
Interactions of Nucleosomes with Acidic Patch-Binding Peptides: A Combined Structural Bioinformatics, Molecular Modeling, Fluorescence Polarization, and Single-Molecule FRET Study
 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Analysis of Nucleosome Complexes from the PDB Database
2.1.1. Analysis of Nucleosome–Peptide Structures
2.1.2. Analysis of Nucleosome–Protein Structures
2.2. Molecular Dynamics Simulations of LANA1-22 and CENP-Cmotif Bound Nucleosomes
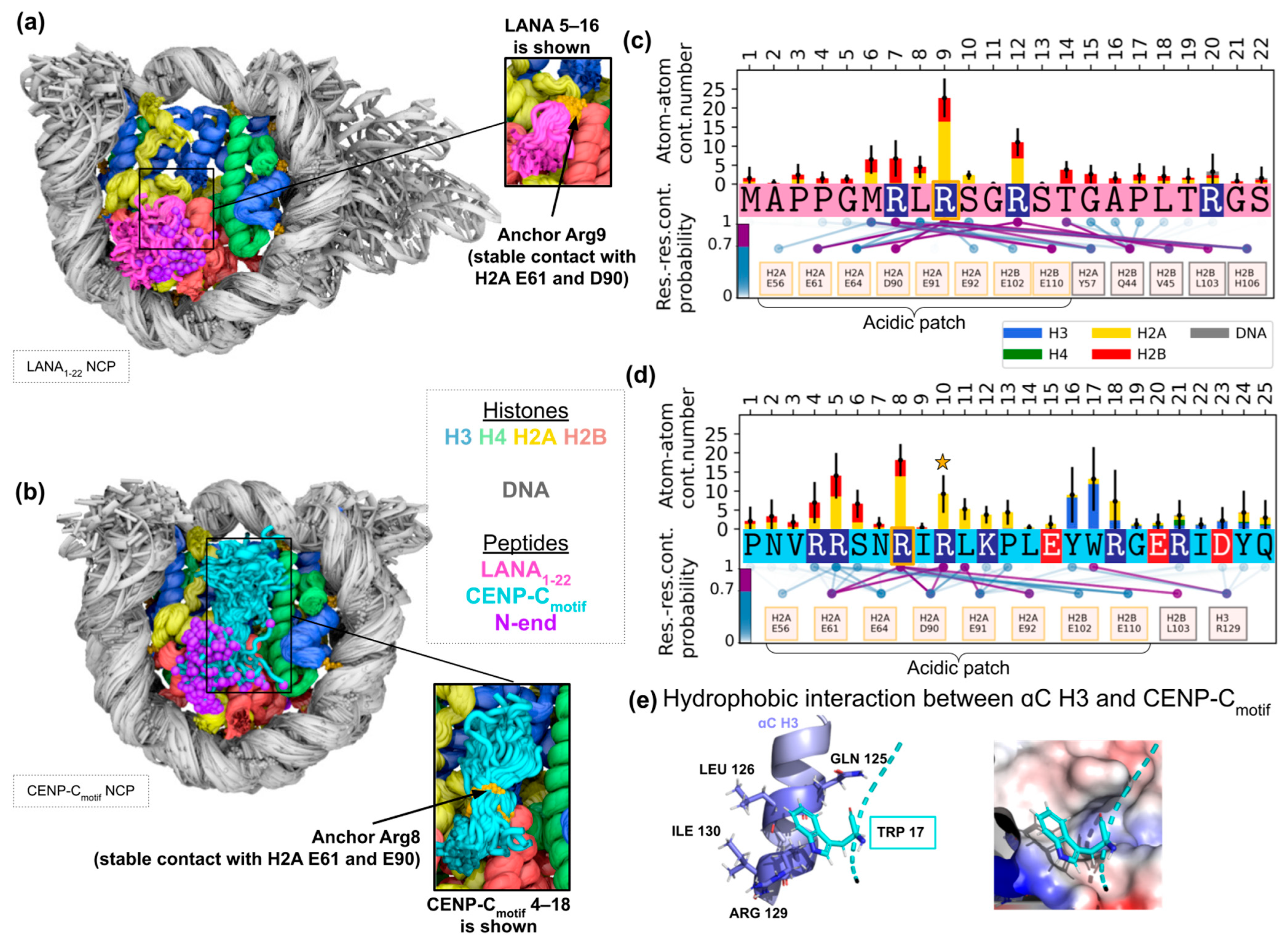
2.2.1. Characterization of Binding Dynamics and Stability of Contacts
2.2.2. Effects on Nucleosome Dynamics and Conformation
2.3. Histone Sequence Variants and Potential Effects on Peptide–Nucleosome Interactions
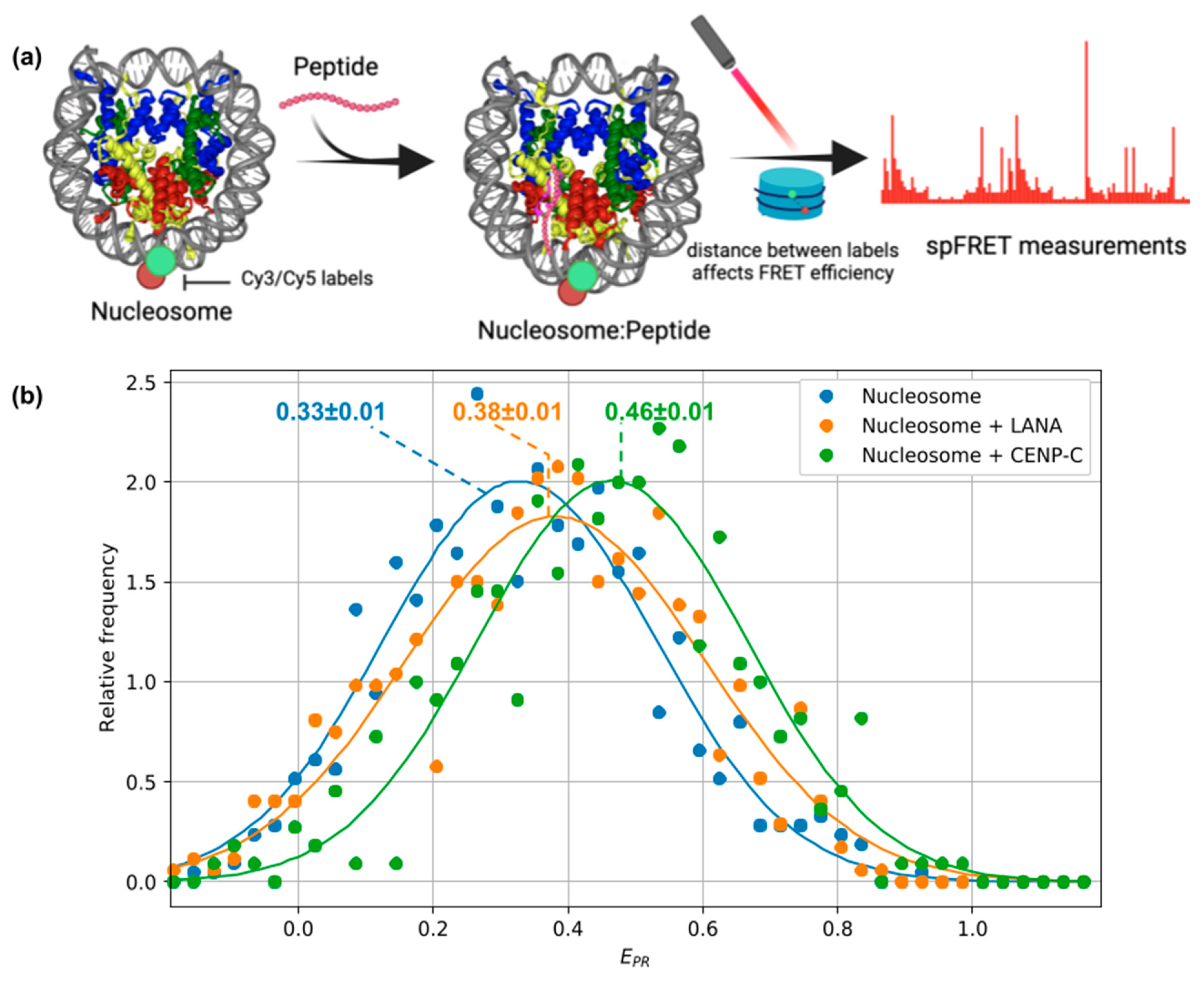
2.4. Experimental Estimation of Peptide-Nucleosome Interactions
2.5. Estimating Effects of Peptide Binding on Nucleosome Geometry Using Single-Molecule FRET
3. Discussion
4. Materials and Methods
4.1. PDB Structure Analysis
4.2. MD Simulations and Analysis
4.3. Bioinformatics Analysis of Histone Variants
4.4. Nucleosomal DNA Preparation
4.5. Histone Expression, Preparation of Histone Octamers
4.6. Nucleosome Assembly
4.7. LANA1-22 and CENP-Cmotif Peptides
4.8. Electrophoretic Assay for Peptide Binding
4.9. Fluorescence Polarization Studies
4.10. Single-Molecule FRET Analysis of Nucleosome–Peptide Interactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal Structure of the Nucleosome Core Particle at 2.8 A Resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Shaytan, A.K.; Xiao, H.; Armeev, G.A.; Wu, C.; Landsman, D.; Panchenko, A.R. Hydroxyl-Radical Footprinting Combined with Molecular Modeling Identifies Unique Features of DNA Conformation and Nucleosome Positioning. Nucleic Acids Res. 2017, 45, 9229–9243. [Google Scholar] [CrossRef]
- Armeev, G.A.; Gribkova, A.K.; Pospelova, I.; Komarova, G.A.; Shaytan, A.K. Linking Chromatin Composition and Structural Dynamics at the Nucleosome Level. Curr. Opin. Struct. Biol. 2019, 56, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone Post-Translational Modifications—Cause and Consequence of Genome Function. Nat. Rev. Genet. 2022, 203, 563–580. [Google Scholar] [CrossRef]
- Draizen, E.J.; Shaytan, A.K.; Mariño-Ramírez, L.; Talbert, P.B.; Landsman, D.; Panchenko, A.R. HistoneDB 2.0: A Histone Database with Variants—An Integrated Resource to Explore Histones and Their Variants. Database 2016, 2016, baw014. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Mueller-Planitz, F. Nucleosome Positioning and Spacing: From Mechanism to Function. J. Mol. Biol. 2021, 433, 166847. [Google Scholar] [CrossRef] [PubMed]
- Skrajna, A.; Goldfarb, D.; Kedziora, K.M.; Cousins, E.M.; Grant, G.D.; Spangler, C.J.; Barbour, E.H.; Yan, X.; Hathaway, N.A.; Brown, N.G.; et al. Comprehensive Nucleosome Interactome Screen Establishes Fundamental Principles of Nucleosome Binding. Nucleic Acids Res. 2020, 48, 9415–9432. [Google Scholar] [CrossRef] [PubMed]
- Kalashnikova, A.A.; Porter-Goff, M.E.; Muthurajan, U.M.; Luger, K.; Hansen, J.C. The Role of the Nucleosome Acidic Patch in Modulating Higher Order Chromatin Structure. J. R. Soc. Interface 2013, 10, 20121022. [Google Scholar] [CrossRef]
- Wagner, F.R.; Dienemann, C.; Wang, H.; Stützer, A.; Tegunov, D.; Urlaub, H.; Cramer, P. Structure of SWI/SNF Chromatin Remodeller RSC Bound to a Nucleosome. Nature 2020, 579, 448–451. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, K.; Zhang, W.; Chen, Z. Structure of Human Chromatin-Remodelling PBAF Complex Bound to a Nucleosome. Nature 2022, 605, 166–171. [Google Scholar] [CrossRef]
- He, S.; Wu, Z.; Tian, Y.; Yu, Z.; Yu, J.; Wang, X.; Li, J.; Liu, B.; Xu, Y. Structure of Nucleosome-Bound Human BAF Complex. Science 2020, 367, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.; Sun, M.; Liu, A.; Sun, Z.; Liu, T.; Cao, L.; Liang, L.; Qu, Q.; Li, Z.; Deng, Z.; et al. H2B Lys34 Ubiquitination Induces Nucleosome Distortion to Stimulate Dot1L Activity. Nat. Chem. Biol. 2022, 18, 972–980. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Henrici, R.C.; Tan, S. Crystal Structure of the PRC1 Ubiquitylation Module Bound to the Nucleosome. Nature 2014, 514, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dienemann, C.; Stützer, A.; Urlaub, H.; Cheung, A.C.M.; Cramer, P. Structure of the Transcription Coactivator SAGA. Nature 2020, 577, 717–720. [Google Scholar] [CrossRef]
- Ariyoshi, M.; Makino, F.; Watanabe, R.; Nakagawa, R.; Kato, T.; Namba, K.; Arimura, Y.; Fujita, R.; Kurumizaka, H.; Okumura, E.; et al. Cryo-EM Structure of the CENP-A Nucleosome in Complex with Phosphorylated CENP-C. EMBO J. 2021, 40, e105671. [Google Scholar] [CrossRef] [PubMed]
- Ali-Ahmad, A.; Bilokapić, S.; Schäfer, I.B.; Halić, M.; Sekulić, N. CENP-C Unwraps the Human CENP-A Nucleosome through the H2A C-terminal Tail. EMBO Rep. 2019, 20, e48913. [Google Scholar] [CrossRef]
- Makde, R.D.; England, J.R.; Yennawar, H.P.; Tan, S. Structure of RCC1 Chromatin Factor Bound to the Nucleosome Core Particle. Nature 2010, 467, 562–566. [Google Scholar] [CrossRef]
- Kato, H.; Van Ingen, H.; Zhou, B.-R.; Feng, H.; Bustin, M.; Kay, L.E.; Bai, Y. Architecture of the High Mobility Group Nucleosomal Protein 2-Nucleosome Complex as Revealed by Methyl-Based NMR. Proc. Natl. Acad. Sci. USA 2011, 108, 12283–12288. [Google Scholar] [CrossRef]
- Roussel, L.; Erard, M.; Cayrol, C.; Girard, J. Molecular Mimicry between IL-33 and KSHV for Attachment to Chromatin through the H2A–H2B Acidic Pocket. EMBO Rep. 2008, 9, 1006–1012. [Google Scholar] [CrossRef]
- Hirai, S.; Tomimatsu, K.; Miyawaki-Kuwakado, A.; Takizawa, Y.; Komatsu, T.; Tachibana, T.; Fukushima, Y.; Takeda, Y.; Negishi, L.; Kujirai, T.; et al. Unusual Nucleosome Formation and Transcriptome Influence by the Histone H3mm18 Variant. Nucleic Acids Res. 2022, 50, 72–91. [Google Scholar] [CrossRef]
- Lagadec, F.; Parissi, V.; Lesbats, P. Targeting the Nucleosome Acidic Patch by Viral Proteins: Two Birds with One Stone? mBio 2022, 13, e01733-21. [Google Scholar] [CrossRef]
- McGinty, R.K.; Tan, S. Recognition of the Nucleosome by Chromatin Factors and Enzymes. Curr. Opin. Struct. Biol. 2016, 37, 54–61. [Google Scholar] [CrossRef]
- Barbera, A.J.; Chodaparambil, J.V.; Kelley-Clarke, B.; Joukov, V.; Walter, J.C.; Luger, K.; Kaye, K.M. The Nucleosomal Surface as a Docking Station for Kaposi’s Sarcoma Herpesvirus LANA. Science 2006, 311, 856–861. [Google Scholar] [CrossRef]
- Kato, H.; Jiang, J.; Zhou, B.-R.; Rozendaal, M.; Feng, H.; Ghirlando, R.; Xiao, T.S.; Straight, A.F.; Bai, Y. A Conserved Mechanism for Centromeric Nucleosome Recognition by Centromere Protein CENP-C. Science 2013, 340, 1110–1113. [Google Scholar] [CrossRef]
- Fang, Q.; Chen, P.; Wang, M.; Fang, J.; Yang, N.; Li, G.; Xu, R.-M. Human Cytomegalovirus IE1 Protein Alters the Higher-Order Chromatin Structure by Targeting the Acidic Patch of the Nucleosome. eLife 2016, 5, e11911. [Google Scholar] [CrossRef]
- Teles, K.; Fernandes, V.; Silva, I.; Leite, M.; Grisolia, C.; Lobbia, V.R.; van Ingen, H.; Honorato, R.; Lopes-de-Oliveira, P.; Treptow, W.; et al. Nucleosome Binding Peptide Presents Laudable Biophysical and In Vivo Effects. Biomed. Pharmacother. 2020, 121, 109678. [Google Scholar] [CrossRef] [PubMed]
- Chodaparambil, J.V.; Barbera, A.J.; Lu, X.; Kaye, K.M.; Hansen, J.C.; Luger, K. A Charged and Contoured Surface on the Nucleosome Regulates Chromatin Compaction. Nat. Struct. Mol. Biol. 2007, 14, 1105–1107. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.D.; Benlekbir, S.; Fradet-Turcotte, A.; Sherker, A.; Julien, J.-P.; McEwan, A.; Noordermeer, S.M.; Sicheri, F.; Rubinstein, J.L.; Durocher, D. The Structural Basis of Modified Nucleosome Recognition by 53BP1. Nature 2016, 536, 100–103. [Google Scholar] [CrossRef]
- Nozaki, T.; Kanai, M. Chemical Catalysis Intervening to Histone Epigenetics. Acc. Chem. Res. 2021, 54, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Corbeski, I.; Horn, V.; van der Valk, R.A.; le Paige, U.B.; Dame, R.T.; van Ingen, H. Microscale Thermophoresis Analysis of Chromatin Interactions. In Bacterial Chromatin: Methods and Protocols; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1837, pp. 177–197. [Google Scholar] [CrossRef]
- Beauchemin, C.; Moerke, N.J.; Faloon, P.; Kaye, K.M. Assay Development and High-Throughput Screening for Inhibitors of Kaposi’s Sarcoma–Associated Herpesvirus N-Terminal Latency-Associated Nuclear Antigen Binding to Nucleosomes. J. Biomol. Screen. 2014, 19, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Novikov, R.V.; Bondarenko, E.A.; Malyuchenko, N.V.; Feofanov, A.V.; Studitsky, V.M.; Shaytan, A.K. Determining the Binding Constant of LANA Protein Fragment with Nucleosome. Mosc. Univ. Biol. Sci. Bull. 2020, 75, 252–256. [Google Scholar] [CrossRef]
- Armeev, G.A.; Kniazeva, A.S.; Komarova, G.A.; Kirpichnikov, M.P.; Shaytan, A.K. Histone Dynamics Mediate DNA Unwrapping and Sliding in Nucleosomes: Insights from Multi-Microsecond Molecular Dynamics Simulations. Biophysics 2021, 12, 2387. [Google Scholar]
- Peng, Y.; Li, S.; Onufriev, A.; Landsman, D.; Panchenko, A.R. Binding of Regulatory Proteins to Nucleosomes Is Modulated by Dynamic Histone Tails. Nat. Commun. 2021, 12, 5280. [Google Scholar] [CrossRef]
- Kniazeva, A.S.; Armeev, G.A.; Shaytan, A.K. H2A-H2B Histone Dimer Plasticity and Its Functional Implications. Cells 2022, 11, 2837. [Google Scholar] [CrossRef]
- Seal, R.L.; Denny, P.; Bruford, E.A.; Gribkova, A.K.; Landsman, D.; Marzluff, W.F.; McAndrews, M.; Panchenko, A.R.; Shaytan, A.K.; Talbert, P.B. A Standardized Nomenclature for Mammalian Histone Genes. Epigenetics Chromatin 2022, 15, 34. [Google Scholar] [CrossRef]
- Doyen, C.-M.; Montel, F.; Gautier, T.; Menoni, H.; Claudet, C.; Delacour-Larose, M.; Angelov, D.; Hamiche, A.; Bednar, J.; Faivre-Moskalenko, C.; et al. Dissection of the Unusual Structural and Functional Properties of the Variant H2A.Bbd Nucleosome. EMBO J. 2006, 25, 4234–4244. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, T.A.; Parker, B.J.; Nekrasov, M.; Hart-Smith, G.; Tay, Y.J.; Tng, W.Q.; Wilkins, M.; Ryan, D.; Tremethick, D.J. A New Link between Transcriptional Initiation and Pre-MRNA Splicing: The RNA Binding Histone Variant H2A.B. PLoS Genet. 2017, 13, 1006633. [Google Scholar] [CrossRef]
- Molaro, A.; Young, J.M.; Malik, H.S. Evolutionary Origins and Diversification of Testis-Specific Short Histone H2A Variants in Mammals. Genome Res. 2018, 28, 460–473. [Google Scholar] [CrossRef]
- Zambrano-Mila, M.S.; Aldaz-Villao, M.J.; Armando Casas-Mollano, J. Canonical Histones and Their Variants in Plants: Evolution and Functions. In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications; Alvarez-Venegas, R., De-la-Peña, C., Casas-Mollano, J.A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 185–222. ISBN 978-3-030-14759-4. [Google Scholar]
- Ueda, K.; Suzuki, M.; Ono, M.; Ide, N.; Tanaka, I.; Inoue, M. Male Gametic Cell-Specific Histone GH2A Gene of Lilium Longiflorum: Genomic Structure and Promoter Activity in the Generative Cell. Plant Mol. Biol. 2005, 59, 229–238. [Google Scholar] [CrossRef]
- Kudryashova, K.S.; Chertkov, O.V.; Nikitin, D.V.; Pestov, N.A.; Kulaeva, O.I.; Efremenko, A.V.; Solonin, A.S.; Kirpichnikov, M.P.; Studitsky, V.M.; Feofanov, A.V. Preparation of Mononucleosomal Templates for Analysis of Transcription with RNA Polymerase Using SpFRET. In Chromatin Protocols; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1288, pp. 395–412. [Google Scholar] [CrossRef]
- Valieva, M.E.; Armeev, G.A.; Kudryashova, K.S.; Gerasimova, N.S.; Shaytan, A.K.; Kulaeva, O.I.; McCullough, L.L.; Formosa, T.; Georgiev, P.G.; Kirpichnikov, M.P.; et al. Large-Scale ATP-Independent Nucleosome Unfolding by a Histone Chaperone. Nat. Struct. Mol. Biol. 2016, 23, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Joo, C.; Ha, T. Single-Molecule FRET with Total Internal Reflection Microscopy. Cold Spring Harb. Protoc. 2012, 2012, pdb.top072058. [Google Scholar] [CrossRef] [PubMed]
- Thåström, A.; Bingham, L.M.; Widom, J. Nucleosomal Locations of Dominant DNA Sequence Motifs for Histone-DNA Interactions and Nucleosome Positioning. J. Mol. Biol. 2004, 338, 695–709. [Google Scholar] [CrossRef]
- Li, G.; Levitus, M.; Bustamante, C.; Widom, J. Rapid Spontaneous Accessibility of Nucleosomal DNA. Nat. Struct. Mol. Biol. 2005, 12, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Falk, S.J.; Guo, L.Y.; Sekulic, N.; Smoak, E.M.; Mani, T.; Logsdon, G.A.; Gupta, K.; Jansen, L.E.T.; Van Duyne, G.D.; Vinogradov, S.A.; et al. CENP-C Reshapes and Stabilizes CENP-A Nucleosomes at the Centromere. Science 2015, 348, 699–703. [Google Scholar] [CrossRef]
- Falk, S.J.; Lee, J.; Sekulic, N.; Sennett, M.A.; Lee, T.H.; Black, B.E. CENP-C Directs a Structural Transition of CENP-A Nucleosomes Mainly through Sliding of DNA Gyres. Nat. Struct. Mol. Biol. 2016, 23, 204–208. [Google Scholar] [CrossRef]
- Davey, G.E.; Adhireksan, Z.; Ma, Z.; Riedel, T.; Sharma, D.; Padavattan, S.; Rhodes, D.; Ludwig, A.; Sandin, S.; Murray, B.S.; et al. Nucleosome Acidic Patch-Targeting Binuclear Ruthenium Compounds Induce Aberrant Chromatin Condensation. Nat. Commun. 2017, 8, 1575. [Google Scholar] [CrossRef]
- Amamoto, Y.; Aoi, Y.; Nagashima, N.; Suto, H.; Yoshidome, D.; Arimura, Y.; Osakabe, A.; Kato, D.; Kurumizaka, H.; Kawashima, S.A.; et al. Synthetic Posttranslational Modifications: Chemical Catalyst-Driven Regioselective Histone Acylation of Native Chromatin. J. Am. Chem. Soc. 2017, 139, 7568–7576. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.J.; Tóth, K.; Mücke, N.; Langowski, J.; Hakusui, A.S.; Olins, A.L.; Olins, D.E. Defining the Epichromatin Epitope. Nucleus 2017, 8, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Perez, A. Ranking Peptide Binders by Affinity with AlphaFold**. Angew. Chem. 2023, 135, e202213362. [Google Scholar] [CrossRef]
- Zsidó, B.Z.; Hetényi, C. Molecular Structure, Binding Affinity, and Biological Activity in the Epigenome. Int. J. Mol. Sci. 2020, 21, 4134. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Yasgar, A.; Peryea, T.; Braisted, J.C.; Jadhav, A.; Simeonov, A.; Coussens, N.P. Fluorescence Polarization Assays in High-Throughput Screening and Drug Discovery: A Review. Methods Appl. Fluoresc. 2016, 4, 022001. [Google Scholar] [CrossRef]
- Zhou, B.-R.; Yadav, K.N.S.; Borgnia, M.; Hong, J.; Cao, B.; Olins, A.L.; Olins, D.E.; Bai, Y.; Zhang, P. Atomic Resolution Cryo-EM Structure of a Native-like CENP-A Nucleosome Aided by an Antibody Fragment. Nat. Commun. 2019, 10, 2301. [Google Scholar] [CrossRef] [PubMed]
- Gansen, A.; Felekyan, S.; Kühnemuth, R.; Lehmann, K.; Tóth, K.; Seidel, C.A.M.; Langowski, J. High Precision FRET Studies Reveal Reversible Transitions in Nucleosomes between Microseconds and Minutes. Nat. Commun. 2018, 9, 4628. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, P.M.; Felthauser, A.; Fletcher, T.M.; Hansen, J.C. Reversible Oligonucleosome Self-Association: Dependence on Divalent Cations and Core Histone Tail Domains. Biochemistry 1996, 35, 4009–4015. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, V.A.; Steele, L.M.; Újvári, A.; Gaykalova, D.A.; Kulaeva, O.I.; Polikanov, Y.S.; Luse, D.S.; Studitsky, V.M. Nucleosomes Can Form a Polar Barrier to Transcript Elongation by RNA Polymerase II. Mol. Cell 2006, 24, 469–479. [Google Scholar] [CrossRef]
- Kujirai, T.; Kurumizaka, H. Transcription through the Nucleosome. Curr. Opin. Struct. Biol. 2020, 61, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Krithivas, A.; Young, D.B.; Liao, G.; Greene, D.; Hayward, S.D. Human Herpesvirus 8 LANA Interacts with Proteins of the MSin3 Corepressor Complex and Negatively Regulates Epstein-Barr Virus Gene Expression in Dually Infected PEL Cells. J. Virol. 2000, 74, 9637–9645. [Google Scholar] [CrossRef]
- Renne, R.; Barry, C.; Dittmer, D.; Compitello, N.; Brown, P.O.; Ganem, D. Modulation of Cellular and Viral Gene Expression by the Latency-Associated Nuclear Antigen of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2001, 75, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Yamanashi, Y.; Fujimura, A.; Sato, Y.; Kujirai, T.; Kurumizaka, H.; Kimura, H.; Yamatsugu, K.; Kawashima, S.A.; Kanai, M. Live-Cell Epigenome Manipulation by Synthetic Histone Acetylation Catalyst System. Proc. Natl. Acad. Sci. USA 2021, 118, e2019554118. [Google Scholar] [CrossRef]
- Xiao, H.; Wang, F.; Wisniewski, J.; Shaytan, A.K.; Ghirlando, R.; FitzGerald, P.C.; Huang, Y.; Wei, D.; Li, S.; Landsman, D.; et al. Molecular Basis of CENP-C Association with the CENP-A Nucleosome at Yeast Centromeres. Genes Dev. 2017, 31, 1958–1972. [Google Scholar] [CrossRef] [PubMed]
- Mardian, J.K.W.; Paton, A.E.; Bunick, G.J.; Olins, D.E. Nucleosome Cores Have Two Specific Binding Sites for Nonhistone Chromosomal Proteins HMG 14 and HMG 17. Science 1980, 209, 1534–1536. [Google Scholar] [CrossRef] [PubMed]
- Armache, J.P.; Gamarra, N.; Johnson, S.L.; Leonard, J.D.; Wu, S.; Narlikar, G.J.; Cheng, Y. Cryo-EM Structures of Remodeler-Nucleosome Intermediates Suggest Allosteric Control through the Nucleosome. eLife 2019, 8, e46057. [Google Scholar] [CrossRef]
- Armeev, G.A.; Gribkova, A.K.; Shaytan, A.K. NucleosomeDB—A Database of 3D Nucleosome Structures and Their Complexes with Comparative Analysis Toolkit. bioRxiv 2023. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Ef, P.; Td, G.; Cc, H.; Gs, C.; Dm, G.; Ec, M.; Te, F. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. Available online: https://pubmed.ncbi.nlm.nih.gov/15264254/ (accessed on 10 September 2020).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–28. [Google Scholar] [CrossRef]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. NGL Viewer: Web-Based Molecular Graphics for Large Complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef]
- McDonald, I.K.; Thornton, J.M. Satisfying Hydrogen Bonding Potential in Proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent Mediated Interactions in the Structure of the Nucleosome Core Particle at 1.9 a Resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches? Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A Refined Force Field for DNA Simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Aksimentiev, A. New Tricks for Old Dogs: Improving the Accuracy of Biomolecular Force Fields by Pair-Specific Corrections to Non-Bonded Interactions. Phys. Chem. Chem. Phys. 2018, 20, 8432–8449. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. More Bang for Your Buck: Improved Use of GPU Nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Beitz, E. TEXshade: Shading and Labeling of Multiple Sequence Alignments Using LATEX2 Epsilon. Bioinformatics 2000, 16, 135–139. [Google Scholar] [CrossRef]
- Dyer, P.N.; Edayathumangalam, R.S.; White, C.L.; Bao, Y.; Chakravarthy, S.; Muthurajan, U.M.; Luger, K. Reconstitution of Nucleosome Core Particles from Recombinant Histones and DNA. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2003; Volume 375, pp. 23–44. ISBN 978-0-12-182779-3. [Google Scholar]
- Kudryashova, K.S.; Nikitin, D.V.; Chertkov, O.V.; Gerasimova, N.S.; Valieva, M.E.; Studitsky, V.M.; Feofanov, A.V. Development of Fluorescently Labeled Mononucleosomes for the Investigation of Transcription Mechanisms by Single Complex Microscopy. Mosc. Univ. Biol. Sci. Bull. 2015, 70, 189–193. [Google Scholar] [CrossRef]
- Valieva, M.; Gerasimova, N.; Kudryashova, K.; Kozlova, A.; Kirpichnikov, M.; Hu, Q.; Botuyan, M.; Mer, G.; Feofanov, A.; Studitsky, V. Stabilization of Nucleosomes by Histone Tails and by FACT Revealed by SpFRET Microscopy. Cancers 2017, 9, 3. [Google Scholar] [CrossRef]
- Klinker, H.; Haas, C.; Harrer, N.; Becker, P.B.; Mueller-Planitz, F. Rapid Purification of Recombinant Histones. PLoS ONE 2014, 9, e104029. [Google Scholar] [CrossRef]
- Gerasimova, N.S.; Korovina, A.N.; Afonin, D.A.; Shaytan, K.V.; Feofanov, A.V.; Studitsky, V.M. Analysis of Structure of Elongation Complexes in Polyacrylamide Gel with Förster Resonance Energy Transfer Technique. Biophysics 2022, 67, 165–170. [Google Scholar] [CrossRef]
- Moerke, N.J. Fluorescence Polarization (FP) Assays for Monitoring Peptide-Protein or Nucleic Acid-Protein Binding. Curr. Protoc. Chem. Biol. 2009, 1, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Roehrl, M.H.A.; Wang, J.Y.; Wagner, G. A General Framework for Development and Data Analysis of Competitive High-Throughput Screens for Small-Molecule Inhibitors of Protein−Protein Interactions by Fluorescence Polarization. Biochemistry 2004, 43, 16056–16066. [Google Scholar] [CrossRef]
- Newville, M.; Stensitzki, T.; Allen, D.B.; Ingargiola, A. LMFIT: Non-Linear Least-Square Minimization and Curve-Fitting for Python. Astrophys. Source Code Libr. 2014, ascl-1606.014. [Google Scholar]
- Ingargiola, A.; Lerner, E.; Chung, S.; Weiss, S.; Michalet, X. FRETBursts: An Open Source Toolkit for Analysis of Freely-Diffusing Single-Molecule FRET. PLoS ONE 2016, 11, e0160716. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oleinikov, P.D.; Fedulova, A.S.; Armeev, G.A.; Motorin, N.A.; Singh-Palchevskaia, L.; Sivkina, A.L.; Feskin, P.G.; Glukhov, G.S.; Afonin, D.A.; Komarova, G.A.; et al. Interactions of Nucleosomes with Acidic Patch-Binding Peptides: A Combined Structural Bioinformatics, Molecular Modeling, Fluorescence Polarization, and Single-Molecule FRET Study. Int. J. Mol. Sci. 2023, 24, 15194. https://doi.org/10.3390/ijms242015194
Oleinikov PD, Fedulova AS, Armeev GA, Motorin NA, Singh-Palchevskaia L, Sivkina AL, Feskin PG, Glukhov GS, Afonin DA, Komarova GA, et al. Interactions of Nucleosomes with Acidic Patch-Binding Peptides: A Combined Structural Bioinformatics, Molecular Modeling, Fluorescence Polarization, and Single-Molecule FRET Study. International Journal of Molecular Sciences. 2023; 24(20):15194. https://doi.org/10.3390/ijms242015194
Chicago/Turabian StyleOleinikov, Pavel D., Anastasiia S. Fedulova, Grigoriy A. Armeev, Nikita A. Motorin, Lovepreet Singh-Palchevskaia, Anastasiia L. Sivkina, Pavel G. Feskin, Grigory S. Glukhov, Dmitry A. Afonin, Galina A. Komarova, and et al. 2023. "Interactions of Nucleosomes with Acidic Patch-Binding Peptides: A Combined Structural Bioinformatics, Molecular Modeling, Fluorescence Polarization, and Single-Molecule FRET Study" International Journal of Molecular Sciences 24, no. 20: 15194. https://doi.org/10.3390/ijms242015194