

Decrease in α-Globin and Increase in the Autophagy-Activating Kinase ULK1 mRNA in Erythroid Precursors from β-Thalassemia Patients Treated with Sirolimus
 , , and
, , and 
Abstract
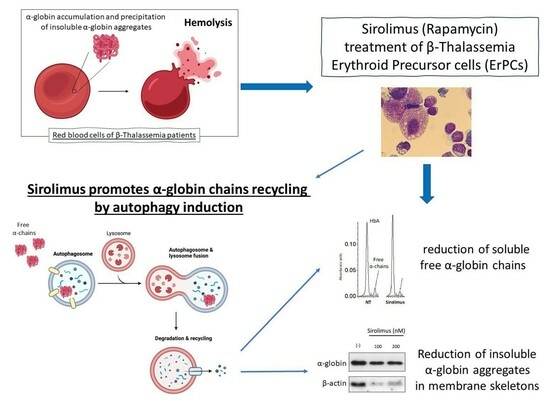
:
1. Introduction
2. Results
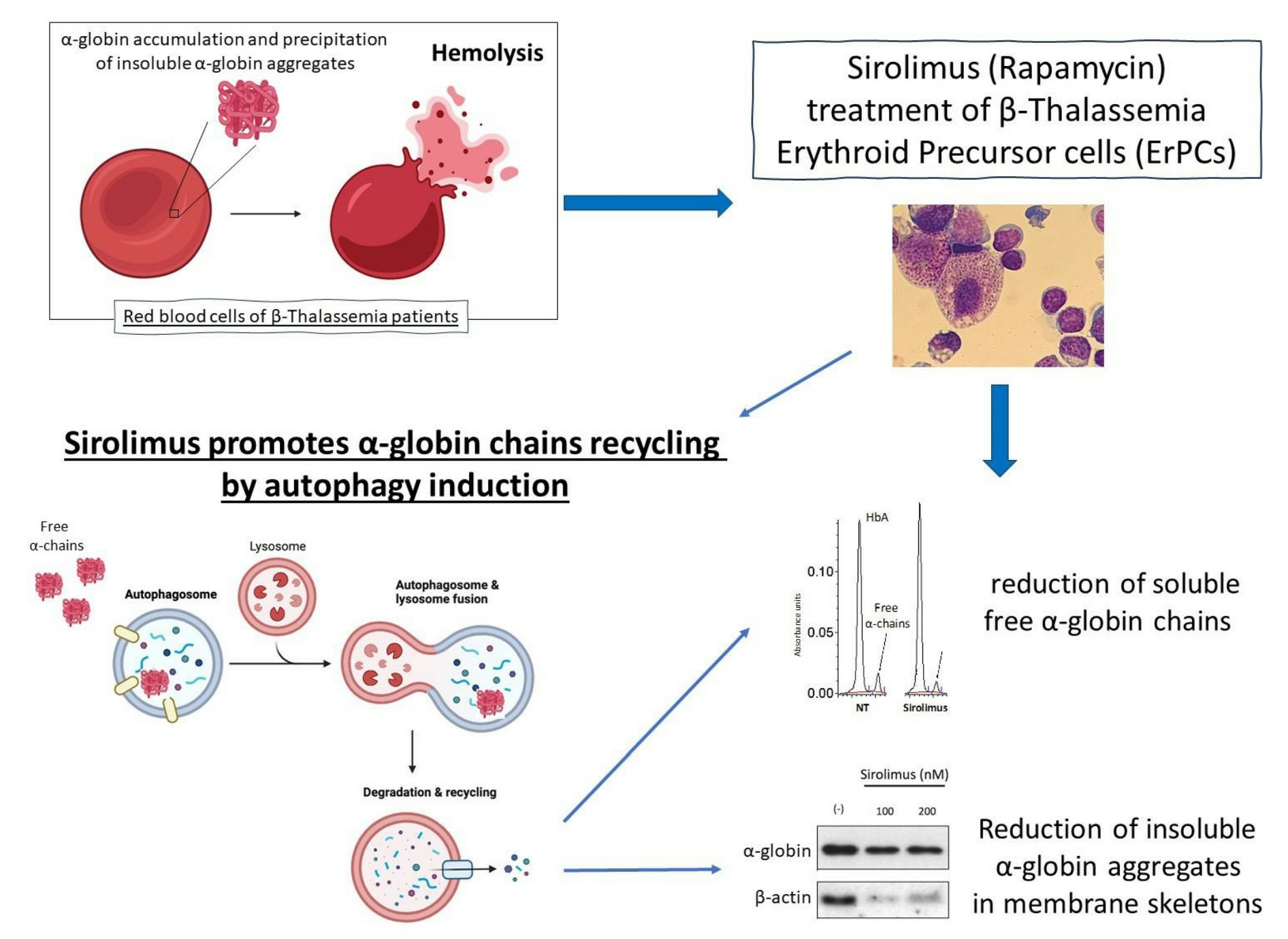
2.1. The Autophagy Program Is Activated in Erythroid Precursor Cells from β-Thalassemia Patients
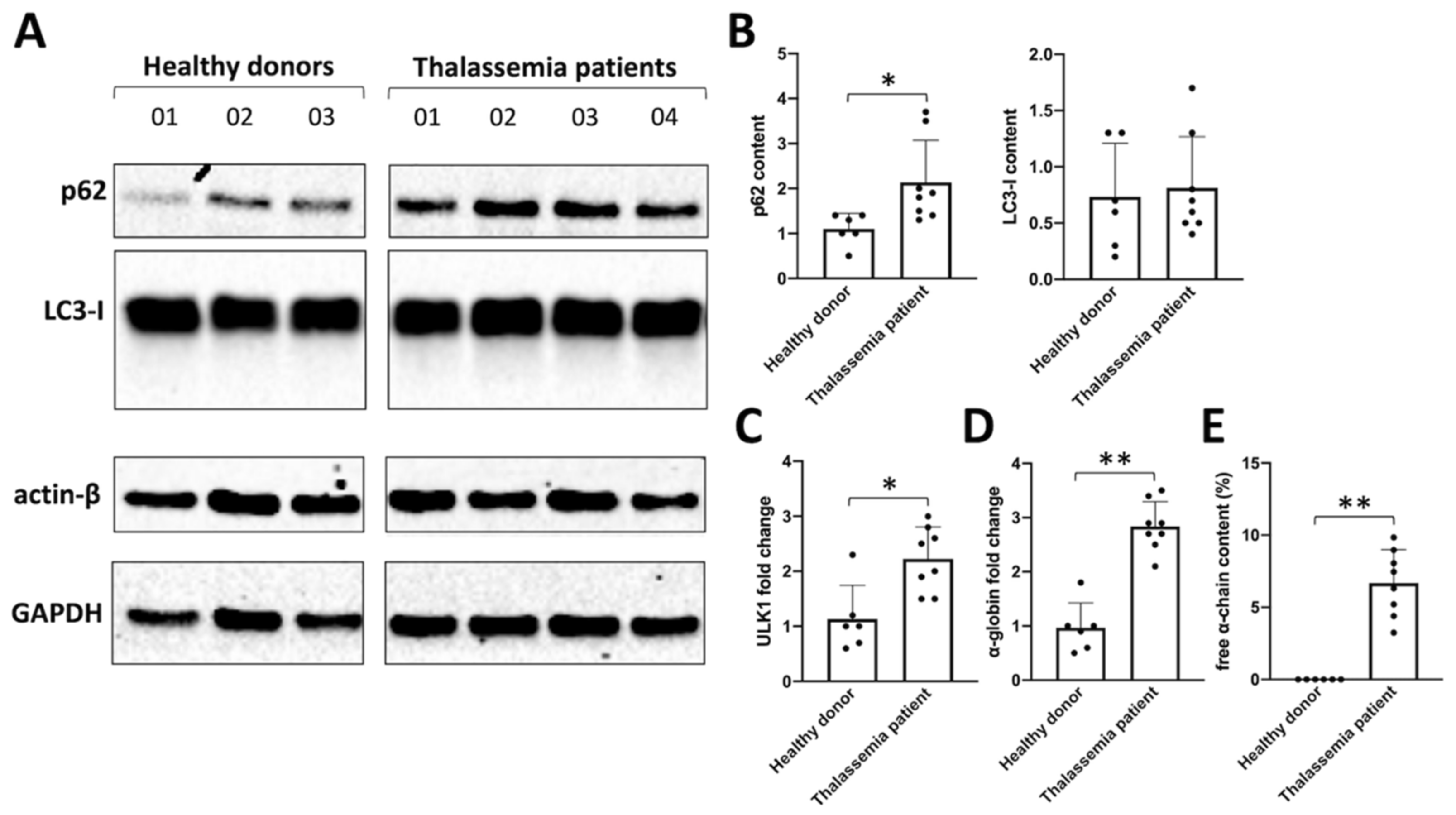
2.2. Sirolimus Potentiates Autophagy in Erythroid Precursor Cells from β-Thalassemia Patients
2.3. Sirolimus-Induced Autophagy Reduces Accumulation of Insoluble α-Chains in Membrane Skeletons of Cultured ErPCs
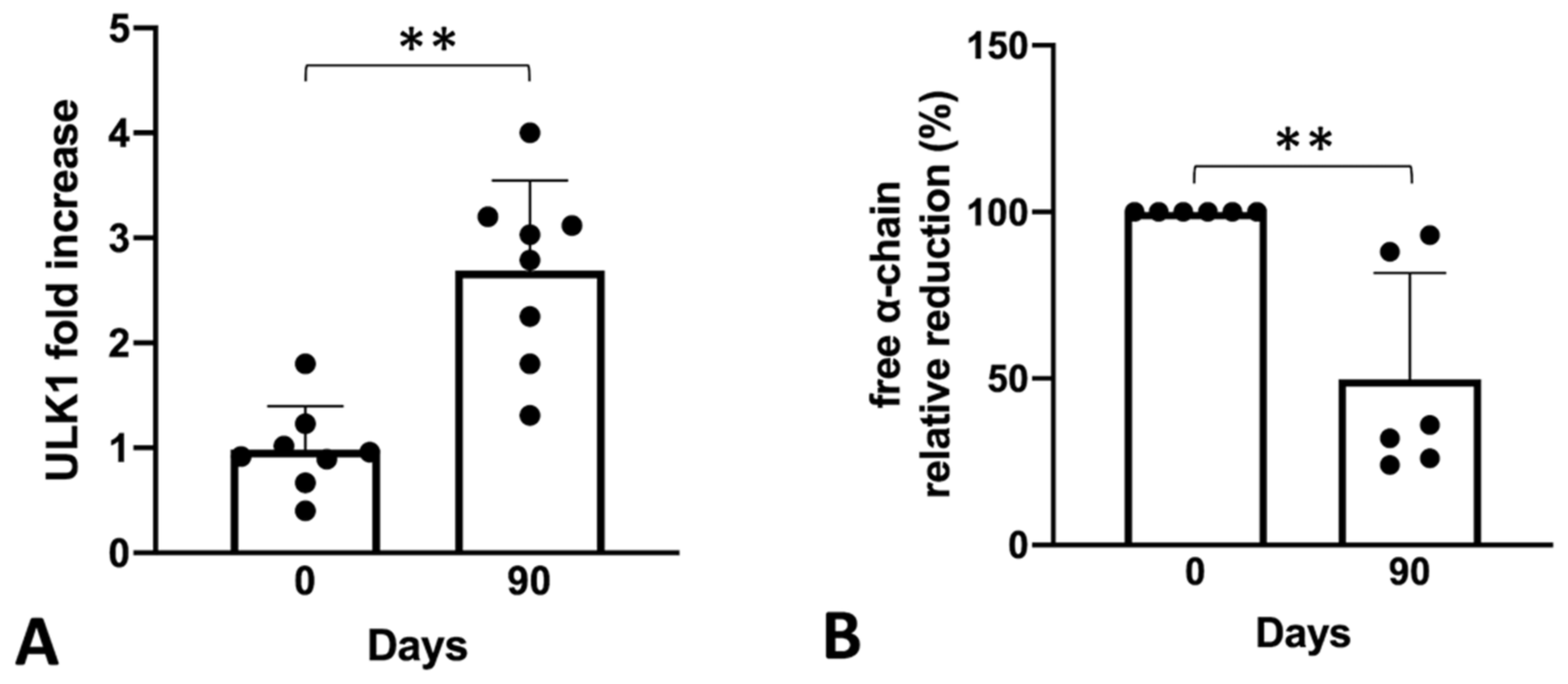
2.4. Sirolimus Treatment In Vivo Leads to a Lower Accumulation of Free α-Chains
3. Discussion
4. Materials and Methods
4.1. Culture and Treatment of Human Erythroid Precursor Cells
4.2. RNA Extraction and RT-qPCR Analysis
4.3. HPLC Analyses of Hemoglobin
4.4. Western Blotting of Soluble Fractions
4.5. Western Blotting of Insoluble Fractions
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weatherall, D.J. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat. Rev. Genet. 2001, 2, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Origa, R. β-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Origa, R. β-Thalassemia. Genet. Med. 2017, 19, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. The molecular basis of β-thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011700. [Google Scholar] [CrossRef] [PubMed]
- Rieder, R.F.; James, G.W., 3rd. Imbalance in alpha and beta globin synthesis associated with a hemoglobinopathy. J. Clin. Investig. 1974, 54, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Villalobos, M.; Blanquer, M.; Moraleda, J.M.; Salido, E.J.; Perez-Oliva, A.B. New Insights into Patho-physiology of β-Thalassemia. Front. Med. 2022, 9, 880752. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.Y.; Ren, Z.R.; Zhang, J.Z.; Guo, X.B.; Wang, Q.X.; Wang, S.; Lin, D.; Gong, X.L.; Li, W.; Huang, S.Z.; et al. Restoration of the balanced alpha/beta-globin gene expression in beta654-thalassemia mice using combined RNAi and antisense RNA approach. Hum. Mol. Genet. 2007, 16, 2616–2625. [Google Scholar] [CrossRef]
- Mettananda, S.; Gibbons, R.J.; Higgs, D.R. α-Globin as a molecular target in the treatment of β-thalassemia. Blood 2015, 125, 3694–3701. [Google Scholar] [CrossRef]
- Mao, B.; Zhang, Q.; Ma, L.; Zhao, D.S.; Zhao, P.; Yan, P. Overview of Research into mTOR Inhibitors. Molecules 2022, 27, 5295. [Google Scholar] [CrossRef]
- Fibach, E.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Finotti, A.; Lampronti, I.; Prus, E.; Mischiati, C.; Gambari, R. Effects of rapamycin on accumulation of alpha-, β- and gamma-globin mRNAs in erythroid precursor cells from β-thalassaemia patients. Eur. J. Haematol. 2006, 77, 437–441. [Google Scholar] [CrossRef]
- Cosenza, L.C.; Breda, L.; Breveglieri, G.; Zuccato, C.; Finotti, A.; Lampronti, I.; Borgatti, M.; Chiavilli, F.; Gamberini, M.R.; Satta, S.; et al. A validated cellular biobank for β-thalassemia. J. Transl. Med. 2016, 14, 255. [Google Scholar] [CrossRef]
- Lechauve, C.; Keith, J.; Khandros, E.; Fowler, S.; Mayberry, K.; Freiwan, A.; Thom, C.S.; Delbini, P.; Romero, E.B.; Zhang, J.; et al. The autophagy-activating kinase ULK1 mediates clearance of free α-globin in β-thalassemia. Sci. Transl. Med. 2019, 11, eaav4881. [Google Scholar] [CrossRef]
- Gaudre, N.; Cougoul, P.; Bartolucci, P.; Dörr, G.; Bura-Riviere, A.; Kamar, N.; Del Bello, A. Improved Fetal Hemoglobin With mTOR Inhibitor-Based Immunosuppression in a Kidney Transplant Recipient With Sickle Cell Disease. Am. J. Transplant. 2017, 17, 2212–2214. [Google Scholar] [CrossRef] [PubMed]
- Al-Khatti, A.A.; Alkhunaizi, A.M. Additive effect of sirolimus and hydroxycarbamide on fetal haemoglobin level in kidney transplant patients with sickle cell disease. Br. J. Haematol. 2019, 185, 959–961. [Google Scholar] [CrossRef]
- Gamberini, M.R.; Prosdocimi, M.; Gambari, R. Sirolimus for Treatment of β-Thalassemia: From Pre-Clinical Studies to the Design of Clinical Trials. Health Educ. Public Health 2021, 4, 425–435. [Google Scholar]
- ClinicalTrials.gov. NCT03877809, a Personalized Medicine Approach for Beta-Thalassemia Transfusion Dependent Patients: Testing SIROLIMUS in a First Pilot Clinical Trial (SIRTHALACLIN). 18 March 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03877809 (accessed on 6 May 2023).
- ClinicalTrials.gov. NCT04247750, Treatment of Beta-Thalassemia Patients with Rapamycin (Sirolimus): From Pre-Clinical Research to a Clinical Trial (THALA-RAP). 30 January 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04247750 (accessed on 6 May 2023).
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Gasparello, J.; Papi, C.; D’Aversa, E.; Breveglieri, G.; Lampronti, I.; Finotti, A.; Borgatti, M.; et al. Expression of γ-globin genes in β-thalassemia patients treated with sirolimus: Results from a pilot clinical trial (Sirthalaclin). Ther. Adv. Hematol. 2022, 13, 20406207221100648. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, M.; Nicoli, F.; Proietto, D.; Dallan, B.; Zuccato, C.; Cosenza, L.C.; Gasparello, J.; Papi, C.; d’Aversa, E.; Borgatti, M.; et al. Effects of Sirolimus treatment on patients with β-Thalassemia: Lymphocyte immunophenotype and biological activity of memory CD4+ and CD8+ T cells. J. Cell. Mol. Med. 2023, 27, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Lee, E.; Lee, K.W. Therapeutic Implications of Autophagy Inducers in Immunological Disorders, Infection, and Cancer. Int. J. Mol. Sci. 2017, 18, 1959. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Fernández-Ramos, A.A.; Loriot, M.A. Impact of Immunosuppressive Drugs on the Metabolism of T Cells. Int. Rev. Cell Mol. Biol. 2018, 341, 169–200. [Google Scholar]
- Rostamzadeh, D.; Yousefi, M.; Haghshenas, M.R.; Ahmadi, M.; Dolati, S.; Babaloo, Z. mTOR Signaling pathway as a master regulator of memory CD8+ T-cells, Th17, and NK cells development and their functional properties. J. Cell. Physiol. 2019, 234, 12353–12368. [Google Scholar] [CrossRef]
- Lin, M.G.; Hurley, J.H. Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 2016, 39, 61–68. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar]
- Mischiati, C.; Sereni, A.; Lampronti, I.; Bianchi, N.; Borgatti, M.; Prus, E.; Fibach, E.; Gambari, R. Rapamycin-mediated induction of gamma-globin mRNA accumulation in human erythroid cells. Br. J. Haematol. 2004, 126, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, A.; Troia, A.; Calzolari, R.; Scazzone, C.; Rigano, P.; Martorana, A.; Sacco, M.; Maggio, A.; Di Marzo, R. Efficacy of Rapamycin as Inducer of Hb F in Primary Erythroid Cultures from Sickle Cell Disease and β-Thalassemia Patients. Hemoglobin 2015, 39, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Acar, M.; Jupelli, M.; MacBeth, K.; Schwickart, M. Rapamycin (Sirolimus) and Rap-536 Increase Red Blood Cell Parameters through Distinct Mechanisms in Wild-Type and Thalassemic Mice. Blood 2020, 136, 17. [Google Scholar] [CrossRef]
- Wang, J.; Tran, J.; Wang, H.; Guo, C.; Harro, D.; Campbell, A.D.; Eitzman, D.T. mTOR Inhibition improves anaemia and reduces organ damage in a murine model of sickle cell disease. Br. J. Haematol. 2016, 174, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Khaibullina, A.; Almeida, L.E.; Wang, L.; Kamimura, S.; Wong, E.C.; Nouraie, M.; Maric, I.; Albani, S.; Finkel, J.; Quezado, Z.M. Rapamycin increases fetal hemoglobin and ameliorates the nociception phenotype in sickle cell mice. Blood Cells Mol. Dis. 2015, 55, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, M.; Gasparello, J.; Cosenza, L.C.; Breveglieri, G.; Papi, C.; Zuccato, C.; Gambari, R.; Finotti, A. Production and Characterization of K562 Cellular Clones Hyper-Expressing the Gene Encoding α-Globin: Preliminary Analysis of Biomarkers Associated with Autophagy. Genes 2023, 14, 556. [Google Scholar] [CrossRef] [PubMed]
- Nurzadeh, M.; Ghalandarpoor-Attar, S.M.; Ghalandarpoor-Attar, S.N.; Rabiei, M. The sequestosome 1 protein: Therapeutic vulnerabilities in ovarian cancer. Clin. Transl. Oncol. 2023, 25, 2783–2792. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Park, J.H.; Chung, K.C. The central regulator p62 between ubiquitin proteasome system and autophagy and its role in the mitophagy and Parkinson’s disease. BMB Rep. 2020, 53, 56–63. [Google Scholar] [CrossRef]
- Khandros, E.; Thom, C.S.; D’Souza, J.; Weiss, M.J. Integrated protein quality-control pathways regulate free α-globin in murine β-thalassemia. Blood 2012, 119, 5265–5275. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, A.T.; Tzounakas, V.L.; Dzieciatkowska, M.; Arvaniti, V.Z.; Papageorgiou, E.G.; Papassideri, I.S.; Stamoulis, K.; D’Alessandro, A.; Kriebardis, A.G.; Antonelou, M.H. Innate Variability in Physiological and Omics Aspects of the Beta Thalassemia Trait-Specific Donor Variation Effects. Front. Physiol. 2022, 13, 907444. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, A.T.; Tzounakas, V.L.; Arvaniti, V.Z.; Dzieciatkowska, M.; Stamoulis, K.; Lekka, M.E.; Papassideri, I.S.; D’Alessandro, A.; Kriebardis, A.G.; Antonelou, M.H. Red Blood Cell Proteasome in Beta-Thalassemia Trait: Topology of Activity and Networking in Blood Bank Conditions. Membranes 2021, 11, 716. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed]
- Osmulski, P.A.; Gaczynska, M. Rapamycin allosterically inhibits the proteasome. Mol. Pharmacol. 2013, 84, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Keith, J.; Christakopoulos, G.E.; Fernandez, A.G.; Yao, Y.; Zhang, J.; Mayberry, K.; Telange, R.; Sweileh, R.B.A.; Dudley, M.; Westbrook, C.; et al. Loss of miR-144/451 alleviates β-thalassemia by stimulating ULK1-mediated autophagy of free α-globin. Blood 2023, 142, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Lampronti, I.; Borgatti, M.; Scapoli, C.; Gambari, R.; Finotti, A. Treatment of Erythroid Precursor Cells from β-Thalassemia Patients with Cinchona Alkaloids: Induction of Fetal Hemoglobin Production. Int. J. Mol. Sci. 2021, 22, 13433. [Google Scholar] [CrossRef]
- Cosenza, L.C.; Gasparello, J.; Romanini, N.; Zurlo, M.; Zuccato, C.; Gambari, R.; Finotti, A. Efficient CRISPR-Cas9-based genome editing of β-globin gene on erythroid cells from homozygous β039-thalassemia patients. Mol. Ther. Methods Clin. Dev. 2021, 21, 507–523. [Google Scholar] [CrossRef]
- Cosenza, L.C.; Zuccato, C.; Zurlo, M.; Gambari, R.; Finotti, A. Co-Treatment of Erythroid Cells from β-Thalassemia Patients with CRISPR-Cas9-Based β039-Globin Gene Editing and Induction of Fetal Hemoglobin. Genes 2022, 13, 1727. [Google Scholar] [CrossRef]
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Breveglieri, G.; Bianchi, N.; Lampronti, I.; Gasparello, J.; Scapoli, C.; Borgatti, M.; Finotti, A.; et al. The rs368698783 (G>A) Polymorphism Affecting LYAR Binding to the Aγ-Globin Gene Is Associated with High Fetal Hemoglobin (HbF) in β-Thalassemia Erythroid Precursor Cells Treated with HbF Inducers. Int. J. Mol. Sci. 2023, 24, 776. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers and Probes | Sequences |
|---|---|
| primer forward α-globin | 5′-CGACAAGACCAACGTCAAGG-3′ |
| primer reverse α-globin | 5′-GGTCTTGGTGGTGGGGAAG-3′ |
| probe α-globin | 5′-HEX-ACATCCTCTCCAGGGCCTCCG-BFQ-3′ |
| primer forward ULK1 | 5′-CTACCTGGTTATGGAGTACTGC-3′ |
| primer reverse ULK1 | 5′-GGAAGAGCCTGATGGTGTC-3′ |
| probe ULK1 | 5′-FAM-CGACTACCT/ZEN/GCACGCCATGC-BFQ-3′ |
| primer forward RPL13A | 5′-GGCAATTTCTACAGAAACAAGTTG-3′ |
| primer reverse RPL13A | 5′-GTTTTGTGGGGCAGCATACC-3′ |
| probe RPL13A | 5′-HEX-CGCACGGTCCGCCAGAAGAT-BFQ-3′ |
| primer forward ACTB | 5′-ACAGAGCCTCGCCTTTG-3′ |
| primer reverse ACTB | 5′-ACGATGGAGGGGAAGACG-3′ |
| probe ACTB | 5′-Cy5-CCTTGCACATGCCGGAGCC-BRQ-3′ |
| primer forward GAPDH | 5′-ACATCGCTCAGACACCATG-3′ |
| primer reverse GAPDH | 5′-TGTAGTTGAGGTCAATGAAGGG-3′ |
| probe GAPDH | 5′-FAM-AAGGTCGGAGTCAACGGATTTGGTC-BFQ-3′ |
| Target | Primary Antibody | Cat. n. | Secondary Antibody | Cat. n. |
|---|---|---|---|---|
| p62 | Rabbit anti-p62/SQSTM1 (Sigma-Aldrich, St. Louis, MO, USA) | P0067 | Mouse Anti-rabbit IgG HRP (Cell Signaling Technology, Danvers, MA, USA | 7074 |
| LC3 | Rabbit anti-LC3B (Sigma-Aldrich, St. Louis, MO, USA) | L7543 | Mouse Anti-rabbit IgG HRP (Cell Signaling Technology, Danvers, MA, USA) | 7074 |
| β-actin | Rabbit anti-β-actin (Cell Signaling Technology, Danvers, MA, USA) | 4967 | Mouse Anti-rabbit IgG HRP (Cell Signaling Technology, Danvers, MA, USA | 7074 |
| GAPDH | Mouse anti-GAPDH (Thermo Fisher, Waltham, MA, USA) | MA1-16783 | Goat Anti-mouse IgG HRP (Thermo Fisher, Waltham, MA, USA) | 32430 |
| Target | Primary Antibody | Cat. n. | Secondary Antibody | Cat. n. |
|---|---|---|---|---|
| α-globin | Mouse anti-hemoglobin α (D-4) (Santa Cruz Biotechnology, Dallas, TX, USA) | sc-514378 | Goat Anti-mouse IgG HRP (Thermo Fisher, Waltham, MA, USA) | 32430 |
| GAPDH | Mouse anti-GAPDH (Thermo Fisher, Waltham, MA, USA) | MA1-16783 | Goat Anti-mouse IgG HRP (Thermo Fisher, Waltham, MA, USA) | 32430 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zurlo, M.; Zuccato, C.; Cosenza, L.C.; Gasparello, J.; Gamberini, M.R.; Stievano, A.; Fortini, M.; Prosdocimi, M.; Finotti, A.; Gambari, R. Decrease in α-Globin and Increase in the Autophagy-Activating Kinase ULK1 mRNA in Erythroid Precursors from β-Thalassemia Patients Treated with Sirolimus. Int. J. Mol. Sci. 2023, 24, 15049. https://doi.org/10.3390/ijms242015049
Zurlo M, Zuccato C, Cosenza LC, Gasparello J, Gamberini MR, Stievano A, Fortini M, Prosdocimi M, Finotti A, Gambari R. Decrease in α-Globin and Increase in the Autophagy-Activating Kinase ULK1 mRNA in Erythroid Precursors from β-Thalassemia Patients Treated with Sirolimus. International Journal of Molecular Sciences. 2023; 24(20):15049. https://doi.org/10.3390/ijms242015049
Chicago/Turabian StyleZurlo, Matteo, Cristina Zuccato, Lucia Carmela Cosenza, Jessica Gasparello, Maria Rita Gamberini, Alice Stievano, Monica Fortini, Marco Prosdocimi, Alessia Finotti, and Roberto Gambari. 2023. "Decrease in α-Globin and Increase in the Autophagy-Activating Kinase ULK1 mRNA in Erythroid Precursors from β-Thalassemia Patients Treated with Sirolimus" International Journal of Molecular Sciences 24, no. 20: 15049. https://doi.org/10.3390/ijms242015049
APA StyleZurlo, M., Zuccato, C., Cosenza, L. C., Gasparello, J., Gamberini, M. R., Stievano, A., Fortini, M., Prosdocimi, M., Finotti, A., & Gambari, R. (2023). Decrease in α-Globin and Increase in the Autophagy-Activating Kinase ULK1 mRNA in Erythroid Precursors from β-Thalassemia Patients Treated with Sirolimus. International Journal of Molecular Sciences, 24(20), 15049. https://doi.org/10.3390/ijms242015049