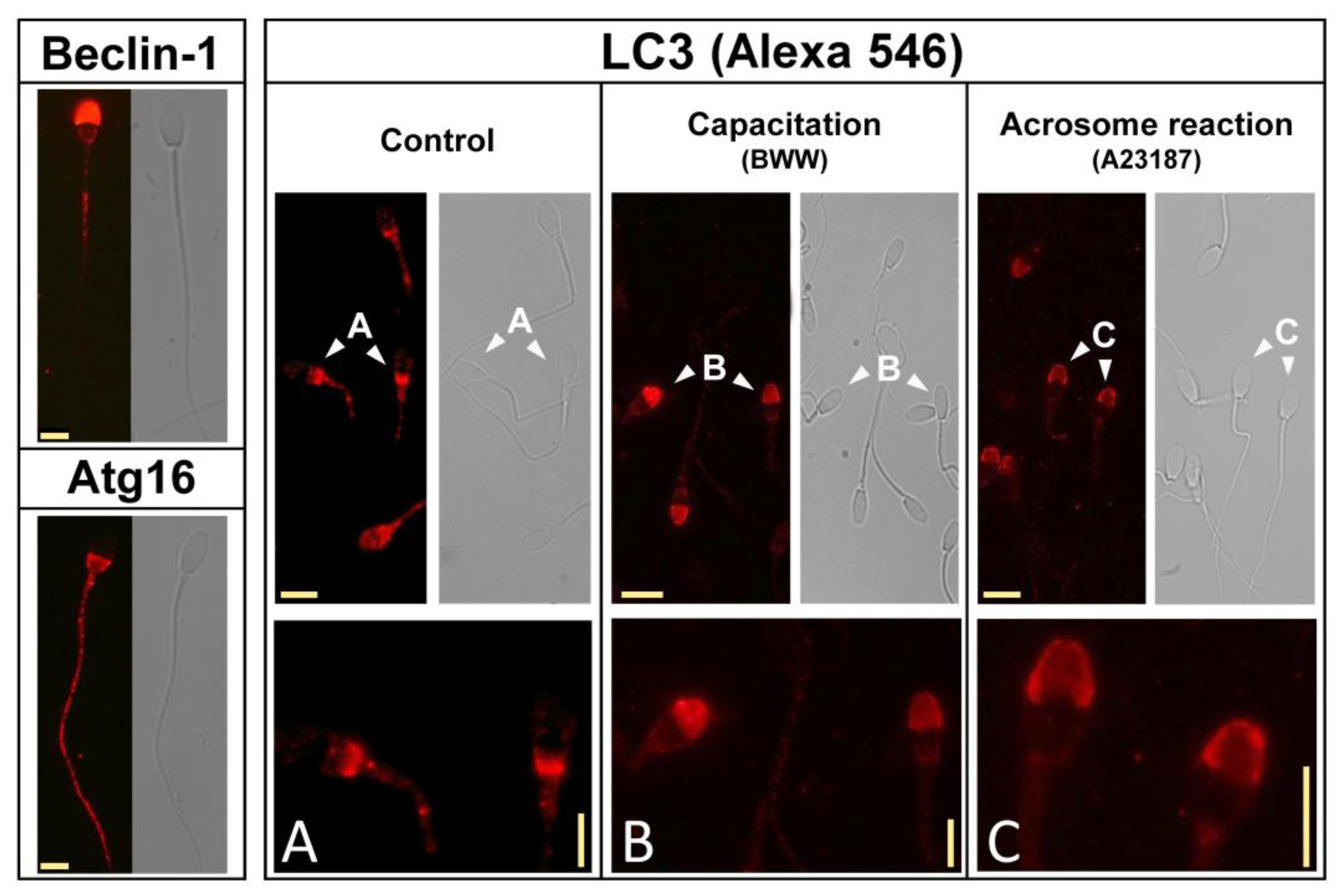
2.3. Identification of Autophagy-Related Proteins in Stallion Sperm and Subcellular Redistribution of LC3 during the Capacitation and the Acrosome Reaction
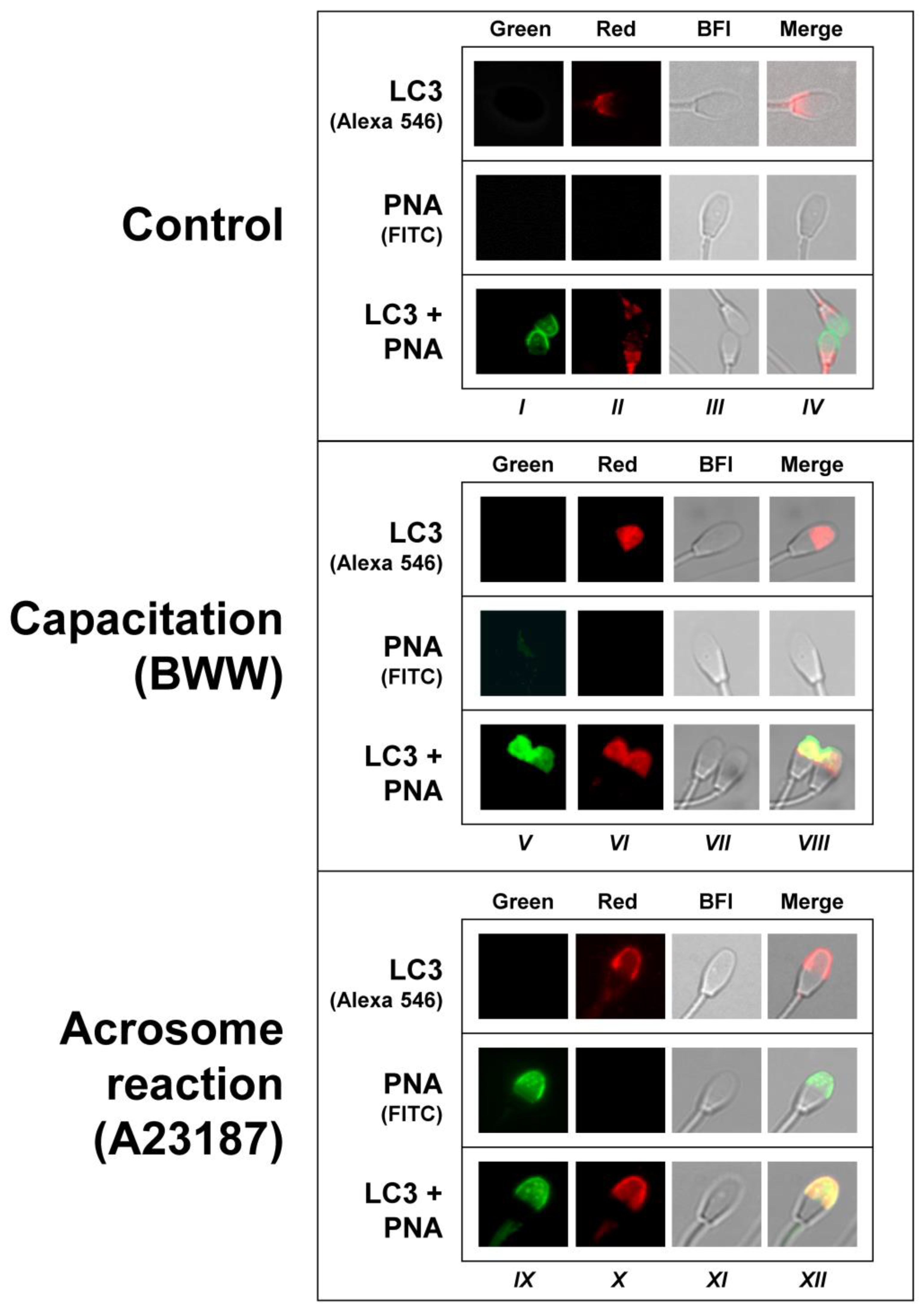
2.4. Simultaneous Detection of LC3 and PNA in Control, Capacitated, and Acrosome-Reacted Stallion Sperm Cells by Confocal Microscopy
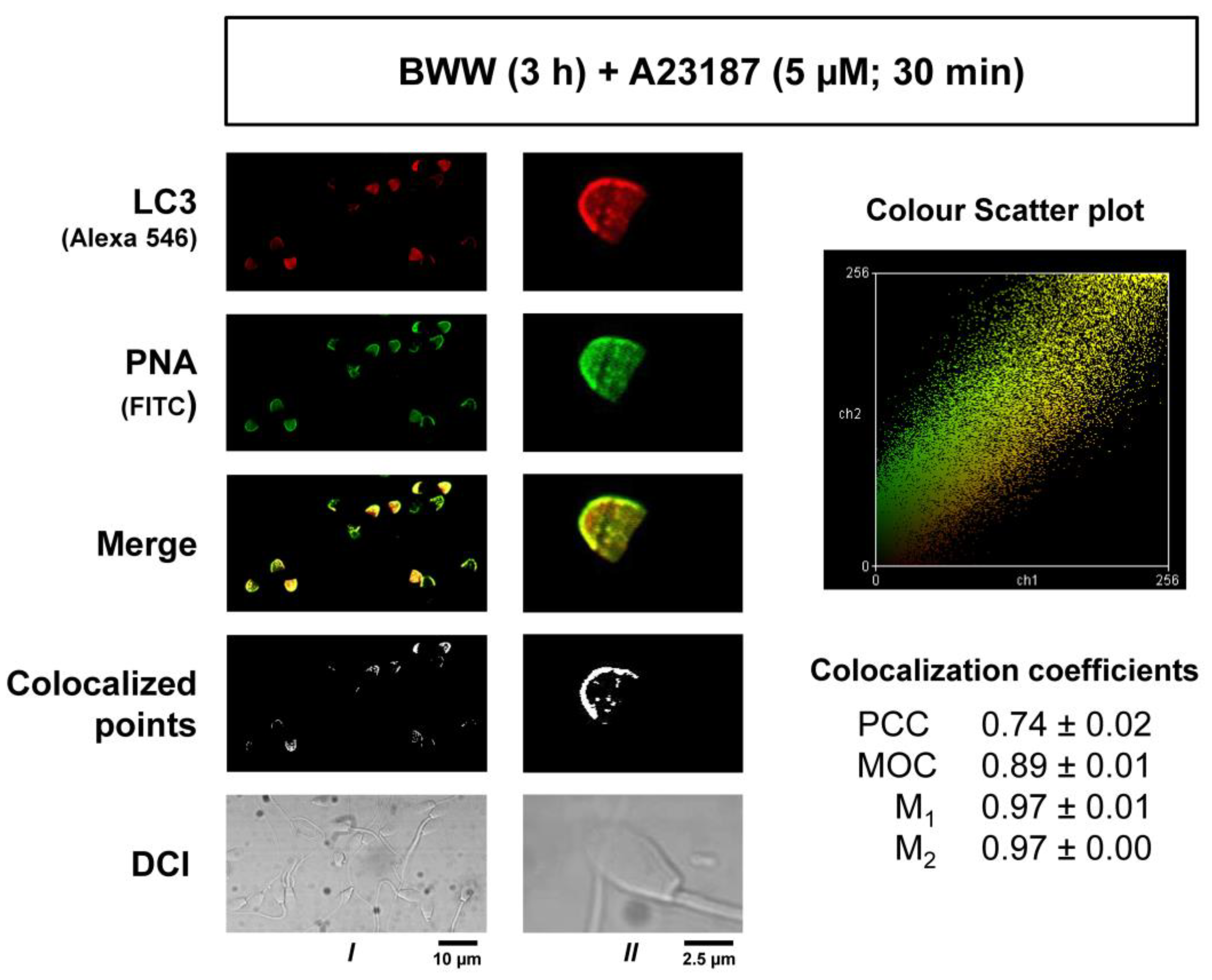
2.5. Colocalization Study of LC3 and PNA in Acrosome-Reacted Spermatozoa
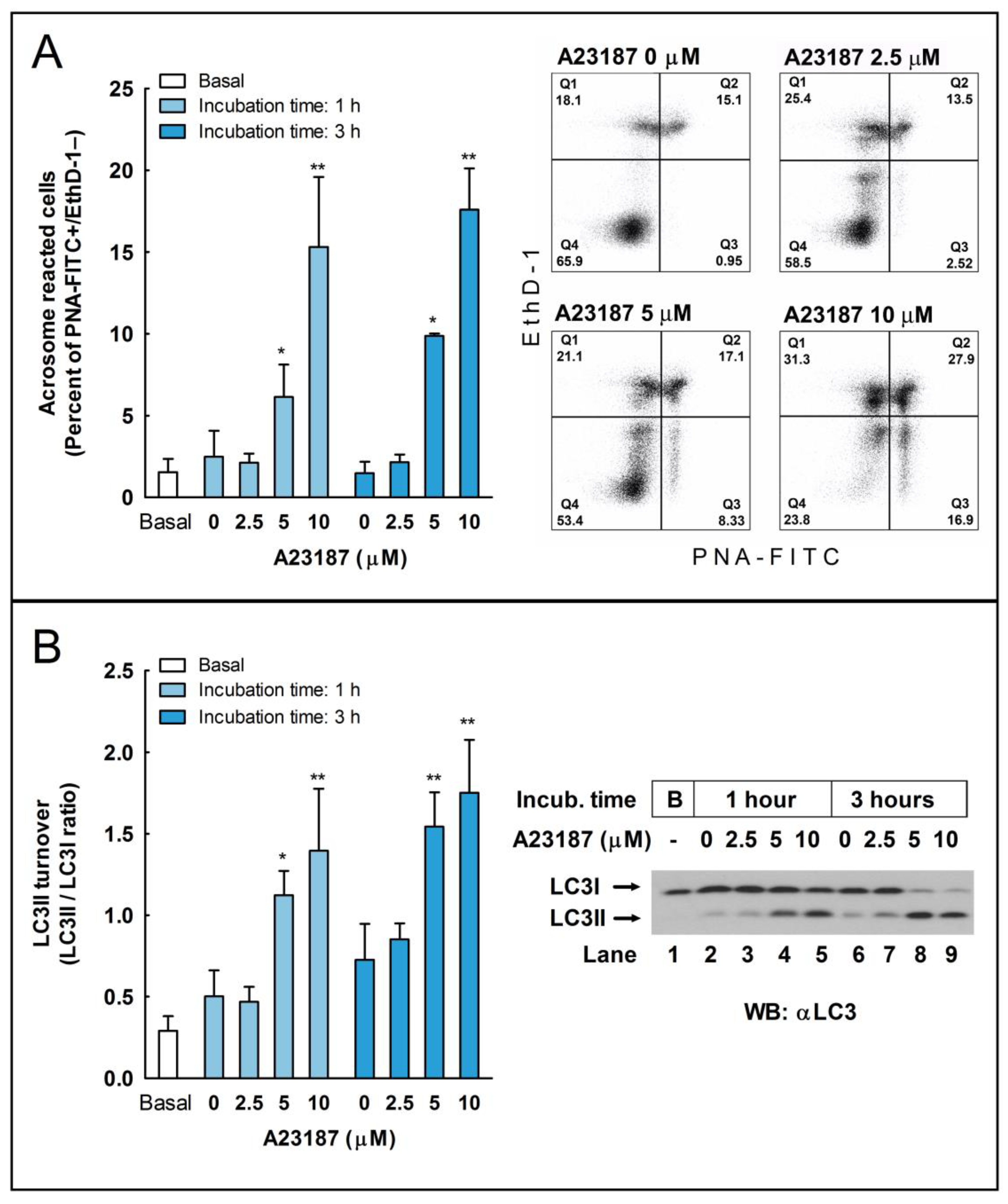
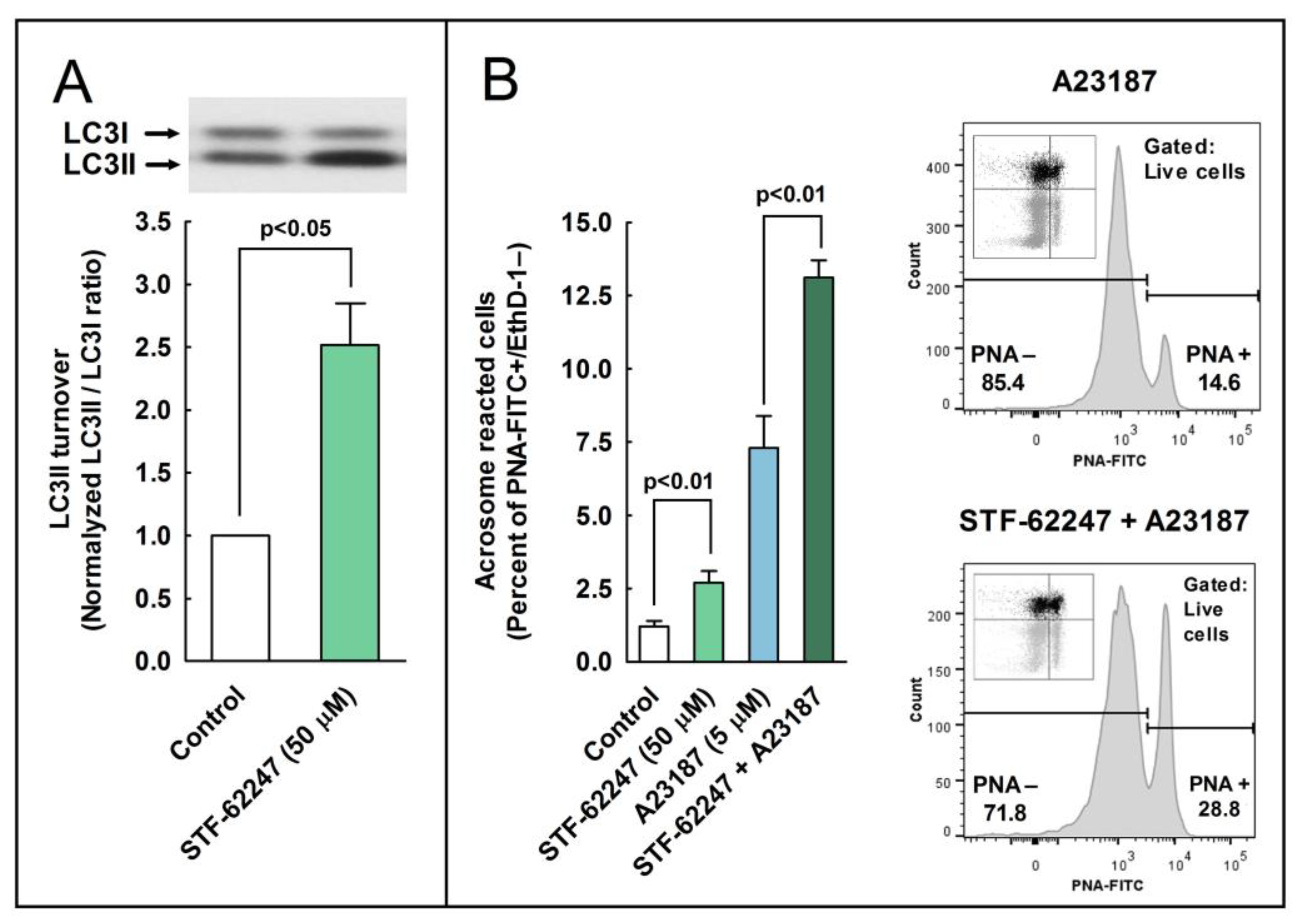
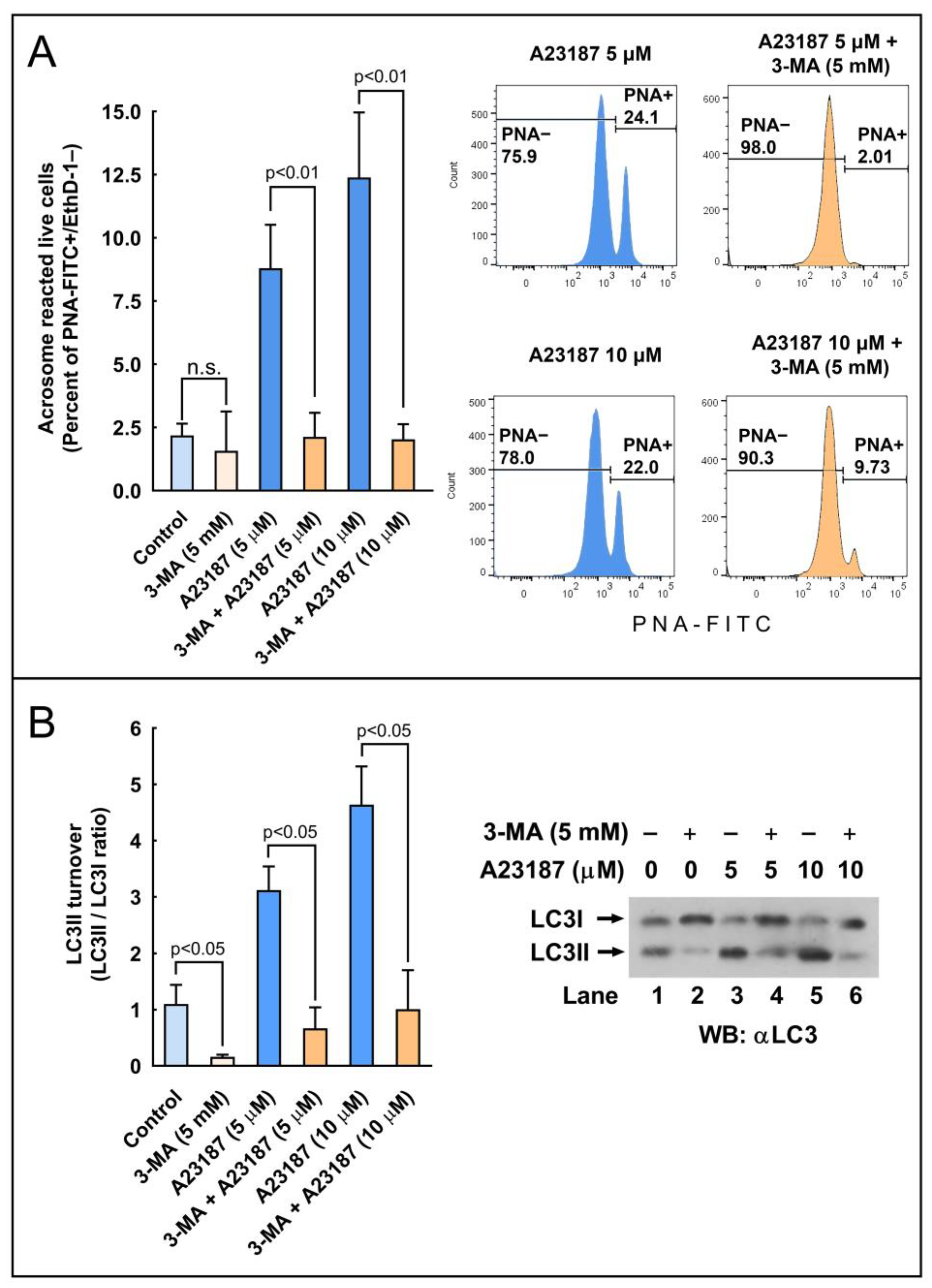
2.6. Effect of the Pharmacological Activation of Autophagy in the Regulation of the Acrosome Reaction
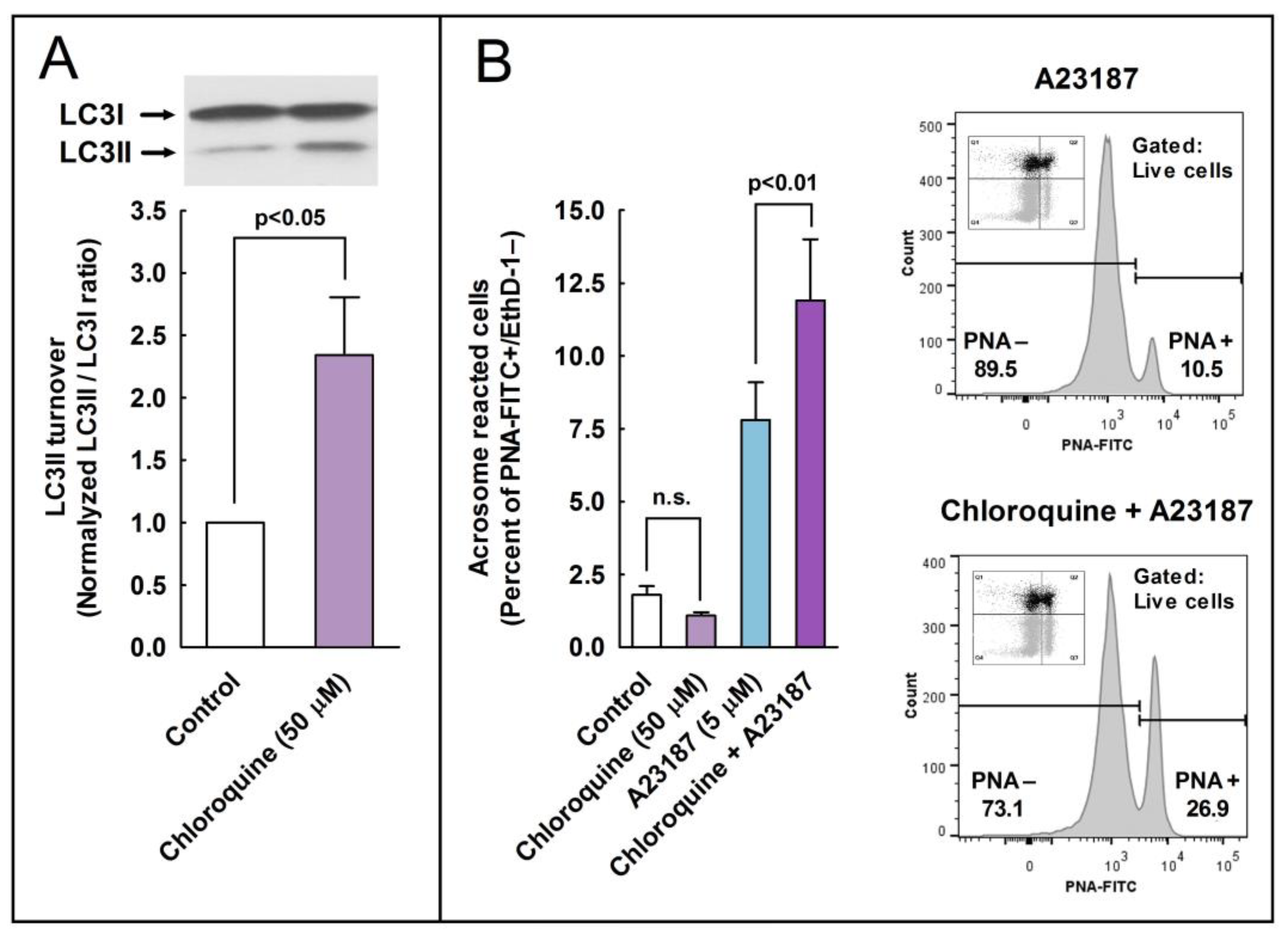
2.7. Effect of the Pharmacological Inhibition of Autophagy in the Regulation of the Acrosome Reaction
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Collection of Equine Semen
4.3. Incubation Media and Treatments
4.4. Autophagy Regulation
4.5. Western Blotting
4.6. Assessment of Acrosome-Reacted Spermatozoa and Cell Viability
4.7. Indirect Immunofluorescence with Classical Microscopy and with Confocal Microscopy
4.8. Colocalization Study
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Schmid, D.; Dengjel, J.; Schoor, O.; Stevanovic, S.; Munz, C. Autophagy in innate and adaptive immunity against intracellular pathogens. J. Mol. Med. 2006, 84, 194–202. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Peña-Martinez, C.; Rickman, A.D.; Heckmann, B.L. Beyond autophagy: LC3-associated phagocytosis and endocytosis. Sci. Adv. 2022, 8, eabn1702. [Google Scholar] [CrossRef] [PubMed]
- Durgan, J.; Lystad, A.H.; Sloan, K.; Carlsson, S.R.; Wilson, M.I.; Marcassa, E.; Ulferts, R.; Webster, J.; Lopez-Clavijo, A.F.; Wakelam, M.J.; et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol. Cell 2021, 81, 2031–2040 e8. [Google Scholar] [CrossRef]
- Wang, M.; Zeng, L.; Su, P.; Ma, L.; Zhang, M.; Zhang, Y.Z. Autophagy: A multifaceted player in the fate of sperm. Hum. Reprod. Update 2022, 28, 200–231. [Google Scholar] [CrossRef]
- Zhang, M.; Jiang, M.; Bi, Y.; Zhu, H.; Zhou, Z.; Sha, J. Autophagy and apoptosis act as partners to induce germ cell death after heat stress in mice. PLoS ONE 2012, 7, e41412. [Google Scholar] [CrossRef] [Green Version]
- Yefimova, M.G.; Messaddeq, N.; Harnois, T.; Meunier, A.C.; Clarhaut, J.; Noblanc, A.; Weickert, J.L.; Cantereau, A.; Philippe, M.; Bourmeyster, N.; et al. A chimerical phagocytosis model reveals the recruitment by Sertoli cells of autophagy for the degradation of ingested illegitimate substrates. Autophagy 2013, 9, 653–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante-Marin, X.; Quiroga, C.; Lavandero, S.; Reyes, J.G.; Moreno, R.D. Apoptosis, necrosis and autophagy are influenced by metabolic energy sources in cultured rat spermatocytes. Apoptosis 2012, 17, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, G.; Wang, Z.; Liu, Y.; Dong, J.; Dong, X.; Liu, J.; Cao, J.; Ao, L.; Zhang, S. The protective effect of autophagy on mouse spermatocyte derived cells exposure to 1800 MHz radiofrequency electromagnetic radiation. Toxicol. Lett. 2014, 228, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Ahmed, N.; Wang, L.; Chen, H.; Waqas, Y.; Liu, T.; Haseeb, A.; Bangulzai, N.; Huang, Y.; Chen, Q. In vivo autophagy and biogenesis of autophagosomes within male haploid cells during spermiogenesis. Oncotarget 2017, 8, 56791–56801. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wan, H.; Li, X.; Liu, W.; Chen, Q.; Wang, Y.; Yang, L.; Tang, H.; Zhang, X.; Duan, E.; et al. Atg7 is required for acrosome biogenesis during spermatogenesis in mice. Cell Res. 2014, 24, 852–869. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Shen, C.; Wang, Z. The molecular mechanisms underlying acrosome biogenesis elucidated by gene-manipulated mice. Biol. Reprod. 2021, 105, 789–807. [Google Scholar] [CrossRef]
- Haseeb, A.; Tarique, I.; Bai, X.; Yang, P.; Ali Vistro, W.; Huang, Y.; Ali Fazllani, S.; Ahmed, Z.; Chen, Q. Inhibition of autophagy impairs acrosome and mitochondrial crista formation during spermiogenesis in turtle: Ultrastructural evidence. Micron 2019, 121, 84–89. [Google Scholar] [CrossRef]
- Al Rawi, S.; Louvet-Vallee, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef]
- Al Rawi, S.; Louvet-Vallee, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Allophagy: A macroautophagic process degrading spermatozoid-inherited organelles. Autophagy 2012, 8, 421–423. [Google Scholar] [CrossRef]
- Sato, M.; Sato, K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011, 334, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.M.; Sun, Q.Y. Autophagy is not involved in the degradation of sperm mitochondria after fertilization in mice. Autophagy 2013, 9, 2156–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, I.M.; Espino, J.; Bejarano, I.; Gallardo-Soler, A.; Campo, M.L.; Salido, G.M.; Pariente, J.A.; Peña, F.J.; Tapia, J.A. Autophagy-related proteins are functionally active in human spermatozoa and may be involved in the regulation of cell survival and motility. Sci. Rep. 2016, 6, 33647. [Google Scholar] [CrossRef]
- Gallardo Bolaños, J.M.; Miro-Moran, A.; Balao da Silva, C.M.; Davila, M.P.; Martin-Muñoz, P.; Aparicio, I.M.; Tapia, J.A.; Ortega-Ferrusola, C.; Peña, F.J. During cooled storage the extender influences processed autophagy marker light chain 3 (LC3B) of stallion spermatozoa. Anim. Reprod. Sci. 2014, 145, 40–46. [Google Scholar] [CrossRef]
- Aparicio, I.M.; Martin Muñoz, P.; Salido, G.M.; Peña, F.J.; Tapia, J.A. The autophagy-related protein LC3 is processed in stallion spermatozoa during short-and long-term storage and the related stressful conditions. Anim. Int. J. Anim. Biosci. 2016, 10, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- McPartlin, L.A.; Littell, J.; Mark, E.; Nelson, J.L.; Travis, A.J.; Bedford-Guaus, S.J. A defined medium supports changes consistent with capacitation in stallion sperm, as evidenced by increases in protein tyrosine phosphorylation and high rates of acrosomal exocytosis. Theriogenology 2008, 69, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pastor, F.; Mata-Campuzano, M.; Alvarez-Rodriguez, M.; Alvarez, M.; Anel, L.; de Paz, P. Probes and techniques for sperm evaluation by flow cytometry. Reprod. Domest. Anim. 2010, 45 (Suppl. 2), 67–78. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224 Pt 3, 213–232. [Google Scholar] [CrossRef]
- Yang, Y.P.; Hu, L.F.; Zheng, H.F.; Mao, C.J.; Hu, W.D.; Xiong, K.P.; Wang, F.; Liu, C.F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Gallardo Bolaños, J.M.; Miro Moran, A.; Balao da Silva, C.M.; Morillo Rodriguez, A.; Plaza Davila, M.; Aparicio, I.M.; Tapia, J.A.; Ortega Ferrusola, C.; Peña, F.J. Autophagy and apoptosis have a role in the survival or death of stallion spermatozoa during conservation in refrigeration. PLoS ONE 2012, 7, e30688. [Google Scholar] [CrossRef]
- Uribe, P.; Meriño, J.; Matus, C.E.; Schulz, M.; Zambrano, F.; Villegas, J.V.; Conejeros, I.; Taubert, A.; Hermosilla, C.; Sánchez, R. Autophagy is activated in human spermatozoa subjected to oxidative stress and its inhibition impairs sperm quality and promotes cell death. Hum. Reprod. 2022, 37, 680–695. [Google Scholar] [CrossRef] [PubMed]
- Foroozan-Boroojeni, S.; Tavalaee, M.; Zakeri, Z.; Lockshin, R.A.; Nasr-Esfahani, M.H. Assessment of Atg7 and LC3II/LC3, as The Markers of Autophagy, in Sperm of Infertile Men with Globozoospermia: A Case-Control Study. Cell J. 2021, 23, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Elzeiny, D.; Monir, R.; El Sabakhawy, K.; Selim, M.K.; Zalata, A. Relationship between DYNLT1 and Beclin1 expression and the fertilising potential of human spermatozoa. Andrologia 2019, 51, e13380. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ma, Y.; Zhang, J.; Jiang, S.; Yuan, G.; Cheng, J.; Lan, T.; Hao, J. Alteration in autophagy gene expression profile correlates with low sperm quality. Reprod. Biol. 2021, 21, 100546. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-haila, A.; Tulsiani, D.R. Signal transduction pathways that regulate sperm capacitation and the acrosome reaction. Arch. Biochem. Biophys. 2009, 485, 72–81. [Google Scholar] [CrossRef]
- Anbalagan, S.; Pires, I.M.; Blick, C.; Hill, M.A.; Ferguson, D.J.; Chan, D.A.; Hammond, E.M. Radiosensitization of renal cell carcinoma in vitro through the induction of autophagy. Radiother. Oncol. 2012, 103, 388–393. [Google Scholar] [CrossRef]
- Long, L.; Yang, X.; Southwood, M.; Lu, J.; Marciniak, S.J.; Dunmore, B.J.; Morrell, N.W. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ. Res. 2013, 112, 1159–1170. [Google Scholar] [CrossRef]
- Moreno, R.D.; Alvarado, C.P. The mammalian acrosome as a secretory lysosome: New and old evidence. Mol. Reprod. Dev. 2006, 73, 1430–1434. [Google Scholar] [CrossRef]
- Carr, D.W.; Newell, A.E. The role of A-kinase anchoring proteins (AKaps) in regulating sperm function. Soc. Reprod. Fertil. Suppl. 2007, 63, 135–141. [Google Scholar] [PubMed]
- Miro-Moran, A.; Jardin, I.; Ortega-Ferrusola, C.; Salido, G.M.; Pena, F.J.; Tapia, J.A.; Aparicio, I.M. Identification and function of exchange proteins activated directly by cyclic AMP (Epac) in mammalian spermatozoa. PLoS ONE 2012, 7, e37713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lybaert, P.; Danguy, A.; Leleux, F.; Meuris, S.; Lebrun, P. Improved methodology for the detection and quantification of the acrosome reaction in mouse spermatozoa. Histol. Histopathol. 2009, 24, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Johannisson, A.; Wallgren, M.; Nagy, S.; Siqueira, A.P.; Rodriguez-Martinez, H. Flow cytometry for the assessment of animal sperm integrity and functionality: State of the art. Asian J. Androl. 2011, 13, 406–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004, 36, 2445–2462. [Google Scholar] [CrossRef]
- Leidal, A.M.; Debnath, J. LC3-dependent extracellular vesicle loading and secretion (LDELS). Autophagy 2020, 16, 1162–1163. [Google Scholar] [CrossRef]
- Delorme-Axford, E.; Klionsky, D.J. The LC3-conjugation machinery specifies cargo loading and secretion of extracellular vesicles. Autophagy 2020, 16, 1169–1171. [Google Scholar] [CrossRef]
- Tomes, C.N. The proteins of exocytosis: Lessons from the sperm model. Biochem. J. 2015, 465, 359–370. [Google Scholar] [CrossRef]
- Khawar, M.B.; Gao, H.; Li, W. Mechanism of Acrosome Biogenesis in Mammals. Front. Cell Dev. Biol. 2019, 7, 195. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, L.; Ortega-Ferrusola, C.; Macias-Garcia, B.; Salido, G.M.; Pena, F.J.; Tapia, J.A. Identification of protein tyrosine phosphatases and dual-specificity phosphatases in mammalian spermatozoa and their role in sperm motility and protein tyrosine phosphorylation. Biol. Reprod. 2009, 80, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aparicio, I.M.; Rojo-Domínguez, P.; Castillejo-Rufo, A.; Peña, F.J.; Tapia, J.A. The Autophagy Marker LC3 Is Processed during the Sperm Capacitation and the Acrosome Reaction and Translocates to the Acrosome Where It Colocalizes with the Acrosomal Membranes in Horse Spermatozoa. Int. J. Mol. Sci. 2023, 24, 937. https://doi.org/10.3390/ijms24020937
Aparicio IM, Rojo-Domínguez P, Castillejo-Rufo A, Peña FJ, Tapia JA. The Autophagy Marker LC3 Is Processed during the Sperm Capacitation and the Acrosome Reaction and Translocates to the Acrosome Where It Colocalizes with the Acrosomal Membranes in Horse Spermatozoa. International Journal of Molecular Sciences. 2023; 24(2):937. https://doi.org/10.3390/ijms24020937
Chicago/Turabian StyleAparicio, Ines M., Patricia Rojo-Domínguez, Alba Castillejo-Rufo, Fernando J. Peña, and Jose A. Tapia. 2023. "The Autophagy Marker LC3 Is Processed during the Sperm Capacitation and the Acrosome Reaction and Translocates to the Acrosome Where It Colocalizes with the Acrosomal Membranes in Horse Spermatozoa" International Journal of Molecular Sciences 24, no. 2: 937. https://doi.org/10.3390/ijms24020937
APA StyleAparicio, I. M., Rojo-Domínguez, P., Castillejo-Rufo, A., Peña, F. J., & Tapia, J. A. (2023). The Autophagy Marker LC3 Is Processed during the Sperm Capacitation and the Acrosome Reaction and Translocates to the Acrosome Where It Colocalizes with the Acrosomal Membranes in Horse Spermatozoa. International Journal of Molecular Sciences, 24(2), 937. https://doi.org/10.3390/ijms24020937