1. Introduction
The most distinguishing feature of Gram-negative bacteria, such as
Escherichia coli, is the presence of an asymmetric outer membrane (OM) [
1]. This asymmetry is due to the characteristic location of lipopolysaccharide (LPS) in the outer leaflet of the cell envelope, with phospholipids facing its inner leaflet [
1,
2]. The maintenance of this OM asymmetry is essential to impart a permeability barrier, which prevents the entry of bulky toxic molecules, such as antibiotics and bile salts, inside the cells. Thus, with few exceptions, LPS is essential for bacterial viability and is one of the major virulence factors in pathogenic Gram-negative bacteria. LPS is a complex glycolipid and, in general, they share a common architecture and can be, for convenience, divided into three parts. The most conserved component is the hydrophobic membrane-anchored lipid A part, which constitutes the endotoxin principal. To the lipid A part is attached a core oligosaccharide, to which an oligosaccharide of variable length called the
O-antigen is attached in bacteria with smooth LPS [
3]. The biosynthesis of LPS at the genetic and biochemical levels is well established, although how LPS amounts are regulated and its final assembly in the OM is not well understood. This is particularly important because bacteria must maintain a strict balance of 1:0.15 between phospholipids and LPS, the two essential components of the cell envelope [
4]. In
E.
coli, this is achieved by regulating the first committed step in LPS biosynthesis, catalyzed by the essential enzyme LpxC, because LPS and phospholipids use the same (
R)-3-hydroxymyristate as the common metabolic precursor [
5]. However, the regulation of LpxC as we understand now turns out to be more complex than it was thought a few years ago, as it responds to multiple physiological cues and a regulated turnover by FtsH and HslVU proteases (
Figure 1) [
5,
6].
In
E.
coli, the biosynthesis and transport of LPS require products of more than thirty genes and many of them are essential for bacterial viability [
2]. Briefly, LPS biosynthesis occurs on the inner leaflet of the inner membrane and begins with the acylation of UDP-GlcNAc, catalyzed by LpxA. In
E.
coli, LpxA is selective for
β-hydroxymyristoyl acyl carrier protein [
7]. This is followed by deacetylation of UDP-3-
O-(acyl)-GlcNAc by the Zn
2+-dependent LpxC enzyme [
8]. As the equilibrium constant for LpxA-mediated acylation is unfavorable, LpxC-catalyzed deacetylation becomes the first committed step in lipid A biosynthesis [
9]. Following the deacetylation step, a second
β-hydroxymyristoyl chain is incorporated by
E. coli LpxD, leading to the synthesis of UDP-2,3-diacyl-GlcN [
10]. Three additional enzymes, LpxH, LpxB and LpxK, act successively to generate the intermediate lipid IV
A [
11]. This intermediate serves as an acceptor for the incorporation of two 3-deoxy-α-D-
manno-oct-2-ulosonic acid (Kdo) residues by WaaA, with CMP-Kdo as the donor, generating Kdo
2-lipid IV
A [
12]. In
E.
coli, up to the step of Kdo
2-lipid IV
A biosynthesis, all enzymes are essential for bacterial viability, although strains synthesizing only a lipid IV
A precursor or Kdo
2-lipid IV
A can be constructed [
13]. The incorporation of Kdo residues serves as a key intermediate to generate hexaacylated lipid A via sequential acylation by lauroyl (LpxL) and myristoyl (LpxM) transferases and further incorporation of sugar residues using specific glycosyltransferases to produce a core oligosaccharide linked to the lipid A part [
13,
14]. Strains synthesizing tetraacylated lipid A can only grow on minimal medium and exhibit an elevated cell envelope stress response [
13,
14]. Interestingly, suppressors mapping to the
msbA gene encoding LPS flippase can allow the growth of strains devoid of LpxL, LpxM and LpxP on rich medium and at elevated temperatures. These observations are in agreement with the known higher selectivity exerted by MsbA for hexaacylated lipid A as compared to the less favored underacylated species. Individually, all three late acyltransferases are nonessential, although Δ
lpxL exhibits a temperature-sensitive growth phenotype. However, LpxL and LpxM become essential when cardiolipin synthase A, encoded by the
clsA gene, is absent, suggesting that MsbA and ClsA could cooperate in LPS trafficking, although the precise mechanism of their interaction remains unknown [
15,
16]. Thus, besides the regulatory control exerted by the regulation of LpxC amounts, the preferential selection of hexaacylated lipid A species by MsbA constitutes early checkpoints in controlling the proper amounts of LPS and its translocation across the inner membrane [
17].
Initial studies on the regulation of LpxC deacetylase revealed that its levels are controlled in a 20-fold range in relation to the lipid A content without any increase in the corresponding mRNA levels of the encoded gene [
18]. Subsequently, it was shown that LpxC is a substrate of FtsH protease and this proteolytic activity might be affected by alterations in acyl-ACP pools [
5]. Such a model of FtsH-mediated proteolysis of LpxC could, in turn, provide a balance between biosynthesis of phospholipids and lipid A since both derive their fatty acyl chains from the same (
R)-3-hydroxyacyl-ACP pool (
Figure 1) [
5]. In an important breakthrough, a new essential inner membrane protein was identified and shown to co-purify with FtsH and several proteins involved in either LPS and phospholipid biosynthesis or LPS translocation, and hence designated the LPS assembly protein LapB [
19]. The absence of LapB could be tolerated either in the presence of a hyperactive
fabZ variant that allowed an
ftsH deletion or when the biosynthesis of LPS was reduced [
19,
20]. The co-purification of LapB with FtsH, FabZ, WaaC and Lpt proteins led to the proposal that LapB could act as a hub for LPS assembly in the IM and further cooperate with FtsH to regulate LpxC turnover [
19]. More recent studies have validated these earlier observations and indeed, LapB was found to interact with LpxA, LpxC, LpxD and FabZ [
21]. Similarly, recent structural and biochemical studies have shown that LpxC degradation by FtsH requires LapB [
22]. However, how such LpxC degradation by FtsH-LapB is adjusted to the cellular demand for LPS remained unknown till the more recent discovery of LapC [
2]. We and other independent research groups have shown that LapC acts as an antagonist of the proteolytic pathway of LpxC mediated by FtsH-LapB [
6,
22,
23,
24,
25,
26]. Our results were further supported by an examination of the properties of strains with chromosomal mutations that cause truncations in the periplasmic domain of LapC [
6]. Such mutant bacteria exhibited a temperature-sensitive (Ts) phenotype, hypersensitivity to the LpxC inhibitor CHIR090, hyperdegradation of LpxC and consequently reduced levels of LPS (
Figure 1). Thus, not only our group, but parallel independent studies, identified extragenic suppressors of
lapC mutant bacteria mapping to the
lapA/
B operon,
lpxC and
ftsH genes [
6,
23,
24,
26,
27]. Furthermore, an additional partner, LapD, which also co-purifies with LapB, was identified [
28]. Δ
lapD bacteria were found to exhibit a Ts phenotype and membrane permeability defects with a synthetic lethality in the absence of either cardiolipin synthase A, or when LpxL or LpxM acyltransferase was absent or when bacteria synthesize LPS composed of only Kdo
2-lipid A due to the absence of WaaC heptosyltransferase [
28,
29]. Moreover, extragenic suppressors that overcome defects of
lapC mutant bacteria were also found to rescue the Ts phenotype or vancomycin sensitivity of Δ
lapD bacteria [
28].
However, how LapB, LapC and LapD sense LPS biosynthetic demand and whether there are additional factors that either inhibit or enhance LpxC degradation remain unknown. Adding to this complexity, LpxC can be degraded in the absence of LapB by HslVU, and levels of LpxC can also be regulated by regulatory small RNAs, although the precise mechanism remains unknown (
Figure 1) [
6]. LPS synthesis can also be stimulated by additional signals that may involve systems that respond to lipid asymmetry, including the activation of outer membrane phospholipase PldA, the Mla system that facilitates retrograde lipid trafficking, and PagP palmitoyltransferase [
30]. LpxC stability can further respond to changes in levels of acyl-CoA, ppGpp and alterations in the levels of saturated fatty acids as compared to unsaturated [
17,
31]. Thus, the regulation of LpxC and, in turn, that of LPS involves several factors. Hence, in this study, we identified additional genes whose products might regulate LpxC or LPS levels by selecting for suppressors whose overexpression can overcome the Ts phenotype of
lapC mutant bacteria (
Figure 2). We show that overexpression of the
lapD gene and some other potential new regulatory factors (MarA, SrrA, DksA and YceJ) can suppress the Ts phenotype of
lapC190 mutant bacteria and, among these, some act by increasing the amount of LpxC. MarA is a well-characterized transcription factor that regulates the expression of several genes involved in regulating antibiotic resistance [
32]. Using transposon mutagenesis, gene(s) whose products are required for MarA- and SrrA-mediated suppression were identified to gain a better understanding of that factors that limit bacterial growth when LapC is dysfunctional and additional players that regulate LpxC amounts (
Figure 2).
3. Discussion
In this study, we identified new additional factors that regulate LpxC levels. Regulation of LpxC is critical for our understanding of the mechanism of balancing biosynthesis of LPS and phospholipids, since they both share the same (
R)-3-hydroxymyristate-ACP as the common metabolic precursor. Recent studies have shown that LpxC stability is regulated by the FtsH-LapB complex, wherein FtsH degrades LpxC, which requires LapB function for its proteolytic activity towards LpxC [
19,
22]. However, this activity is counteracted by the essential LapC (YejM) protein [
6,
23,
25]. In addition to FtsH-LapB and LapC, many additional factors participate in the regulation of LpxC amounts and, hence, the overall LPS content. Mutations in either the
ftsH or
lapB genes lead to an increased abundance of LPS due to the stabilization of LpxC, whereas loss-of-function mutations in the
lapC gene exhibit enhanced LpxC degradation with a concomitant reduction in the overall LPS content. During earlier work, we isolated temperature-sensitive
lapC190,
lapC377fs and
lapC F349S mutant bacteria [
6]. Because
lapC190 mutant bacteria exhibit a tight Ts phenotype, we exploited such a property to isolate multicopy suppressors that restore growth at elevated temperatures to identify additional factors that regulate LpxC amounts. Such an approach should also reveal what are the limiting factors that confer the Ts phenotype to
lapC190 mutant bacteria. Using such a procedure, thirteen genes were identified whose mild induction overcomes the Ts phenotype. As
lapC190 bacteria exhibit permeability defects and reduced LpxC and LPS levels, we examined whether overexpression of any of these genes also suppress such defects. Quite satisfactorily, a major proportion of such genes can be implicated either directly or indirectly in functions related to LPS, phospholipoid/fatty acid biosynthesis and regulation of LpxC protease FtsH amount (
lapD,
pldA,
acpP,
acpT,
accB,
gnsA,
yfgM,
marA) or envelope stress response (
srrA,
dksA, ymgG). These thirteen genes were thus grouped into different categories based on their known or predicted functions. Three out of them (
marA,
dksA and
srrA) act as transcriptional factors and should contribute by their regulation of downstream target gene(s), whose gene products either directly regulate LPS or phospholipid biosynthesis. Another group of suppressors include genes encoding the LPS assembly protein LapD, the essential AcpP protein that shuttles fatty acid chains in the lipid A and fatty acid/phospholipid biosynthesis, the outer membrane phospholipase A encoded by the
pldA gene. Isolation of these genes is interesting since this shows that
lapC mutant bacteria have imbalanced phospholipid and LPS biosynthesis. Importantly, PldA degrades phospholipids that have been mislocalized to the outer leaflet in the OM, which signal stabilization of LpxC and an increased production of LPS [
29], explaining its isolation as a multicopy suppressor. However, it should be noted that overexpression of the
pldA gene does not overcome the permeability defects of
lapC190 bacteria, as reflected by an inability to restore growth on bile-salt-containing MacConkey agar. Interestingly, up to now, the sole dosage-dependent suppressor of
lapC mutant bacteria expressing only the periplasmic domain has been the
acpT gene, whose product encodes a phosphopantetheinyl transferase [
54]. In this work, we also isolated the
acpT gene as a multicopy suppressor; however, this suppressing phenotype was much weaker than the others. Surprisingly, it has been shown that AcpT-mediated suppression of
lapC (
yejM) mutant bacteria for the Ts phenotype does not require its enzymatic activity [
54].
The global transcriptomes of MarA and DksA are known [
55,
56,
57], but only sketchy information about SrrA is available. Thus, in this study, we systematically attempted to identify genes whose presence is required for multicopy suppression by MarA and SrrA. In
E.
coli, MarA acts as a transcriptional activator and is known to activate the expression of
acrAB-
tolC-encoded efflux pump, thereby contributing to resistance against antimicrobial compounds [
58,
59,
60]. More recently, approximately 30 transcriptional units have been identified that could be regulated by MarA [
61]. Here, we used saturated transposon mutagenesis to identify genes whose inactivation abrogates the
marA- and
srrA-mediated restoration of the growth of
lapC190 mutant bacteria at high temperatures. The vast majority of transposon mutants that prevented MarA-mediated multicopy suppression mapped to the
mla operon, whose products are implicated in lipid trafficking, leading to the removal of the mislocalized phospholipids from the OM [
29]. Consistent with our results, a MarA-binding motif has been identified in the promoter region of the
mla operon [
61]. Thus, our results provide a rationale for the identification of the
marA gene as a multicopy suppressor of
lapC190 mutant bacteria, and this suppression requires the functionality of
mla genes. In accordance with these findings, the
pldA gene was isolated as a multicopy suppressor since both PldA and Mla pathways sense OM disturbance due to the accumulation of phospholipids in the OM when LPS is limiting.
In a similar manner, we addressed the pathway of SrrA-mediated suppression. Our results show that the overexpression of the
srrA gene efficiently restores the growth at high temperatures, LpxC levels as well as permeability defects. This suppression was found to be contingent on the presence of the functional copy of the
clsA gene encoding cardiolipin synthase A and the envelope-responsive two-component system CpxA/R. The CpxA/R system, along with the RpoE sigma factor, responds to severe defects in LPS assembly and biosynthesis as well as protein misfolding in the periplasm [
13,
19,
39,
62]. We have previously shown that ClsA is required for the viability of strains synthesizing either penta- or tetraacylated lipid A, or when LapD assembly protein is missing [
15,
28]. Since LPS is presumably sensed in the inner membrane by LapC, the absence of ClsA could further accentuate this defect as it is required to assist MsbA-mediated LPS translocation. However, this requires further studies because the
clsA gene is per se essential in
lapC190 bacteria. Our gene expression studies did not show that SrrA regulates transcription of the
clsA gene based on q-RT-PCR results. Thus, we are initiating a global transcriptosome study to identify genes that are regulated by SrrA, and initial results, which require further in-depth experimentation, suggest that SrrA acts as a transcriptional repressor of the envelope stress response and fatty acid metabolism in
E.
coli.
Another important finding from this study is the partial overlap between LapD and LapC functions in the regulation of LpxC and LPS levels. This is based on the following results: (i) Some of the multicopy suppressors identified in this work that relieve the Ts phenotype of
lapC190 bacteria were earlier also identified as suppressors of Δ
lapD. These include
dksA,
srrA,
acpP,
accB and
yfgM [
28]. (ii) Of significance is the isolation of the
lapD gene as a multicopy suppressor of
lapC mutant bacteria for the Ts phenotype. We recently proposed that LapD could also act upstream of LapB-FtsH in a manner similar to LapC, since suppressors mapping to
lapB,
ftsH and
lpxC that stabilize LpxC, can suppress Δ
lapD and
lapC190 bacteria. (iii) However, some suppressors, like the
marA gene and the
acpT gene cannot relieve the Ts phenotype of Δ
lapD bacteria upon their overexpression and are specific to
lapC mutant bacteria. Consistent with these results, the overexpression of the
lapC gene does not relieve the Ts phenotype of Δ
lapD bacteria. These results are not surprising because, at the biochemical level, despite the LapC and LapD co-purification with LapB, several other interacting partners of LapD are unique to it and were not found to be part of the LapC complex [
6,
24,
28].
To further understand LapD-mediated multicopy suppression of
lapC mutant bacteria, we constructed a series of mutations in the
lapD gene to identify the essential domains of the encoded protein. This allowed us to express such mutant versions in a controlled manner from a regulated inducible promoter in
lapC mutant bacteria. This revealed that the N-terminal membrane anchor and the last 32 conserved C-terminal amino residues are critical for LapD function. Furthermore, several residues that were predicted for LapD to interact with its partners [
50] were also found to be essential for its function since their replacement by Ala amino acid residues abolished LapD function (inability to suppress the Ts phenotype of either Δ
lapD or
lapC190 mutant bacteria). This includes a motif constituted by the N93 P94 F96 triplet, which we show is essential for the LapD function and has been predicted to interact with LapA. This conserved NFP motif is located after the long cytoplasmic helical domain.
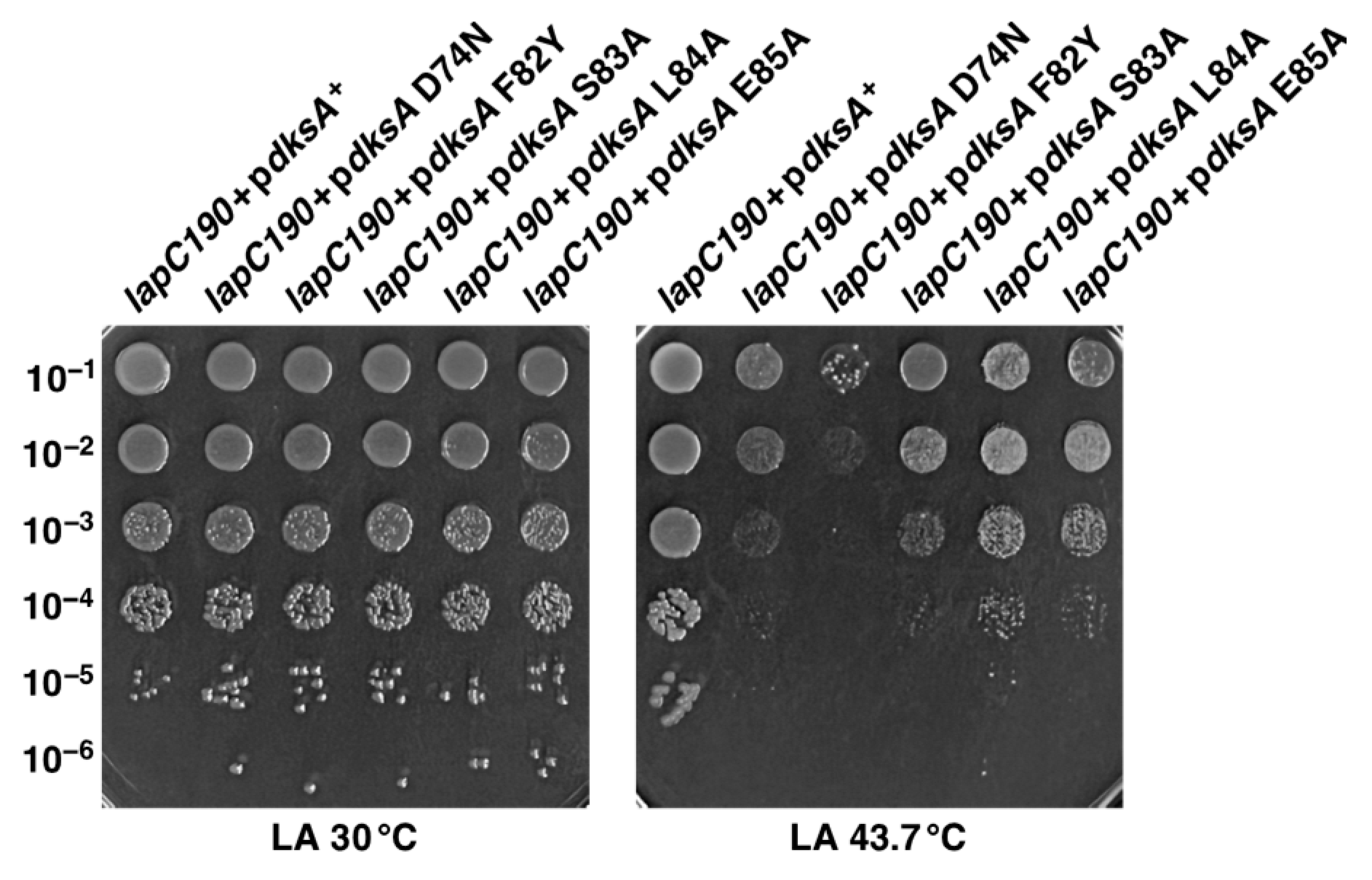
Regarding the mechanism of suppression by overproduction of the global transcriptional regulator DksA, we show that, most critically, its PPIase activity is required for this activity. However, the replacement of conserved amino acid residues in the tip region of the coiled-coil domain of DksA which are known to be essential for its role in interaction with RNA polymerase and hence the transcriptional regulation caused a highly attenuated ability to confer suppressing ability of
lapC190 bacteria. However, the most severe phenotype was the replacement of F82 by Y, which resulted in the total abolishment of the multicopy-suppressing ability of DksA. This amino acid residue is important for conferring the PPIase activity of DksA but is not essential for transcriptional function [
36]. Hence, the PPIase activity of DksA is critical for its multicopy-suppressing ability, and its transcriptional activity also plays an important role. Further studies are required to determine the mechanism(s) of the restoration of growth at elevated temperatures by other multicopy suppressors that were identified in this study.
It would be interesting to determine whether the suppression by
gnsA overexpression is due to an increase in the unsaturated fatty acid synthesis that could lead to the stabilization of LpxC. The
gnsA gene was initially identified as a multicopy suppressor of the
secG null mutant and was shown to increase the acidic phospholipid content and inhibit phosphatidylethanolamine accumulation via an unknown mechanism [
63]. Furthermore, overexpression of the
gnsA gene also suppresses the Ts phenotype of
fabA6 mutation and causes an increase in the unsaturated fatty acid content, such as
cis-vaccenic acid (18:1), at the expense of palmitic acid (16:0), particularly at low temperatures [
64]. At present, the molecular basis of the suppression of either
lapC190 bacteria or the
fabA6 Ts mutation or the ascribed increase in the unsaturated fatty acid content is not known. To this end, we have started to measure the impact of
gnsA overexpression on fatty acid and phospholipid content in various
lapC mutant bacteria. It is also intriguing why the central cofactor acyl carrier protein (AcpP) is limiting in
lapC190 bacteria. AcpP is the fourth most abundant protein in
E.
coli, its excess is usually toxic in wild-type bacteria [
65], and yet its mild induction quite effectively overcomes the various growth defects of
lapC mutant bacteria without increasing LpxC stability or restoring LPS amounts. AcpP is involved in not only LPS and phospholipid biosynthesis, but its interactome contains more than 35 proteins, some of which are unrelated to LPS and fatty acid synthesis [
66]. Since the replacement of catalytically active site residues confers a toxic growth phenotype due to the accumulation of
apo-ACP [
67], we could not address if the catalytic activity of AcpP is required for the suppressing phenotype. However, AcpP overproduction does not enhance either LPS or LpxC amounts, and thus, its action could be either rebalancing fatty acid composition or via some another mechanism. Isolation of other multicopy suppressors, such as the
yfgM gene, can be rationalized, since its product has been found to be a substrate of FtsH protease [
68]. Overexpression of the
yfgM gene could potentially titrate out FtsH, and this could lead to the stabilization of LpxC. The function of YceJ is currently unknown. However, the expression of the
yceJ gene is induced by a variety of stress conditions such as heat shock, the induction of extracytoplasmic responsive CpxR regulon and when the Lol system (lipoprotein sorting) is inhibited [
69]. Thus, YceJ may be involved in stress response management that could directly or indirectly modulate LpxC levels and hence more studies are required to address its function.
In summary, in this study, we identified new proteins, such as the transcriptional activator MarA, factors that are required to maintain OM asymmetry (PldA, Mla system), and the essentiality of cardiolipins which participate in the regulation of LPS assembly summarized in the
Figure 2. To date, MarA has not been shown to directly regulate LpxC stability, which we have now established here. Isolation of the
srrA gene as a multicopy suppressor is interesting because its overexpression stabilizes LpxC. We also identified essential amino acid residues that are required for LapD function and its partial overlap with LapC in regulating LpxC amounts.

 and
and 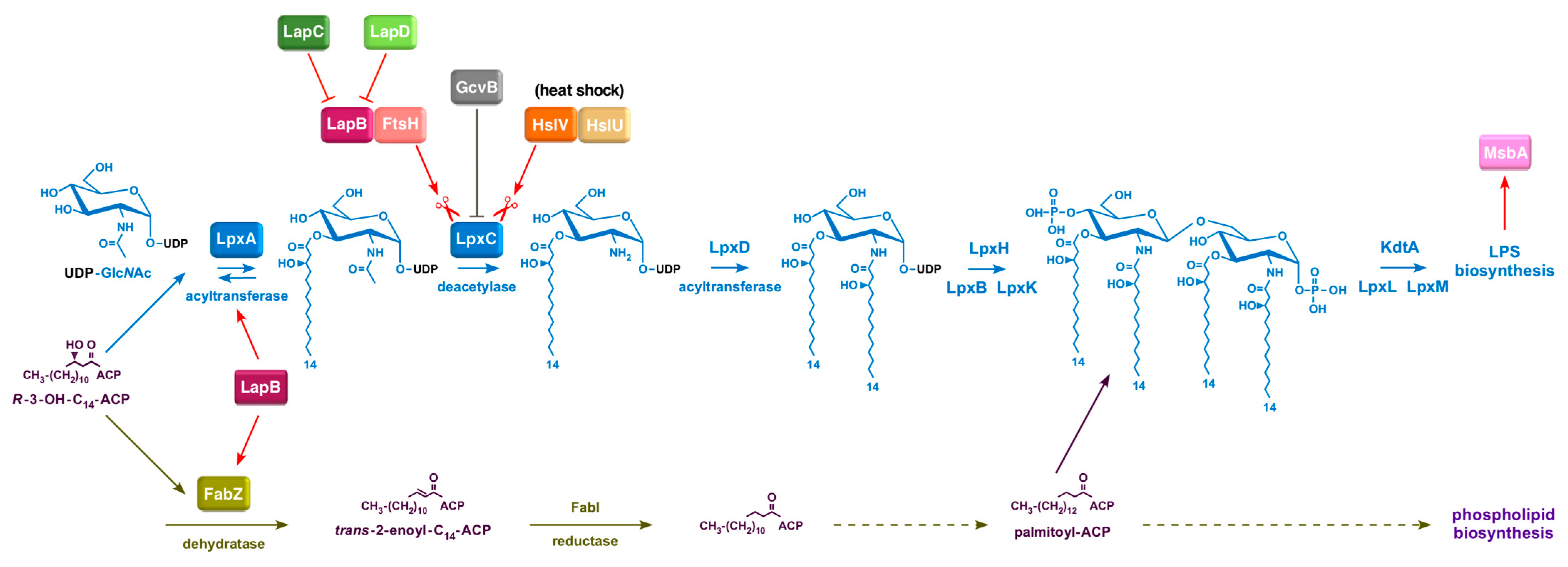









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}