
PANoptosis: Mechanism and Role in Pulmonary Diseases




Abstract
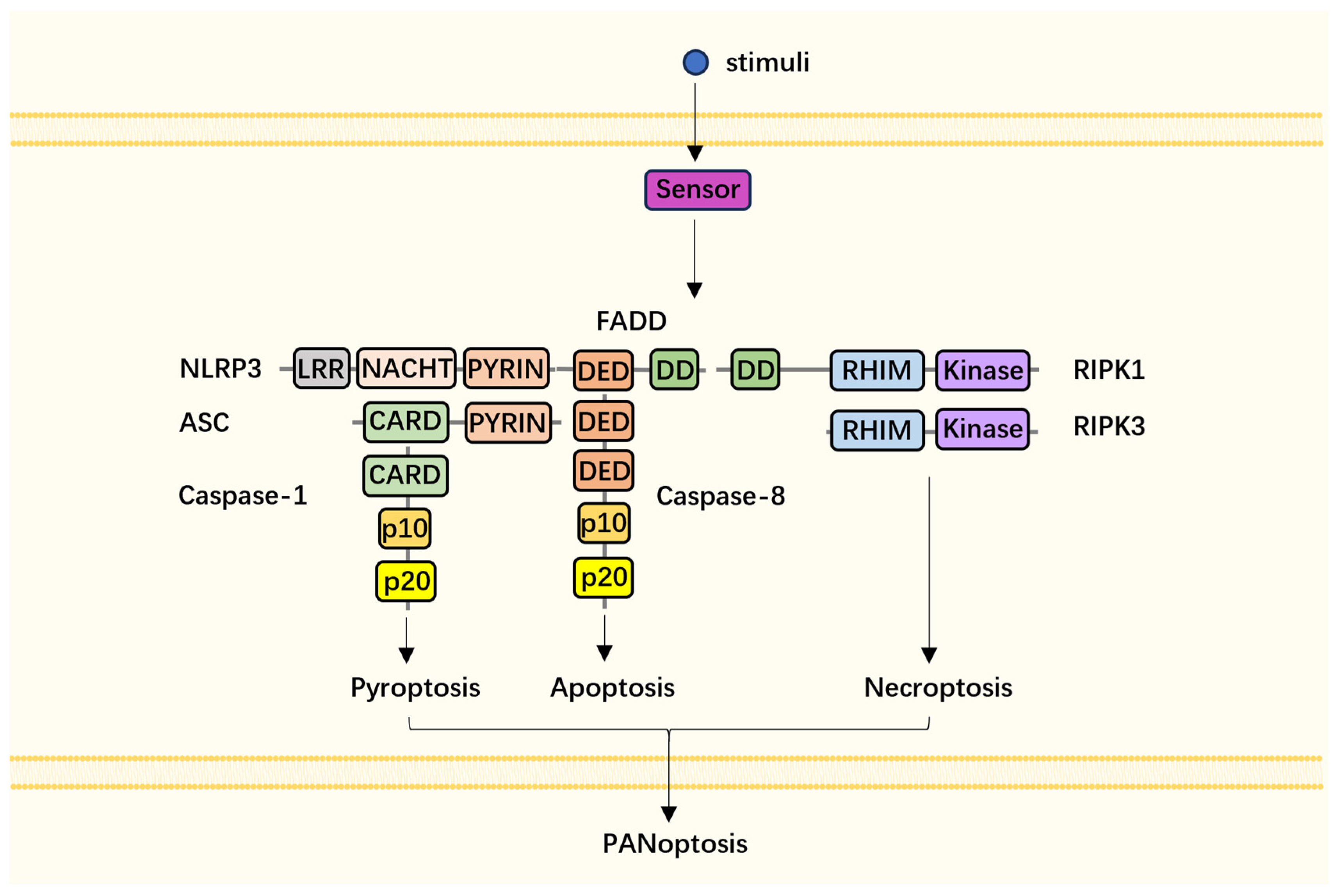
:1. Introduction
2. General Crosstalk between Apoptosis, Pyroptosis, and Necroptosis
3. Components and Assembly of PANoptosome
4. PANoptosome in Pulmonary Disease
4.1. Acute Lung Injury (ALI)/Acute Respiratory Distress Syndrome (ARDS)
4.2. Asthma
4.3. Idiopathic Pulmonary Fibrosis (IPF)
4.4. Chronic Obstructive Pulmonary Syndrome (COPD)
4.5. Lung Cancer
4.6. Other Pulmonary Diseases

{kind=link}
{kind=link}
| Involved PANoptosome Component | Trigger | Disease | Reference |
|---|---|---|---|
| ZBP1-PANoptosome | IAV, SARS-CoV-2, diABZI (STING agonist) | ALI/ARDS | [8,48,69] |
| RIPK1/3-FADD-caspase-8 | SARS-CoV-2 | ALI/ARDS | [6] |
| AIM2-PANoptosome | HSV-1, F. novicida | ALI/ARDS | [9] |
| RIPK1-PANoptosome | Y. pestis | ALI/ARDS | [5] |
| ZBP1 | self-mtDNA | SLE | [81,82,85] |
| NLRP3 | TDI | asthma | [89,90] |
| AIM2 | PFAS | asthma | [91,92] |
| NLRP3 | bleomycin | IPF | [104,105,106] |
| RIPK1, NLRP3 | CSE | COPD | [115,116,119,120] |
| ZBP1-PANoptosome | ADAR1 knockdown or IFNs plus nuclear transport inhibitors KPT-330 | cancer | [135,136] |
5. Potential Inhibitors of PANoptosome
| Target | Compound | Potential Mechanism | Reference |
|---|---|---|---|
| GSDMD | Necrosulfonamide | binds to Cys191 of human GSDMD, inhibits GSDMD-NT oligomerization | [151] |
| LDC7559/2618 | unknown | [152] | |
| Disulfiram | binds to Cys191 of human GSDMD | [153] | |
| Dimethyl fumarate | binds to Cys192 of mouse GSDMD | [154] | |
| MLKL | GW806742X | binds to nucleotide-binding site of the MLKL, suppresses the conformational change of MLKL, and retards MLKL membrane translocation | [156] |
| NBC1 | covalently conjugates cysteine of HSP70 to block its protein chaperone function and inhibits MLKL polymerization | [157] | |
| NSA | binds to Cys86 of human MLKL and blocks its translocation to the cell membrane | [158] | |
| RIPK1 | GSK481/547/963 | inhibits Ser166 phosphorylation of RIPK1 | [159] |
| Nec-1 | inhibits kinase domain of RIPK1 | [161] | |
| RIPA-56 | inhibits kinase domain of RIPK1 | [159] | |
| RIPK3 | GSK872 | binds to kinase domain of RIPK3 | [160] |
| Caspase proteins | qVD-OPh | substrate analogue that binds to catalytic units | [119] |
| Z-VAD-FMK | substrate analogue that binds to catalytic units | [162] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Kesavardhana, S.; Karki, R.; Kancharana, B.; Burton, A.R.; Kanneganti, T.D. RIPK1 Distinctly Regulates Yersinia-Induced Inflammatory Cell Death, PANoptosis. Immunohorizons 2020, 4, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef]
- Karki, R.; Lee, S.; Mall, R.; Pandian, N.; Wang, Y.; Sharma, B.R.; Malireddi, R.S.; Yang, D.; Trifkovic, S.; Steele, J.A.; et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci. Immunol. 2022, 7, eabo6294. [Google Scholar] [CrossRef]
- Messaoud-Nacer, Y.; Culerier, E.; Rose, S.; Maillet, I.; Rouxel, N.; Briault, S.; Ryffel, B.; Quesniaux, V.F.J.; Togbe, D. STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS). Cell Death Dis. 2022, 13, 269. [Google Scholar] [CrossRef]
- Lee, S.; Karki, R.; Wang, Y.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 2021, 597, 415–419. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.-N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Lupfer, C.R.; Anand, P.K.; Liu, Z.; Stokes, K.L.; Vogel, P.; Lamkanfi, M.; Kanneganti, T.D. Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical NLRP3 inflammasome during enteric pathogen infection. PLoS Pathog. 2014, 10, e1004410. [Google Scholar] [CrossRef] [PubMed]
- Zasłona, Z.; Flis, E.; Wilk, M.M.; Carroll, R.G.; Palsson-McDermott, E.M.; Hughes, M.M.; Diskin, C.; Banahan, K.; Ryan, D.G.; Hooftman, A.; et al. Caspase-11 promotes allergic airway inflammation. Nat. Commun. 2020, 11, 1055. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef]
- Vercammen, D.; Brouckaert, G.; Denecker, G.; Van de Craen, M.; Declercq, W.; Fiers, W.; Vandenabeele, P. Dual Signaling of the Fas Receptor: Initiation of Both Apoptotic and Necrotic Cell Death Pathways. J. Exp. Med. 1998, 188, 919–930. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.-L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef]
- Maelfait, J.; Liverpool, L.; Bridgeman, A.; Ragan, K.B.; Upton, J.W.; Rehwinkel, J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017, 36, 2529–2543. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Wachsmuth, L.; Kumari, S.; Schwarzer, R.; Lin, J.; Eren, R.O.; Fisher, A.; Lane, R.; Young, G.R.; Kassiotis, G.; et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 2020, 580, 391–395. [Google Scholar] [CrossRef]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli-Jain, P.; Tait, S.W.G.; Gale, M., Jr.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1β Independently of Gasdermin-D. J. Immunol. 2017, 198, 2156–2164. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Demarco, B.; Grayczyk, J.P.; Bjanes, E.; Le Roy, D.; Tonnus, W.; Assenmacher, C.-A.; Radaelli, E.; Fettrelet, T.; Mack, V.; Linkermann, A.; et al. Caspase-8–dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci. Adv. 2020, 6, eabc3465. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol. 2017, 24, 507–514.e504. [Google Scholar] [CrossRef]
- He, K.; Wan, T.; Wang, D.; Hu, J.; Zhou, T.; Tao, W.; Wei, Z.; Lu, Q.; Zhou, R.; Tian, Z.; et al. Gasdermin D licenses MHCII induction to maintain food tolerance in small intestine. Cell 2023, 186, 3033–3048.E20. [Google Scholar] [CrossRef]
- Li, M.; Yang, D.; Yan, H.; Tang, Z.; Jiang, D.; Zhang, J.; Chi, Z.; Nie, W.; Zhen, W.; Yu, W.; et al. Gasdermin D maintains bone mass by rewiring the endo-lysosomal pathway of osteoclastic bone resorption. Dev. Cell 2022, 57, 2365–2380.e2368. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Thi Nguyen, D.; Hattori, T.; Manh Le, T.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef] [PubMed]
- Rosalie, H.; Marisa, D.; Dave, B.; Kaiwen, W.C.; Dora, H.; Benjamin, D.; Kateryna, S.; Petr, B. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci. Alliance 2020, 3, e202000735. [Google Scholar] [CrossRef]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486.e475. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kanneganti, T.-D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput. Struct. Biotechnol. J. 2021, 19, 4641–4657. [Google Scholar] [CrossRef]
- Herbert, A. Z-DNA and Z-RNA in human disease. Commun. Biol. 2019, 2, 7. [Google Scholar] [CrossRef]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef]
- Pothlichet, J.; Meunier, I.; Davis, B.K.; Ting, J.P.; Skamene, E.; von Messling, V.; Vidal, S.M. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 2013, 9, e1003256. [Google Scholar] [CrossRef]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef]
- Szczesny, B.; Marcatti, M.; Ahmad, A.; Montalbano, M.; Brunyánszki, A.; Bibli, S.-I.; Papapetropoulos, A.; Szabo, C. Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci. Rep. 2018, 8, 914. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.-D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687.e613. [Google Scholar] [CrossRef] [PubMed]
- Henry, T.; Brotcke, A.; Weiss, D.S.; Thompson, L.J.; Monack, D.M. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 2007, 204, 987–994. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Sasai, M.; Place, D.E.; Kesavardhana, S.; Temirov, J.; Frase, S.; Zhu, Q.; Malireddi, R.K.S.; Kuriakose, T.; et al. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 2016, 167, 382–396. [Google Scholar] [CrossRef]
- Meunier, E.; Wallet, P.; Dreier, R.F.; Costanzo, S.; Anton, L.; Ruhl, S.; Dussurgey, S.; Dick, M.S.; Kistner, A.; Rigard, M.; et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol. 2015, 16, 476–484. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Malireddi, R.K.; Neale, G.; Vogel, P.; Yamamoto, M.; Lamkanfi, M.; Kanneganti, T.D. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol. 2015, 16, 467–475. [Google Scholar] [CrossRef]
- Kist, M.; Kőműves, L.G.; Goncharov, T.; Dugger, D.L.; Yu, C.; Roose-Girma, M.; Newton, K.; Webster, J.D.; Vucic, D. Impaired RIPK1 ubiquitination sensitizes mice to TNF toxicity and inflammatory cell death. Cell Death Differ. 2021, 28, 985–1000. [Google Scholar] [CrossRef]
- Mihaly, S.R.; Ninomiya-Tsuji, J.; Morioka, S. TAK1 control of cell death. Cell Death Differ. 2014, 21, 1667–1676. [Google Scholar] [CrossRef]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef]
- Liu, X.; Tang, A.L.; Chen, J.; Gao, N.; Zhang, G.; Xiao, C. RIPK1 in the inflammatory response and sepsis: Recent advances, drug discovery and beyond. Front. Immunol. 2023, 14, 1114103. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020, 217, 1644. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Gurung, P.; Mavuluri, J.; Dasari, T.K.; Klco, J.M.; Chi, H.; Kanneganti, T.D. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 2018, 215, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, R.; Tomii, K.; Seo, R.; Nagata, K.; Otsuka, K.; Nakagawa, A.; Otsuka, K.; Hashimoto, H.; Watanabe, K.; Shimizu, N. Detection of herpes viruses by multiplex and real-time polymerase chain reaction in bronchoalveolar lavage fluid of patients with acute lung injury or acute respiratory distress syndrome. Respiration 2014, 87, 279–286. [Google Scholar] [CrossRef]
- Groeneveld, A.B.; Vandenbroucke-Grauls, C.M. One swallow does not make a summer: Can herpes simplex virus-1 cause pneumonia and acute lung injury? Am. J. Respir. Crit. Care Med. 2007, 175, 865–866. [Google Scholar] [CrossRef]
- Lim, H.K.; Huang, S.X.L.; Chen, J.; Kerner, G.; Gilliaux, O.; Bastard, P.; Dobbs, K.; Hernandez, N.; Goudin, N.; Hasek, M.L.; et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 2019, 216, 2038–2056. [Google Scholar] [CrossRef]
- Labbé, K.; Saleh, M. Cell death in the host response to infection. Cell Death Differ. 2008, 15, 1339–1349. [Google Scholar] [CrossRef]
- Simpson, D.S.; Pang, J.; Weir, A.; Kong, I.Y.; Fritsch, M.; Rashidi, M.; Cooney, J.P.; Davidson, K.C.; Speir, M.; Djajawi, T.M.; et al. Interferon-γ primes macrophages for pathogen ligand-induced killing via a caspase-8 and mitochondrial cell death pathway. Immunity 2022, 55, 423–441.e429. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Ye, M.; Li, H.; You, M.; Zhou, Z.; Zhang, C.; Zhang, F.; Lu, B.; et al. SARS-CoV-2 Z-RNA activates the ZBP1-RIPK3 pathway to promote virus-induced inflammatory responses. Cell Res. 2023, 33, 201–214. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, M.; Huang, J.; Zeng, Q.; Zhu, Q.; Xu, S.; Chen, H. H1N1 Influenza A Virus Protein NS2 Inhibits Innate Immune Response by Targeting IRF7. Viruses 2022, 14, 2411. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fang, P.; He, R.; Li, M.; Yu, H.; Zhou, L.; Yi, Y.; Wang, F.; Rong, Y.; Zhang, Y.; et al. O-GlcNAc transferase promotes influenza A virus–induced cytokine storm by targeting interferon regulatory factor–5. Sci. Adv. 2020, 6, eaaz7086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Song, J.W.; Huang, H.H.; Fan, X.; Huang, L.; Deng, J.N.; Tu, B.; Wang, K.; Li, J.; Zhou, M.J.; et al. NLRP3 inflammasome induces CD4+ T cell loss in chronically HIV-1-infected patients. J. Clin. Investig. 2021, 131, e138861. [Google Scholar] [CrossRef]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; Zepeda, O.; Yang, Z.; Santiago, M.L.; Hebbeler, A.M.; Greene, W.C. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Blot, F.; Herrmann, J.L.; Brunengo, P.; Marsal, L.; Bekka, R.; Lang, M.P.; Laaban, J.P. Septic shock and adult respiratory distress syndrome due to Listeria monocytogenes. Intensive Care Med. 1994, 20, 83–84. [Google Scholar] [CrossRef]
- Grousd, J.A.; Rich, H.E.; Alcorn, J.F. Host-Pathogen Interactions in Gram-Positive Bacterial Pneumonia. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef]
- Ugas, M.B.; Carroll, T.; Kovar, L.; Chavez-Bueno, S. Salmonella Typhi-Induced Septic Shock and Acute Respiratory Distress Syndrome in a Previously Healthy Teenage Patient Treated With High-Dose Dexamethasone. J. Investig. Med. High Impact Case Rep. 2016, 4, 2324709616652642. [Google Scholar] [CrossRef]
- Nizet, V. Bacteria and phagocytes: Mortal enemies. J. Innate Immun. 2010, 2, 505–507. [Google Scholar] [CrossRef]
- Underhill, D.M.; Goodridge, H.S. Information processing during phagocytosis. Nat. Rev. Immunol. 2012, 12, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.J.; Arruda, A.; Reyes, C.N.; Kaplan, A.T.; Shimada, T.; Shimada, K.; Arditi, M.; Liu, G.; Underhill, D.M. Phagosomal degradation increases TLR access to bacterial ligands and enhances macrophage sensitivity to bacteria. J. Immunol. 2011, 187, 6002–6010. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Guan, Y.; Lv, M.; Zhang, R.; Guo, Z.; Wei, X.; Du, X.; Yang, J.; Li, T.; Wan, Y.; et al. Manganese Increases the Sensitivity of the cGAS-STING Pathway for Double-Stranded DNA and Is Required for the Host Defense against DNA Viruses. Immunity 2018, 48, 675–687.e677. [Google Scholar] [CrossRef]
- Caielli, S.; Cardenas, J.; de Jesus, A.A.; Baisch, J.; Walters, L.; Blanck, J.P.; Balasubramanian, P.; Stagnar, C.; Ohouo, M.; Hong, S.; et al. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human SLE. Cell 2021, 184, 4464–4479.e4419. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.S.; Kang, Y.; Lee, N.; Wahl, E.R.; Kim, S.H.; Kang, K.S.; Lazova, R.; Kang, I. Self double-stranded (ds)DNA induces IL-1β production from human monocytes by activating NLRP3 inflammasome in the presence of anti-dsDNA antibodies. J. Immunol. 2013, 190, 1407–1415. [Google Scholar] [CrossRef]
- Sharapova, T.N.; Romanova, E.A.; Soshnikova, N.V.; Belogurov, A.A., Jr.; Lomakin, Y.A.; Sashchenko, L.P.; Yashin, D.V. Autoantibodies from SLE patients induce programmed cell death in murine fibroblast cells through interaction with TNFR1 receptor. Sci. Rep. 2020, 10, 11144. [Google Scholar] [CrossRef]
- Zhang, M.; Jie, H.; Wu, Y.; Han, X.; Li, X.; He, Y.; Shi, X.; Luo, Y.; Sun, Y.; Yang, J.; et al. Increased MLKL mRNA level in the PBMCs is correlated with autoantibody production, renal involvement, and SLE disease activity. Arthritis Res. Ther. 2020, 22, 239. [Google Scholar] [CrossRef]
- Yuanjiu, L.; Jordyn, J.V.; Yi-Fan, C.; Joshua, D.B.; Ying, L.; Danielle, F.; Sylvia, T.-O.; Katherine, B.R.; Jingti, D.; Armaan, M.; et al. Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. bioRxiv 2023, 2022, 493783. [Google Scholar] [CrossRef]
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis. Primers 2015, 1, 15025. [Google Scholar] [CrossRef]
- Song, J.; Wang, J. SIRT3 regulates bronchial epithelium apoptosis and aggravates airway inflammation in asthma. Mol. Med. Rep. 2022, 25, 144. [Google Scholar] [CrossRef]
- Zhou, C.; Yin, G.; Liu, J.; Liu, X.; Zhao, S. Epithelial apoptosis and loss in airways of children with asthma. J. Asthma 2011, 48, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Cui, H.; Zhuang, L.; Zhai, Z.; Yang, F.; Luo, G.; He, J.; Zhao, H.; Zhao, W.; He, Y.; et al. Bronchial epithelial pyroptosis promotes airway inflammation in a murine model of toluene diisocyanate-induced asthma. Biomed. Pharmacother. 2020, 125, 109925. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Christenson, S.A.; Modena, B.; Li, H.; Busse, W.W.; Castro, M.; Denlinger, L.C.; Erzurum, S.C.; Fahy, J.V.; Gaston, B.; et al. Genetic analyses identify GSDMB associated with asthma severity, exacerbations, and antiviral pathways. J. Allergy Clin. Immunol. 2021, 147, 894–909. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Guo, Y.; Zhou, Q.; Wang, Q.; Pan, S.; Xu, S.; Li, L. Perfluoroalkyl substance exposure is associated with asthma and innate immune cell count in US adolescents stratified by sex. Environ. Sci. Pollut. Res. 2023, 30, 52535–52548. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Q.; Liu, T.; Yang, S.; Sun, L.; Zhao, Z.Y.; Li, L.Y.; She, Y.C.; Zheng, Y.Y.; Ye, X.Y.; Bao, Q.; et al. Perfluoroalkyl substance pollutants activate the innate immune system through the AIM2 inflammasome. Nat. Commun. 2021, 12, 2915. [Google Scholar] [CrossRef]
- He, A.; Chen, J.; Guan, J.; Huang, Y.; Xie, H.; Chen, H.; Wen, Y.; Chen, Q.; Xie, S.; Li, H.; et al. Selective eosinophil necroptosis contributes to airway inflammation and remodeling in asthma. Allergy 2022, 77, 3456–3459. [Google Scholar] [CrossRef]
- Rich, H.E.; Antos, D.; Melton, N.R.; Alcorn, J.F.; Manni, M.L. Insights Into Type I and III Interferons in Asthma and Exacerbations. Front. Immunol. 2020, 11, 574027. [Google Scholar] [CrossRef]
- Porsbjerg, C.; Nieto-Fontarigo, J.J.; Cerps, S.; Ramu, S.; Menzel, M.; Hvidtfeldt, M.; Silberbrandt, A.; Frøssing, L.; Klein, D.; Sverrild, A.; et al. Phenotype and severity of asthma determines bronchial epithelial immune responses to a viral mimic. Eur. Respir. J. 2022, 60, 2102333. [Google Scholar] [CrossRef]
- Huang, X.; Tan, X.; Liang, Y.; Hou, C.; Qu, D.; Li, M.; Huang, Q. Differential DAMP release was observed in the sputum of COPD, asthma and asthma-COPD overlap (ACO) patients. Sci. Rep. 2019, 9, 19241. [Google Scholar] [CrossRef]
- Chen, W.; Gullett, J.M.; Tweedell, R.E.; Kanneganti, T.-D. Innate immune inflammatory cell death: PANoptosis and PANoptosomes in host defense and disease. Eur. J. Immunol. 2023, e2250235. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Toews, G.B.; White, E.S.; Lynch, J.P., 3rd; Martinez, F.J. Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 2004, 55, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Alizadeh, J.; Juarez, M.; Samali, A.; Halayko, A.J.; Kenyon, N.J.; Ghavami, S.; Zeki, A.A. Autophagy, Apoptosis, the Unfolded Protein Response, and Lung Function in Idiopathic Pulmonary Fibrosis. Cells 2021, 10, 1642. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-β on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Maeyama, T.; Kuwano, K.; Kawasaki, M.; Kunitake, R.; Hagimoto, N.; Matsuba, T.; Yoshimi, M.; Inoshima, I.; Yoshida, K.; Hara, N. Upregulation of Fas-signalling molecules in lung epithelial cells from patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2001, 17, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wen, L.; Shi, Q.F.; Gao, F.; Huang, B.; Meng, J.; Hu, C.P.; Wang, C.M. Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis. 2020, 11, 978. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Zhu, Y.; Yao, J.; Meng, X.; Wang, J.; Xie, H.; Wang, R. NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp. Cell Res. 2017, 357, 328–334. [Google Scholar] [CrossRef]
- Jäger, B.; Seeliger, B.; Terwolbeck, O.; Warnecke, G.; Welte, T.; Müller, M.; Bode, C.; Prasse, A. The NLRP3-Inflammasome-Caspase-1 Pathway Is Upregulated in Idiopathic Pulmonary Fibrosis and Acute Exacerbations and Is Inducible by Apoptotic A549 Cells. Front. Immunol. 2021, 12, 642855. [Google Scholar] [CrossRef]
- Terlizzi, M.; Molino, A.; Colarusso, C.; Donovan, C.; Imitazione, P.; Somma, P.; Aquino, R.P.; Hansbro, P.M.; Pinto, A.; Sorrentino, R. Activation of the Absent in Melanoma 2 Inflammasome in Peripheral Blood Mononuclear Cells From Idiopathic Pulmonary Fibrosis Patients Leads to the Release of Pro-Fibrotic Mediators. Front. Immunol. 2018, 9, 670. [Google Scholar] [CrossRef]
- Trachalaki, A.; Tsitoura, E.; Mastrodimou, S.; Invernizzi, R.; Vasarmidi, E.; Bibaki, E.; Tzanakis, N.; Molyneaux, P.L.; Maher, T.M.; Antoniou, K. Enhanced IL-1β Release Following NLRP3 and AIM2 Inflammasome Stimulation Is Linked to mtROS in Airway Macrophages in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 661811. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Bu, E.; Zhang, C.; Lai, R.; He, J.; Guo, B.; Guo, W.; Liu, L.; Pan, H. Deciphering the molecular mechanisms of Maxing Huoqiao Decoction in treating pulmonary fibrosis via transcriptional profiling and circRNA-miRNA-mRNA network analysis. Phytomedicine 2023, 115, 154754. [Google Scholar] [CrossRef] [PubMed]
- Chua, F.; Dunsmore, S.E.; Clingen, P.H.; Mutsaers, S.E.; Shapiro, S.D.; Segal, A.W.; Roes, J.; Laurent, G.J. Mice lacking neutrophil elastase are resistant to bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 2007, 170, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.D.; Kliment, C.R.; Metz, H.E.; Kim, K.H.; Kargl, J.; Agostini, B.A.; Crum, L.T.; Oczypok, E.A.; Oury, T.A.; Houghton, A.M. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J. Leukoc. Biol. 2015, 98, 143–152. [Google Scholar] [CrossRef]
- Warheit-Niemi, H.I.; Huizinga, G.P.; Edwards, S.J.; Wang, Y.; Murray, S.K.; O’Dwyer, D.N.; Moore, B.B. Fibrotic Lung Disease Alters Neutrophil Trafficking and Promotes Neutrophil Elastase and Extracellular Trap Release. Immunohorizons 2022, 6, 817–834. [Google Scholar] [CrossRef]
- Yan, S.; Li, M.; Liu, B.; Ma, Z.; Yang, Q. Neutrophil extracellular traps and pulmonary fibrosis: An update. J. Inflamm. 2023, 20, 2. [Google Scholar] [CrossRef]
- Christenson, S.A.; Smith, B.M.; Bafadhel, M.; Putcha, N. Chronic obstructive pulmonary disease. Lancet 2022, 399, 2227–2242. [Google Scholar] [CrossRef]
- Van Eeckhoutte, H.P.; Donovan, C.; Kim, R.Y.; Conlon, T.M.; Ansari, M.; Khan, H.; Jayaraman, R.; Hansbro, N.G.; Dondelinger, Y.; Delanghe, T.; et al. RIPK1 kinase-dependent inflammation and cell death contribute to the pathogenesis of COPD. Eur. Respir. J. 2023, 61. [Google Scholar] [CrossRef]
- Lu, Z.; Van Eeckhoutte, H.P.; Liu, G.; Nair, P.M.; Jones, B.; Gillis, C.M.; Nalkurthi, B.C.; Verhamme, F.; Buyle-Huybrecht, T.; Vandenabeele, P.; et al. Necroptosis Signaling Promotes Inflammation, Airway Remodeling, and Emphysema in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 667–681. [Google Scholar] [CrossRef]
- Song, Q.; Chen, P.; Liu, X.M. The role of cigarette smoke-induced pulmonary vascular endothelial cell apoptosis in COPD. Respir. Res. 2021, 22, 39. [Google Scholar] [CrossRef]
- Tverezovskyi, V.M.; Kapustnyk, V.A.; Shelest, B.O.; Sadovenko, O.L. Prognostic Potential of Lymphocyte-to-Monocyte Ratio and Caspase-8 in Prediction of Chronic Obstructive Pulmonary Disease Development. Wiad. Lek. 2022, 75, 2677–2682. [Google Scholar] [CrossRef] [PubMed]
- Huot-Marchand, S.; Nascimento, M.; Culerier, E.; Bourenane, M.; Savigny, F.; Panek, C.; Serdjebi, C.; Le Bert, M.; Quesniaux, V.F.J.; Ryffel, B.; et al. Cigarette smoke-induced gasdermin D activation in bronchoalveolar macrophages and bronchial epithelial cells dependently on NLRP3. Front. Immunol. 2022, 13, 918507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Y.; Jiang, Y.-X.; Yang, Y.-C.; Liu, J.-Y.; Huo, C.; Ji, X.-L.; Qu, Y.-Q. Cigarette smoke extract induces pyroptosis in human bronchial epithelial cells through the ROS/NLRP3/caspase-1 pathway. Life Sci. 2021, 269, 119090. [Google Scholar] [CrossRef]
- Panzner, P.; Lafitte, J.-J.; Tsicopoulos, A.; Hamid, Q.; Tulic, M.K. Marked Up-regulation of T Lymphocytes and Expression of Interleukin-9 in Bronchial Biopsies From Patients With Chronic Bronchitis With Obstruction*. Chest 2003, 124, 1909–1915. [Google Scholar] [CrossRef] [PubMed]
- Southworth, T.; Metryka, A.; Lea, S.; Farrow, S.; Plumb, J.; Singh, D. IFN-γ synergistically enhances LPS signalling in alveolar macrophages from COPD patients and controls by corticosteroid-resistant STAT1 activation. Br. J. Pharmacol. 2012, 166, 2070–2083. [Google Scholar] [CrossRef]
- Collinson, N.; Snape, N.; Beagley, K.; Fantino, E.; Spann, K. COPD Is Associated with Elevated IFN-β Production by Bronchial Epithelial Cells Infected with RSV or hMPV. Viruses 2021, 13, 911. [Google Scholar] [CrossRef]
- García-Valero, J.; Olloquequi, J.; Montes, J.F.; Rodríguez, E.; Martín-Satué, M.; Texidó, L.; Ferrer Sancho, J. Deficient pulmonary IFN-β expression in COPD patients. PLoS ONE 2019, 14, e0217803. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef]
- Abulaiti, A.; Maimaiti, A.; Yiming, N.; Fu, Q.; Li, S.; Li, Y.; Wang, Y.; Zhou, Q. Molecular subtypes based on PANoptosis-related genes and tumor microenvironment infiltration characteristics in lower-grade glioma. Funct. Integr. Genom. 2023, 23, 84. [Google Scholar] [CrossRef]
- Mall, R.; Bynigeri, R.R.; Karki, R.; Malireddi, R.K.S.; Sharma, B.R.; Kanneganti, T.D. Pancancer transcriptomic profiling identifies key PANoptosis markers as therapeutic targets for oncology. NAR Cancer 2022, 4, zcac033. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Pan, J.; Li, P.; Gao, J. Characterization of PANoptosis patterns predicts survival and immunotherapy response in gastric cancer. Clin. Immunol. 2022, 238, 109019. [Google Scholar] [CrossRef] [PubMed]
- Qiang, S.; Fu, F.; Wang, J.; Dong, C. Definition of immune molecular subtypes with distinct immune microenvironment, recurrence, and PANoptosis features to aid clinical therapeutic decision-making. Front. Genet. 2022, 13, 1007108. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, Q.; Lan, L.; Xu, X. PANoptosis-related prognostic signature predicts overall survival of cutaneous melanoma and provides insights into immune infiltration landscape. Sci. Rep. 2023, 13, 8449. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, R.; Chan, S.; Meng, L.; Xu, Y.; Zuo, X.; Wang, Z.; Hu, X.; Han, Q.; Dai, L.; et al. PANoptosis-based molecular clustering and prognostic signature predicts patient survival and immune landscape in colon cancer. Front. Genet. 2022, 13, 955355. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, J.; Zhang, N.; Zhu, Y.; Zhong, Y.; Wang, Z.; Jin, H.; Wang, X. A Novel Defined PANoptosis-Related miRNA Signature for Predicting the Prognosis and Immune Characteristics in Clear Cell Renal Cell Carcinoma: A miRNA Signature for the Prognosis of ccRCC. Int. J. Mol. Sci. 2023, 24, 9392. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.-D. ADAR1 and ZBP1 in innate immunity, cell death, and disease. Trends Immunol. 2023, 44, 201–216. [Google Scholar] [CrossRef]
- Karki, R.; Sundaram, B.; Sharma, B.R.; Lee, S.; Malireddi, R.K.S.; Nguyen, L.N.; Christgen, S.; Zheng, M.; Wang, Y.; Samir, P.; et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021, 37, 109858. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Fedorov, A.; Qiao, L.; Bao, H.; Beknazarov, N.; Wang, S.; Gautam, A.; Williams, R.M.; Crawford, J.C.; et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 2022, 606, 594–602. [Google Scholar] [CrossRef]
- Lin, J.F.; Hu, P.S.; Wang, Y.Y.; Tan, Y.T.; Yu, K.; Liao, K.; Wu, Q.N.; Li, T.; Meng, Q.; Lin, J.Z.; et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct. Target. Ther. 2022, 7, 54. [Google Scholar] [CrossRef]
- Liu, L.X.; Heng, J.H.; Deng, D.X.; Zhao, H.; Zheng, Z.Y.; Liao, L.D.; Lin, W.; Xu, X.E.; Li, E.M.; Xu, L.Y. Sulconazole Induces PANoptosis by Triggering Oxidative Stress and Inhibiting Glycolysis to Increase Radiosensitivity in Esophageal Cancer. Mol. Cell. Proteom. 2023, 22, 100551. [Google Scholar] [CrossRef]
- Jiang, M.; Qi, L.; Li, L.; Wu, Y.; Song, D.; Li, Y. Caspase-8: A key protein of cross-talk signal way in “PANoptosis” in cancer. Int. J. Cancer 2021, 149, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Lee, E.; Banoth, B.; Malireddi, R.K.S.; Samir, P.; Tuladhar, S.; Mummareddy, H.; Burton, A.R.; Vogel, P.; et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI Insight 2020, 5, e136720. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Wang, L.; Jin, M.; Jiang, M.; Li, L.; Li, Y. Caspase-6 is a key regulator of cross-talk signal way in PANoptosis in cancer. Immunology 2023, 169, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell. Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef]
- Jarabicová, I.; Horváth, C.; Veľasová, E.; Bies Piváčková, L.; Vetešková, J.; Klimas, J.; Křenek, P.; Adameová, A. Analysis of necroptosis and its association with pyroptosis in organ damage in experimental pulmonary arterial hypertension. J. Cell Mol. Med. 2022, 26, 2633–2645. [Google Scholar] [CrossRef]
- Liu, M.; Li, W.; Xiang, X.; Xie, J. Mycobacterium tuberculosis effectors interfering host apoptosis signaling. Apoptosis 2015, 20, 883–891. [Google Scholar] [CrossRef]
- Rong, W.; Liu, C.; Li, X.; Wan, N.; Wei, L.; Zhu, W.; Bai, P.; Li, M.; Ou, Y.; Li, F.; et al. Caspase-8 Promotes Pulmonary Hypertension by Activating Macrophage-Associated Inflammation and IL-1β (Interleukin 1β) Production. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 613–631. [Google Scholar] [CrossRef]
- Stutz, M.D.; Allison, C.C.; Ojaimi, S.; Preston, S.P.; Doerflinger, M.; Arandjelovic, P.; Whitehead, L.; Bader, S.M.; Batey, D.; Asselin-Labat, M.-L.; et al. Macrophage and neutrophil death programs differentially confer resistance to tuberculosis. Immunity 2021, 54, 1758–1771.e1757. [Google Scholar] [CrossRef]
- Saiga, H.; Kitada, S.; Shimada, Y.; Kamiyama, N.; Okuyama, M.; Makino, M.; Yamamoto, M.; Takeda, K. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int. Immunol. 2012, 24, 637–644. [Google Scholar] [CrossRef]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3, eaat2738. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhu, F.; Zhao, M.; Shao, F.; Yu, D.; Ma, J.; Zhang, X.; Li, W.; Qian, Y.; Zhang, Y.; et al. SARS-CoV-2 nucleocapsid suppresses host pyroptosis by blocking Gasdermin D cleavage. EMBO J. 2021, 40, e108249. [Google Scholar] [CrossRef]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.M.; Dobson, R.C.; Webb, A.I.; et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef]
- Johnston, A.N.; Ma, Y.; Liu, H.; Liu, S.; Hanna-Addams, S.; Chen, S.; Chen, C.; Wang, Z. Necroptosis-blocking compound NBC1 targets heat shock protein 70 to inhibit MLKL polymerization and necroptosis. Proc. Natl. Acad. Sci. USA 2020, 117, 6521–6530. [Google Scholar] [CrossRef]
- Bai, Y.; Lam, H.C.; Lei, X. Dissecting Programmed Cell Death with Small Molecules. Acc. Chem. Res. 2020, 53, 1034–1045. [Google Scholar] [CrossRef]
- Ren, Y.; Su, Y.; Sun, L.; He, S.; Meng, L.; Liao, D.; Liu, X.; Ma, Y.; Liu, C.; Li, S.; et al. Discovery of a Highly Potent, Selective, and Metabolically Stable Inhibitor of Receptor-Interacting Protein 1 (RIP1) for the Treatment of Systemic Inflammatory Response Syndrome. J. Med. Chem. 2017, 60, 972–986. [Google Scholar] [CrossRef]
- Cui, Y.R.; Qu, F.; Zhong, W.J.; Yang, H.H.; Zeng, J.; Huang, J.H.; Liu, J.; Zhang, M.Y.; Zhou, Y.; Guan, C.X. Beneficial effects of aloperine on inflammation and oxidative stress by suppressing necroptosis in lipopolysaccharide-induced acute lung injury mouse model. Phytomedicine 2022, 100, 154074. [Google Scholar] [CrossRef]
- Mou, F.; Mou, C. Necrostatin-1 Alleviates Bleomycin-Induced Pulmonary Fibrosis and Extracellular Matrix Expression in Interstitial Pulmonary Fibrosis. Med. Sci. Monit. 2020, 26, e919739. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Jiang, K.; Zeng, C.; Zhu, R.; Chu, H.; Liu, H.; Du, J. Synergism of TNF-α and IFN-β triggers human airway epithelial cells death by apoptosis and pyroptosis. Mol. Immunol. 2023, 153, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Li, R.; Negro, R.; Cheng, J.; Vora, S.M.; Fu, T.M.; Wang, A.; He, K.; Andreeva, L.; Gao, P.; et al. Phase separation drives RNA virus-induced activation of the NLRP6 inflammasome. Cell 2021, 184, 5759–5774.e5720. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, L.; Dai, T.; Qin, Z.; Lu, H.; Zhang, L.; Zhou, F. Liquid-liquid phase separation in human health and diseases. Signal Transduct. Target. Ther. 2021, 6, 290. [Google Scholar] [CrossRef]
- Xie, J.; He, H.; Kong, W.; Li, Z.; Gao, Z.; Xie, D.; Sun, L.; Fan, X.; Jiang, X.; Zheng, Q.; et al. Targeting androgen receptor phase separation to overcome antiandrogen resistance. Nat. Chem. Biol. 2022, 18, 1341–1350. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, P.; Vora, S.M.; Wu, H. Higher-order assemblies in innate immune and inflammatory signaling: A general principle in cell biology. Curr. Opin. Cell Biol. 2020, 63, 194–203. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Jiang, J.; Li, T.; Huang, L. PANoptosis: Mechanism and Role in Pulmonary Diseases. Int. J. Mol. Sci. 2023, 24, 15343. https://doi.org/10.3390/ijms242015343
Chen S, Jiang J, Li T, Huang L. PANoptosis: Mechanism and Role in Pulmonary Diseases. International Journal of Molecular Sciences. 2023; 24(20):15343. https://doi.org/10.3390/ijms242015343
Chicago/Turabian StyleChen, Shiyi, Jiacheng Jiang, Tongfu Li, and Longshuang Huang. 2023. "PANoptosis: Mechanism and Role in Pulmonary Diseases" International Journal of Molecular Sciences 24, no. 20: 15343. https://doi.org/10.3390/ijms242015343
APA StyleChen, S., Jiang, J., Li, T., & Huang, L. (2023). PANoptosis: Mechanism and Role in Pulmonary Diseases. International Journal of Molecular Sciences, 24(20), 15343. https://doi.org/10.3390/ijms242015343