To a solution with 0.2 g (1.0 mmol) ynone derivative of pyridine dissolved in 15 mL of anhydrous CH2Cl2 (under an inert atmosphere at −78 °C), 0.19 mL (1.2 mmol) of triflic anhydride was added with a syringe, the mixture was stirred constantly during 3 h. Subsequently, 1.2 mmol of the corresponding ketene acetal was added and stirring continued at −78 °C for another 8 h. The reaction was then allowed to reach 25 °C before being transferred to a separatory funnel and washed with water (3 × 30 mL). The organic layers were combined and dried up with anhydrous Na2SO4; then, the solvent was removed by vacuum evaporation. Finally, the reaction crude was purified by column chromatography using silica gel 60 (0.063–0.200 mm, 70–230 mesh ASTM, acquired from Merck-millipore, Germany) with different n-hexane/ethyl acetate mixtures as eluents, obtaining the title pure compounds 3a–p.
Spectroscopical Characterization of Dihydropyridine Carboxylic Acids 3a–p
2-methyl-2-(3-(3-phenylpropioloyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-propanoic acid (3a), white solid was obtained with 91% isolated yield (0.37g); mp 122–124 °C; IR (cm−1): 3086 (COOH), 2199 (C≡C), 1705 (C=O), 1625 (C=C); 1H NMR (CDCl3) δ (ppm): 11.28 (s, 1H, OH), 8.20 (s, 1H, H5), 7.63 (d, J = 6.6 Hz, 2H, H15, H19), 7.49 (m, 1H, H17), 7.45 (d, J = 7.5 Hz, 2H, H16, H18), 6.77 (d, J = 5.4 Hz, 1H, H9), 5.55 (dd, J = 1.8 Hz, J = 5.4 Hz, 1H, H8), 4.16 (d, J = 5.4 Hz, 1H, H7), 1.17 (s, 3H, H10), 1.16 (s, 3H, H3); 13C NMR (CDCl3) δ (ppm): 182.4 (C1, C=O), 176.1 (C11, C=O), 137.5 (C5), 133.5 (C15, C19), 131.0 (C17), 128.8 (C16, C18), 122.7 (C9), 122.9 (C6), 121.3 (C14), 119.1 (q, JC-F = 321 Hz, C21, CF3), 112.2 (C8), 92.5 (C13), 84.6 (C12), 47.8 (C2), 37.5 (C7), 22.6 (C10), 19.8 (C3); HRMS-DART+ (19 eV): elemental composition C19H17F3NO5S, [M+1]+, correlation (error of −1.37 ppm) exact value 428.0779 Daltons and precise value of 428.0773 Daltons; complementarily, the provided unsaturation data (11.5) were in agreement with the structure.
1-(3-(3-phenylpropioloyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-cyclobutane-1-carboxylic acid (3b), white solid was obtained with 82% isolated yield (0.34 g); mp 134–136 °C; IR (cm−1): 3069 (COOH), 2199 (C≡C), 1703 (C=O), 1640 (C=C); 1H NMR (CDCl3) δ(ppm): 9.87 (s, 1H, OH), 8.18 (s, 1H, H3), 7.63 (d, J= 6.9 Hz, 2H, H16, H20), 7.48 (m, 1H, H18), 7.43 (d, J= 6.9 Hz, 2H, H17, H19), 6.68 (d, J= 8.1 Hz, 1H, H7), 5.47 (dd, J= 5.4 Hz, J= 7.8 Hz, 1H, H6), 4.12 (d, J= 5.4 Hz, 1H, H5), 2.35–1.96 (m, 6H, H9, H10, H11); 13C NMR (CDCl3) δ(ppm): 181.6 (C1, C=O), 176.2 (C12, C=O), 137.2 (C3), 133.1 (C16, C20), 131.0 (C18), 128.8 (C17, C19), 122.8 (C4), 122.0 (C7), 119.5 (C15), 119.2 (q, JC-F= 321 Hz, C22, CF3), 111.8 (C6), 92.7 (C14), 84.7 (C13), 53.5 (C8), 36.4 (C5), 28.4 (C11), 28.0 (C9), 16.3 (C10); HRMS-DART+ (19 eV): elemental composition C20H17F3NO5S, [M+1]+, correlation (error of −0.17 ppm) exact value 440.0779 Daltons and precise value of 440.0778 Daltons; complementarily, the provided unsaturation data (12.5) were in agreement with the structure.
1-(3-(3-phenylpropioloyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-cyclohexane-1-carboxylic acid (3c), white solid was obtained with 93% isolated yield (0.49 g); mp 140–142 °C; IR (cm−1): 2959 (COOH), 2198 (C≡C), 1678 (C=O), 1619 (C=C); 1H NMR (CDCl3) δ (ppm): 8.17 (s, 1H, H3), 7.62 (d, J = 9.0 Hz, 2H, H18, H22), 7.50–7.40 (m, 3H, H19, H20, H21), 6.77 (d, J = 6.9 Hz, 1H, H7), 5.53 (dd, J = 6.0 Hz, J = 7.8 Hz, 1H, H6), 4.02 (d, J = 5.7 Hz, 1H, H5), 2.08–2.05 (m, 2H, H9), 1.67–1.63 (m, 4H, H10, H13), 1.33–1.24 (m, 2H, H12), 1.20–1.05 (m, 2H, H11); 13C NMR (CDCl3) δ (ppm): 178.9 (C1, C=O), 176.0 (C14, C=O), 137.1 (C3), 133.1 (C18, C22), 131.0 (C20), 128.7 (C19, C21), 122.9 (C7), 122.5 (C4), 119.5 (C17), 119.2 (q, JC-F = 321 Hz, C24, CF3), 111.6 (C6), 92.4 (C15), 84.6 (C16), 53.4 (C8), 38.6 (C5), 30.0 (C9), 29.7 (C13), 29.5 (C10), 25.4 (C12), 23.3 (C11); HRMS-DART+ (19 eV): elemental composition C22H21F3NO5S, [M+1]+, correlation (error of −2.12 ppm) exact value 468.1092 Daltons and precise value of 468.1082 Daltons; complementarily, the provided unsaturation data (12.5) were in agreement with the structure.
2-methyl-2-(3-(3-(4-(trifluoromethyl)phenyl)propioloyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-propanoic acid (3d), red solid was obtained with 82% isolated yield (0.31g); mp 118–120 °C; IR (cm−1): 2985 (COO-H), 2200 (C≡C), 1680 (C=O), 1596 (C=C); 1H NMR (CDCl3) δ (ppm): 8.13 (s, 1H, H5), 7.72–7.65 (m, 4H, H15, H16, H18, H29), 6.73 (d, J = 7.5 Hz, 1H, H9), 5.47 (dd, J = 7.8 Hz, J = 5.4 Hz, 1H, H8), 4.20 (m, 1H, H7), 1.12 (s, 3H, H3), 1.10 (s, 3H, H10); 13C NMR (CDCl3) δ (ppm): 179.9 (C1, C=O), 175.9 (C11, C=O), 167.96 (C17), 138.0 (C5), 133.3 (C15, C19), 131.02 (C16, C18), 129.9 (d, JC-F = 164 Hz, C27, CF3), 125.85 (C9), 122.9 (C6), 119.1 (q, JC-F = 321 Hz, C21, CF3), 112.2 (C8), 90.1 (C12), 85.8 (C13), 63.7 (C2), 47.5 (C7), 22.1 (C3), 20.4 (C10); HRMS-DART+ (19 eV): elemental composition C20H16F6NO5S, [M+1]+, correlation (error of −1.01 ppm) exact value 496.0653 Daltons and precise value of 496.0648 Daltons; complementarily, the provided unsaturation data (11.5) were in agreement with the structure.
2-(3-(3-(4-methoxyphenyl)propioloyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-2-methylpropanoic acid (3e), red solid was obtained with 76% isolated yield (0.27g); mp 146–148 °C; IR (cm−1): 2923 (COO-H), 2193 (C≡C), 1683 (C=O), 1600 (C=C); 1H NMR (CDCl3) δ (ppm): 9.48 (s, 1H, OH), 8.14 (s, 1H, H5), 7.54 (d, J = 9.0 Hz, 2H, H15, H19), 6.91 (d, J = 8.7 Hz, 2H, H16, H18), 6.75 (d, J = 5.4 Hz, 1H, H9), 5.48–5.46 (dd, J = 7.8 Hz, J = 5.7 Hz, 1H, H8), 4.13 (d, J = 5.7 Hz, 1H, H7), 1.24 (s, 3H, H3), 1.12 (s, 3H, H10); 13C NMR (CDCl3) δ (ppm): 181.4 (C1, C=O), 176.2 (C11, C=O), 162.0 (C17), 137.5 (C5), 135.2 (C15, C19), 122.9 (C9), 122.9 (C6), 119.1 (q, JC-F = 321 Hz, C22, CF3), 114.6 (C16, C18), 112.3 (C8), 111.3 (C14), 93.8 (C12), 84.7 (C13), 55.5 (C2), 37.5 (C7), 22.7 (C3), 19.9 (C10); HRMS-DART+ (19 eV): elemental composition C20H19F3NO5S, [M+1]+, correlation (error of −3.57 ppm) exact value 458.0885 Daltons and precise value of 458.0868 Daltons; complementarily, the provided unsaturation data (11.5) were in agreement with the structure.
2-(3-(hept-2-ynoyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-2-methylpropanoic acid (3f), yellow solid was obtained with 77% isolated yield (0.33 g); mp 128–130 °C; IR (cm−1): 2937 (COOH), 2200 (C≡C), 1686 (C=O), 1614 (C=C); 1H NMR (CDCl3) δ (ppm): 11.2 (s, 1H, OH), 7.94 (s, 1H, H5), 6.63 (d, J = 7.8 Hz, 1H, H9), 5.42 (dd, J = 5.4 Hz, J = 7.5 Hz, 1H, H8), 3.97 (d, J = 5.7 Hz, 1H, H7), 2.36 (t, 2H, H14), 1.57–1.47 (m, 2H, H15), 1.44–1.31 (m, 2H, H16), 1.01 (s, 3H, H3), 0.99 (s, 3H, H10), 0.85 (t, 3H, H17); 13C NMR (CDCl3) δ (ppm): 182.2 (C1, C=O), 176.2 (C11, C=O), 137.1 (C5), 122.7 (C9), 119.1 (q, JC-F = 321 Hz, C19, CF3), 112.0 (C8) 95.8 (C13), 77.4 (C12), 47.6 (C2), 37.3 (C7), 22.3 (C3), 21.8 (C), 19.7 (C10), 18.5 (C14), 13.2 (C17); HRMS-DART+ (19 eV): elemental composition C17H21F3NO5S, [M+1]+, correlation (error of −2.97 ppm) exact value 408.1092 Daltons and precise value of 408.1080 Daltons; complementarily, the provided unsaturation data (7.5) were in agreement with the structure.
1-(3-(hept-2-ynoyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-cyclobutane-1-carboxylic acid (3g), yellow solid was obtained with 72% isolated yield (0.32 g); mp 116–118 °C; IR (cm−1): 2930 (COOH), 2221 (C≡C), 1720 (C=O), 1632 (C=C); 1H NMR (CDCl3) δ (ppm): 7.95 (s, 1H, H3), 6.06 (s, 1H, H7), 4.71 (s, 1H, H6), 3.8 (s, 1H, H5), 2.49–2.36 (m, 6H, H9, H11, H15), 2.13–2.10 (m, 2H, H16), 1.64–1.59 (m, 2H, H27), 1.49–1.46 (m, 2H, H10), 0.95 (t, 3H, H18); 13C NMR (CDCl3) δ (ppm): 181.7 (C1, C=O), 176.4 (C12, C=O), 136.9 (C3), 122.8 (C4), 121.9 (C7), 119.8 (q, JC-F = 321 Hz, C20, CF3), 111.7 (C6), 96.1 (C13), 77.4 (C12), 53.4 (C8), 37.8 (C5), 35.8 (C9), 29.5 (C11), 28.2 (C14), 28.0 (C16), 25.0 (C10), 21.8 (C17), 18.5 (C15), 13.2 (C18); HRMS-DART+ (19 eV): elemental composition C18H21F3NO5S, [M+1]+, correlation (error of +2.60 ppm) exact value 420.1092 Daltons and precise value of 420.1081 Daltons; complementarily, the provided unsaturation data (8.5) were in agreement with the structure.
1-(3-(hept-2-ynoyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-cyclopentane-1-carboxylic acid (3h), yellow solid was obtained with 74% isolated yield (0.34 g); mp 126–128 °C; IR (cm−1): 2937 (COOH), 2224 (C≡C), 1682 (C=O), 1613 (C=C); 1H NMR (CDCl3) δ (ppm): 8.00 (s, 1H, H3), 6.66 (d, J = 7.8 Hz, 1H, H7), 5.50 (dd, J = 5.4 Hz, J = 7.8 Hz, 1H, H6), 4.12 (d, J = 5.4 Hz, 1H, H5), 2.46 (t, 2H, H16), 2.10–2.05 (m, 2H, H9), 1.63–1.28 (m, 12H, H10, H11, H12, H17, H18), 0.96 (t, 3H, H19); 13C NMR (CDCl3) δ (ppm): 181.6 (C1, C=O), 176.4 (C13, C=O), 137.1 (C3), 123.3 (C4), 122.2 (C7), 119.1 (q, JC-F = 321 Hz, C21, CF3), 112.8 (C6), 96.4 (C15), 77.5 (C14), 60.5 (C8), 35.9 (C5), 33.1 (C9), 31.5 (C12), 29.6 (C17), 24.0 (C10), 23.9 (C11), 22.0 (C18), 18.7 (C16), 13.4 (C19); HRMS-DART+ (19 eV): elemental composition C19H23F3NO5S, [M+1]+, correlation (error of +1.85 ppm) exact value 434.1249 Daltons and precise value of 434.1257 Daltons; complementarily, the provided unsaturation data (8.5) were in agreement with the structure.
1-(3-(hept-2-ynoyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-cyclohexane-1-carboxylic acid (3i), yellow solid was obtained with 87% isolated yield (0.41 g); mp 130–132 °C; IR (cm−1): 2938 (COOH), 2223 (C≡C), 1680 (C=O), 1613 (C=C); 1H NMR (CDCl3) δ (ppm): 8.01 (s, 1H, H3), 6.73 (d, J = 7.8 Hz, 1H, H7), 5.51 (dd, J = 5.7 Hz, J = 7.8 Hz, 1H, H6), 3.94 (d, J = 5.7 Hz, 1H, H5), 2.44 (t, 2H, H17), 2.04–2.00 (m, 2H, H9), 1.66–0.92 (m, 15H, H10-H13, H18-H20); 13C NMR (CDCl3) δ (ppm): 180.5 (C1, C=O), 176.3 (C14, C=O), 137.0 (C3), 122.8 (C7), 122.7 (C4), 119.5 (q, JC-F = 321 Hz, C22, CF3), 111.7 (C6), 95.8 (C16), 77.4 (C15), 53.4 (C8), 38.6 (C5), 38.4 (C9), 29.9 (C13), 29.6 (C18), 25.4 (C10), 23.2 (C12), 21.9 (C11), 18.7 (C19), 18.6 (C17), 13.4 (C20); HRMS-DART+ (19 eV): elemental composition C20H25F3NO5S, [M+1]+, correlation (error of −1.92 ppm) exact value 448.1405 Daltons and precise value of 448.1396 Daltons; complementarily, the provided unsaturation data (8.5) were in agreement with the structure.
2-methyl-2-(3-(non-2-ynoyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-propanoic acid (3j), yellow solid was obtained with 68% isolated yield (0.37g); mp 122–124 °C; IR (cm−1): 2930 (COO-H), 2223 (C≡C), 1678 (C=O), 1613 (C=C); 1H NMR (CDCl3) δ (ppm): 10.19 (s, 1H, OH), 7.79 (s, 1H, H5), 6.47 (d, J = 7.8 Hz, 1H, H9), 5.24 (dd, J = 5.7 Hz, J = 7.8 Hz, 1H, H8), 3.82 (d, J = 5.4 Hz, 1H, 7H), 2.20 (t, 2H, H14), 1.43–1.33 (m, 2H, H15), 1.22–1.02 (m, 3H, H3, H16-H18), 0.86 (s, 3H, H10); 0.85 (s, 3H, H19); 13C NMR (CDCl3) δ (ppm): 182.2 (C1, C=O), 176.3 (C11, C=O), 137.2 (C5), 122.8 (C9), 122.6 (C6) 119.3 (q, JC-F = 321 Hz, C21 CF3), 112.1 (C8), 96.1 (C13), 77.4 (C12), 47.7 (C2), 37.3 (C7), 31.1 (C17), 28.5 (C16), 27.6 (C15), 22.5 (C3), 22.3 (C18), 19.8 (C10), 19.0 (C14), 13.9 (C19); HRMS-DART+ (19 eV): elemental composition C19H25F3NO5S, [M+1]+, correlation (error of −0.89 ppm) exact value 436.1405 Daltons and precise value of 436.1401 Daltons; complementarily, the provided unsaturation data (7.5) were in agreement with the structure.
1-(3-(non-2-ynoyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-cyclobutane-1-carboxylic acid (3k), yellow solid was obtained with 60% isolated yield (0.37g); mp 122–124 °C; IR (cm−1): 2930 (COOH), 2216 (C≡C), 1697 (C=O), 1613 (C=C); 1H NMR (CDCl3) δ (ppm): 7.99 (s, 1H, H3), 6.64 (d, J = 8.1 Hz, 1H, H7), 5.49 (dd, J = 5.4 Hz, J = 7.8 Hz, 1H, H6), 4.10 (d, J = 5.4 Hz, 1H, H5), 2.44 (t, 2H, H20), 2.07–2.03 (m, 2H, H9), 1.64–1.57 (m, 4H, H10, H11), 1.48–1.41 (m, 4H, H16, H17), 1.33–1.25 (m, 4H, H18, H19), 0.89 (t, 3H, H20); 13C NMR (CDCl3) δ (ppm): 182.1 (C1, C=O), 176.2 (C12, C=O), 137.2 (C3), 122.8 (C7), 122.6 (C4) 119.2 (q, JC-F = 321 Hz, C22, CF3), 112.1 (C6), 96.1 (C14), 77.4 (C13), 47.7 (C8), 37.3 (C5), 31.1 (C17), 28.5 (C9), 27.6 (C11), 22.5 (C16), 22.3 (C18), 19.8 (C19), 19.0 (C10), 18.3 (C15), 13.9 (C20); HRMS-DART+ (19 eV): elemental composition C20H25F3NO5S, [M+1]+, correlation (error of −3.64 ppm) exact value 448.1405 Daltons and precise value of 448.1389 Daltons; complementarily, the provided unsaturation data (8.5) were in agreement with the structure.
1-(3-(non-2-ynoyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-cyclopentane-1-carboxylic acid (3l), yellow solid was obtained with 65% isolated yield (0.37g); mp 134–136 °C; IR (cm−1): 2927 (COOH), 2223 (C≡C), 1679 (C=O), 1614 (C=C); 1H NMR (CDCl3) δ (ppm): 10.49 (s, 1H, OH), 7.99 (s, 1H, H3), 6.64 (d, J = 8.1 Hz, 1H, H7), 5.49 (dd, J = 5.4 Hz, J = 7.8 Hz, 1H, H6), 4.10 (d, J = 5.4 Hz, 1H, H5), 2.44 (t, 2H, H16), 2.07–2.03 (m, 2H, H9), 1.64–1.57 (m, 4H, H12, H17), 1.48–1.41 (m, 4H, H18, H19), 1.33–1.25 (m, 6H, H10, H11, H20), 0.89 (t, 3H, H21); 13C NMR (CDCl3) δ (ppm): 181.9 (C1, C=O), 176.5 (C13, C=O), 137.1 (C3), 123.3 (C4), 122.1 (C7), 119.3 (q, JC-F = 321 Hz, C23, CF3), 112.8 (C6), 96.1 (C15), 77.5 (C14), 60.6 (C8), 43.6 (C5), 35.9 (C9), 33.1 (C12), 31.5 (C19), 31.1 (C17), 29.9 (C18), 28.5 (C10), 27.5 (C11), 23.9 (C20), 19.8 (C16), 13.9 (C21); HRMS-DART+ (19 eV): elemental composition C21H27F3NO5S, [M+1]+, correlation (error of −3.46 ppm) exact value 462.1562 Daltons and precise value of 462.1546 Daltons; complementarily, the provided unsaturation data (8.5) were in agreement with the structure.
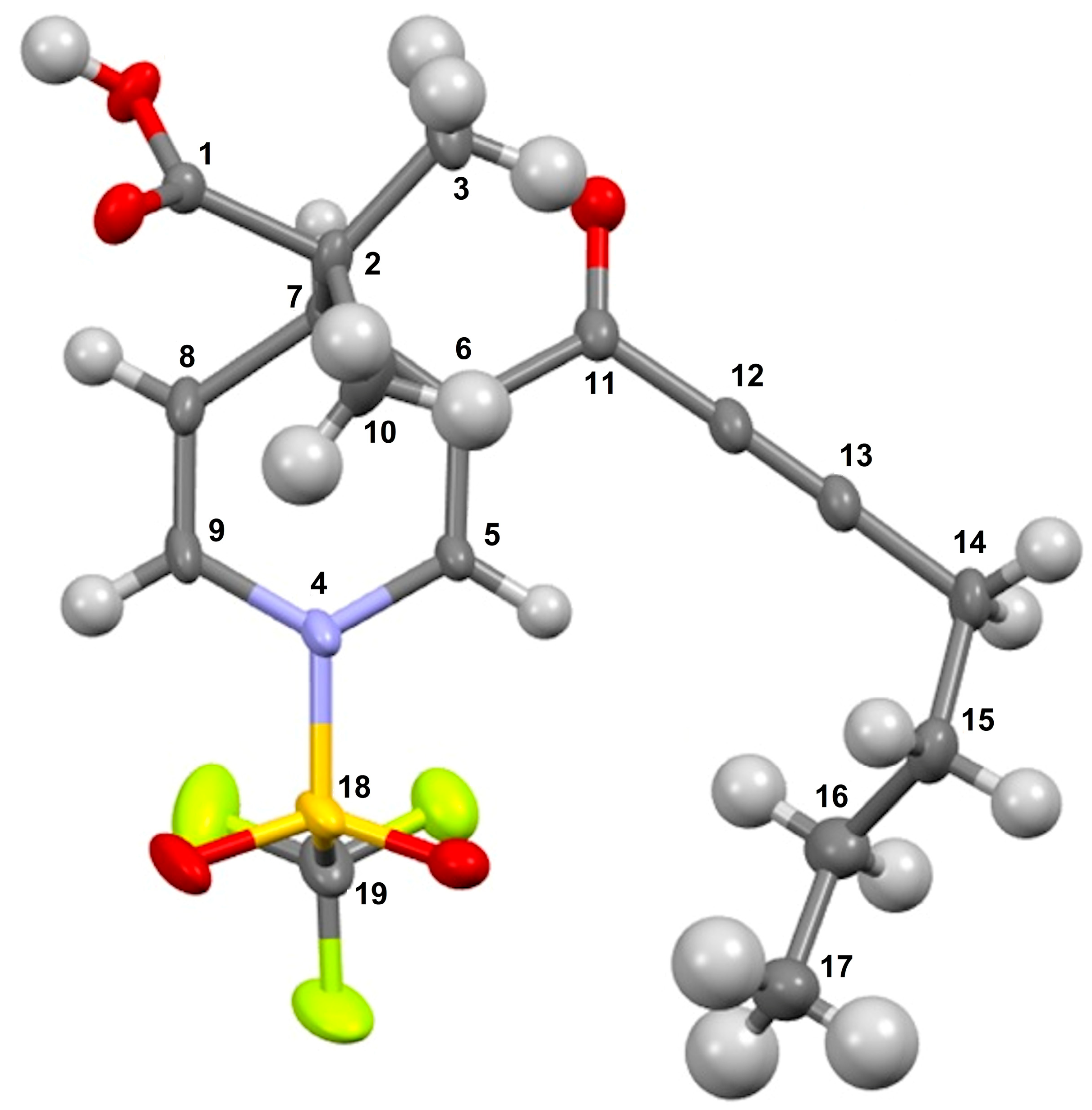
1-(3-(non-2-ynoyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-cyclohexane-1-carboxylic acid (3m), yellow solid was obtained with 74% isolated yield (0.37g); mp 110–112 °C; IR (cm−1): 2932 (COOH), 2217 (C≡C), 1760 (C=O), 1626 (C=C); 1H NMR (CDCl3) δ (ppm): 7.99 (s, 1H, H3), 6.71 (d, J = 7.8 Hz, 1H, H7), 5.46 (dd, J = 5.7 Hz, J = 7.8 Hz, 1H, H6), 3.92 (d, J = 5.7 Hz, 1H, H5), 2.42 (t, 2H, H17), 2.02–1.98 (m, 2H, H9), 1.65–1.56 (m, 4H, H13, H10), 1.44–1.39 (m, 4H, H12, H18), 1.31–1.24 (m, 8H, H11, H19, H20, H21), 0.88 (t, 3H, H22); 13C NMR (CDCl3) δ (ppm): 180.0 (C1, C=O), 176.3 (C14, C=O), 137.0 (C3), 122.8 (C4), 122.5 (C7), 119.3 (q, JC-F = 321, C24, CF3), 111.6 (C6), 96.0 (C16), 77.4 (C15), 53.3 (C8), 38.6 (C5), 38.4 (C20), 31.2 (C9), 29.9 (C13), 29.3 (C18), 28.6 (C19), 27.6 (C10), 25.4 (C12), 23.2 (C11), 22.4 (C21), 19.8 (C17), 13.9 (C22); HRMS-DART+ (19 eV): elemental composition C22H29F3NO5S, [M+1]+, correlation (error of −3.34 ppm) exact value 476.1718 Daltons and precise value of 476.1702 Daltons; complementarily, the provided unsaturation data (8.5) were in agreement with the structure.
2-(3-(3-cyclopentylpropioloyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-2-methylpropanoic acid (3n), yellow oil was obtained with 74% isolated yield (0.31 g); IR (cm−1): 2939 (COOH), 2206 (C≡C), 1701 (C=O), 1613 (C=C); 1H NMR (CDCl3) δ (ppm): 10.02 (s, 1H, OH), 7.97 (s, 1H, H5), 6.66 (d, J = 7.8 Hz, 1H, H9), 5.43 (dd, J = 5.7 Hz, J = 7.8 Hz, 1H, H8), 3.99 (d, J = 5.4 Hz, 1H, H7), 2.80–2.78 (m, 1H, H14), 1.97–1.66 (m, 4H, H15, H18), 1.58–1.56 (m, 4H, H16, H17), 1.04 (s, 3H, H3), 1.02 (s, 3H, H10); 13C NMR (CDCl3) δ (ppm): 182.1 (C1, C=O), 176.6 (C11, C=O), 137.4 (C5), 123.0 (C9), 122.9 (C6), 119.3 (q, JC-F = 321 Hz, C20, CF3), 112.1 (C8), 100.3 (C13), 94.5 (C12), 47.7 (C2), 37.4 (C7), 33.2 (C14), 33.2 (C15), 30.0 (C18), 25.2 (C16), 25.1 (C17), 22.4 (C3), 19.8 (C10); HRMS-DART+ (19 eV): elemental composition C18H21F3NO5S, [M+1]+, correlation (error of +4.13 ppm) exact value 420.1092 Daltons and precise value of 420.1109 Daltons; complementarily, the provided unsaturation data (8.5) were in agreement with the structure.
2-methyl-2-(3-(3-(thiophen-3-yl)propioloyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-propanoic acid (3o), white solid was obtained with 72% isolated yield (0.29 g); mp 156–158 °C; IR (cm−1): 2927 (COOH), 2197 (C≡C), 1758 (C=O), 1623 (C=C); 1H NMR (CDCl3) δ (ppm): 8.12 (s, 1H, H5), 7.79 (d, J = 3.0 Hz, 1H, H18), 7.36 (dd, J = 3.0 Hz, J = 5.1 Hz, 1H, H17), 7.25 (s, 1H, H16) 6.76 (d, J = 7.8 Hz, 1H, 9H), 5.50 (dd, J = 6.0 Hz, J = 8.4 Hz, 1H, H8), 4.12 (d, J = 5.7 Hz, 1H, H7), 1.12 (s, 6H, H3, H10); 13C NMR (CDCl3) δ (ppm): 181.9 (C1, C=O), 176.2 (C11, C=O), 137.4 (C5), 134.3 (C18), 130.2 (C16), 126.5 (C17), 123.0 (C9), 122.7 (C6), 119.3 (q, JC-F = 321 Hz, C20, CF3), 118.8 (C14), 112.2 (C8), 88.1 (C12), 85.0 (C13), 47.8 (C2), 37.5 (C7), 22.6 (C3), 19.8 (C10); HRMS-DART+ (19 eV): elemental composition C17H15F3NO5S2, [M+1]+, correlation (error of −2.71 ppm) exact value 434.0343 Daltons and precise value of 434.0332 Daltons; complementarily, the provided unsaturation data (11.5) were in agreement with the structure.
2-(3-(3-(ferrocenyl-1-yl)propioloyl)-1-((trifluoromethyl)sulfonyl)-1,4-dihydropyridin-4-yl)-propanoic acid (3p), red solid was obtained with 48% isolated yield (0.16 g); mp 148–150 °C; IR (cm−1): 2929 (COOH), 2188 (C≡C), 1725 (C=O), 1614 (C=C).;1H NMR (CDCl3) δ (ppm): 8.10 (s, 1H, H5), 6.75 (d, J = 8.1 Hz, 1H, H9), 5.50 (d, J = 6.3 Hz, 1H, H8), 4.62 (s, 2H, H16, H17, Fc), 4.41 (s, 2H, H15, H18, Fc), 4.27 (s, 5H, H19-H23, Fc) 4.12 (d, J = 5.7 Hz, 1H, H7), 1.13 (s, 6H, H3, H10); 13C NMR (CDCl3) δ (ppm): 181.1 (C1, C=O), 175.9 (C11, C=O), 136.7 (C5), 123.0 (C9), 119.3 (q, JC-F = 321 Hz, C25, CF3), 112.2 (C8), 96.1 (C13), 83.0 (C12), 73.1 (C15, C18, Fc), 73.0 (C14, Fc), 70.5 (C19-C23, Fc), 59.6 (C2), 37.6 (C7), 29.7 (C16, C18, Fc), 22.7 (C3), 20.1 (C10); HRMS-DART+ (19 eV): elemental composition C23H21F3FeNO5S, [M+1]+, correlation (error of −1.43 ppm) exact value 536.0441 Daltons and precise value of 536.0434 Daltons; complementarily, the provided unsaturation data (14.0) were in agreement with the structure.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










