1. Introduction
Cancer is a severe disease causing a considerable number of deaths globally [
1]. It is characterized by uncontrolled and aberrant cell growth, rapid proliferation, or invasion into the human body, constituting formidable illnesses [
2]. According to diagnostic and reporting data from international cancer research institutions [
3], there have been over 19.3 million new cases of cancer worldwide, resulting in approximately 10 million deaths by the year 2020. The global cancer burden is expected to be 28.4 million cases in 2040. Conventional cancer treatment methods include radiation therapy, chemotherapy, surgery, as well as targeted drugs and immunotherapy [
4]. However, commonly employed techniques such as radiation therapy and chemotherapy have detrimental effects on healthy cells, exhibiting noticeable side effects, low success rates, and carrying the risk of relapse. Additionally, these methods are financially burdensome [
5]. Although targeted drug therapies do not harm normal cells, they may still induce certain side effects such as skin inflammation, fatigue, nausea, and vomiting [
6]. Furthermore, traditional drug treatments often lead to the development of drug resistance in cancer cells [
7]. Therefore, there is an urgent need to develop novel anticancer drugs that can effectively inhibit the rapid proliferation of cancer cells.
The emergence of anticancer peptides has opened up new avenues for cancer treatments. Anticancer peptides are naturally occurring small-molecule peptides composed of 5–40 amino acids, known for their high biocompatibility and low toxicity [
8]. The identification and characterization of these peptides in tumor biology experiments are time-consuming, labor-intensive, costly, and challenging. Therefore, there is an urgent need to seek rapid and accurate methods for identifying anticancer peptides. Anticancer peptides have demonstrated promising therapeutic effects in cancer treatments and have been recognized as potential anticancer agents [
9,
10,
11]. Currently, a growing number of anticancer peptides have been identified and validated from protein sequences through clinical experiments. Peelle et al. [
12] showcased the effectiveness of intracellular protein scaffold-mediated random peptide libraries using mammalian cell phenotypic screening methods. Norman et al. [
13], on the other hand, employed genetic approaches to select and inhibit bio-pathway peptides. However, the use of these identification methods is time-consuming, labor-intensive, costly, and challenging. Hence, there is an urgent need to explore rapid and accurate methods for identifying anticancer peptides [
14].
Many computational techniques have been widely applied in the field of bioinformatics to solve various problems [
14]. In the recognition of anticancer peptides (ACPs), machine learning has demonstrated absolute advantages and prospects [
15,
16,
17,
18,
19]. Over the past few years, a series of traditional machine learning methods have been proposed for ACPs identification. These traditional methods require manual design of features to classify protein sequences. As a result, various methods for extracting effective features have emerged, among which the support vector machine (SVM) model is the most commonly used method. Tyagi et al. [
9] first proposed the use of machine learning models for ACPs identification. They developed the AntiCP model, which selected amino acid composition (AAC) [
20], split AAC (using N-terminal and C-terminal residues), dipeptide composition (DPC) [
21,
22], and binary profiles feature (BPF) [
22] as features of peptide sequences. These features were used as inputs to an SVM classifier to distinguish ACPs from non-ACPs sequences. Hajisharifi et al. [
23] proposed two SVM-based methods for ACPs identification. The first method employed pseudo-amino acid composition (PAAC) [
24,
25,
26,
27,
28] to extract combination features of six physicochemical properties of amino acids. The second method extracted features from peptide sequences using the core local alignment technique and utilized SVM for binary classification. Vijayakumar et al. [
29] developed the ACPP model, which selected amino acid distribution measurement-based features and centroid composition information as features, combined with an SVM model for ACPs identification. Chen et al. [
30] developed the iACP model, which utilized g-gap dipeptide composition (g-gap DPC) for feature extraction of peptide sequences and employed radial basis function (RBF) kernel supported SVM for classification. The Random Forest (RF) [
31] model is also a commonly used method for identifying ACPs. Manavalan et al. [
2] developed the MLACP model, which selected AAC, DPC, ATC, and physicochemical properties of residues for feature extraction, and utilized SVM and RF classifiers for ACPs recognition. Akbar et al. [
32] proposed the iACP-GAEnsc model, which selected g-gap DPC, reduced amino acid alphabet composition (RAAAC), and PAAC based on amino acid hydrophobicity for feature extraction, and applied a combination of SVM, RF, probability neural network (PNN), generalized regression neural network (GRNN), and k-nearest neighbors (KNN) classification models for ACPs identification. Wei et al. [
33] proposed a PEPred-Suite model based on RF, which further improves the feature representation of ACPs to predict anticancer peptides. Boopathi et al. [
34] proposed an mACPpred model, which uses seven specific types of encoding features, including AAC, DPC, composition-transition-distribution (CTD), quasi-sequence-order (QSO), amino acid index (AAIF), binary profile (NC5), and conjoint triad (CTF) to represent a peptide sequence and cooperate with an SVM model to predict ACPs. Li et al. [
35] selected AAC, PAAC, and grouped amino acid composition (GAAC) features to construct a low dimensional feature model to identify anticancer peptides. Xu et al. [
36] proposed a sequence-based hybrid model that transformed polypeptides into feature vectors using g-gap DPC and employed SVM and RF as classifiers. Schaduangrat et al. [
37] introduced the ACPred model, which selected AAC, DPC, PAAC, amphiphilic pseudo amino acid composition (Am-PAAC), and physicochemical properties as features of peptide sequences, and used SVM and RF for ACPs identification. Meanwhile, Wei et al. [
38] developed a sequence-based anticancer predictor called ACPred-FL, which employed a two-step feature selection technique and selected peptide length, BPF, overlap property feature (OPF), twenty-one-bit feature (TOBF), CTD, AAC, g-gap DPC, and adaptive skip dipeptide composition (AKDC) as seven representation methods of features.
However, with the rapid development of the big data era in recent years, there has been an explosive increase in biological big data, making traditional machine learning algorithms inadequate for handling complex and diverse data. Deep learning methods, known for their ability to efficiently process unstructured data, have been widely applied in the field of bioinformatics. An increasing number of deep neural network models have been employed for ACPs recognition [
19,
39,
40]. Wu et al. [
41] developed PTPD, which utilized word2vec to represent k-mer sparse matrixers [
42] and employed convolutional neural networks (CNN) for ACPs recognition. Yi et al. [
43] proposed ACP-DL, which selected BPF, a reduced amino acid alphabet, and the k-mer sparse matrix as features, and applied long short-term memory (LSTM) models for ACPs prediction. Cao et al. [
44] presented the DLFF-ACP model, using AAC, DPC, k-spaced amino acid group pairs (CKSAAGP), and Geary as features, and integrating deep learning and multi-view feature fusion for ACPs identification. Ahmed et al. [
40] recently developed APC-MHCNN, a computational model for predicting anticancer peptides that utilizes a multi-headed deep CNN. In their study, they selected sequence, physicochemical, and evolutionary features as inputs to the model. By employing a deep learning approach, the ACP-MHCNN demonstrated promising performance in peptide prediction. Similarly, Sun et al. [
45] introduced ACPNet, a novel framework for identifying anticancer peptides. ACPNet incorporates peptide sequence information, physicochemical properties, and self-encoding features into its architecture. The model employs fully connected networks and recurrent neural networks to achieve accurate ACPs classification. Wang et al. [
46] proposed CL-ACP, which introduces the anticancer peptides secondary structures as additional features and uses a combined network and attention mechanism to predict anticancer peptides. Chen et al. [
47] proposed ACP-DA, which integrates BPF and k-mer sparse matrix features to represent peptide sequences and uses data augmentation to improve the predictive performance of anticancer peptides. Rao et al. [
48] proposed the ACP-GCN model, which leverages one-hot encoding and graph convolutional networks (GCN) to predict anticancer peptides. By utilizing the unique characteristics of peptide sequences and considering their structural relationships through GCN, the ACP-GCN model achieves high accuracy in ACPs identification. Zhu et al. [
49] developed the ACP-check model, which uses LSTM networks to extract time-dependent information from peptide sequences for anticancer peptides to be identified effectively. You et al. [
50] fused the sparse matrix features of BPF and the k-mer sparse matrix to construct a new bidirectional short-term memory network, which achieves the prediction of anticancer peptides through two sets of dense network layers. The aforementioned studies demonstrate significant advancements in the field of computational peptide-based cancer research. The development of these computational models provides valuable tools for predicting and identifying potential anticancer peptides, thereby facilitating the discovery of novel therapeutic agents for combating cancer.
Although the above studies have made some progress, there is still room for improvement. For instance, the above methods only consider the information derived from the amino acid primary sequence and do not take into account the spatial structural information of amino acids. In this study, we propose a novel deep learning model for ACPs prediction called ACP-BC, which is an end-to-end model that combines sequence and chemical information to predict whether a protein sequence is an ACP. The features extracted by ACP-BC are divided into three channels. The first channel extracts features through a three-layer bidirectional long short-term memory (Bi-LSTM) [
51,
52]. The original sequence is first mapped to a 256-dimensional vector through an embedding layer fused within the model, and then input into the Bi-LSTM for feature extraction. The second channel utilizes information from the chemical bidirectional encoder representation transformer (BERT) [
53,
54]. We convert the entire sequence into the form of a chemical molecular formula and then use the Simplified Molecular Input Line Entry System (SMILES) [
55,
56,
57] to further simplify it. This SMILES-encoded sequence is input into a pre-trained BERT model for fine-tuning, resulting in abstract features at a deeper level. The third channel consists of manually crafted features known to be effective, including BPF, DPC, PAAC, and k-mer sparse matrix features. These four types of features are fused together to collectively extract features at different levels of an amino acid sequence.
Our proposed method can be divided into three steps, as shown in
Figure 1. Firstly, data collection is conducted by inputting the given peptide sequences and expanding the data using two combination methods. Then, feature construction is carried out, and peptide sequences are processed by the previously mentioned Bi-LSTM, pre-trained BERT, and feature engineering methods, respectively, to extract features from the three channels. Finally, feature classification connects the features of these three channels and uses a fully connected layer to classify peptide sequences, train the model, and evaluate the trained model. The experimental results indicate that our designed model can better extract deep features, utilizing better representation of peptide sequences and a reasonable model structure. ACP-BC can achieve high accuracy and can be more effectively applied to ACPs prediction.
3. Discussion
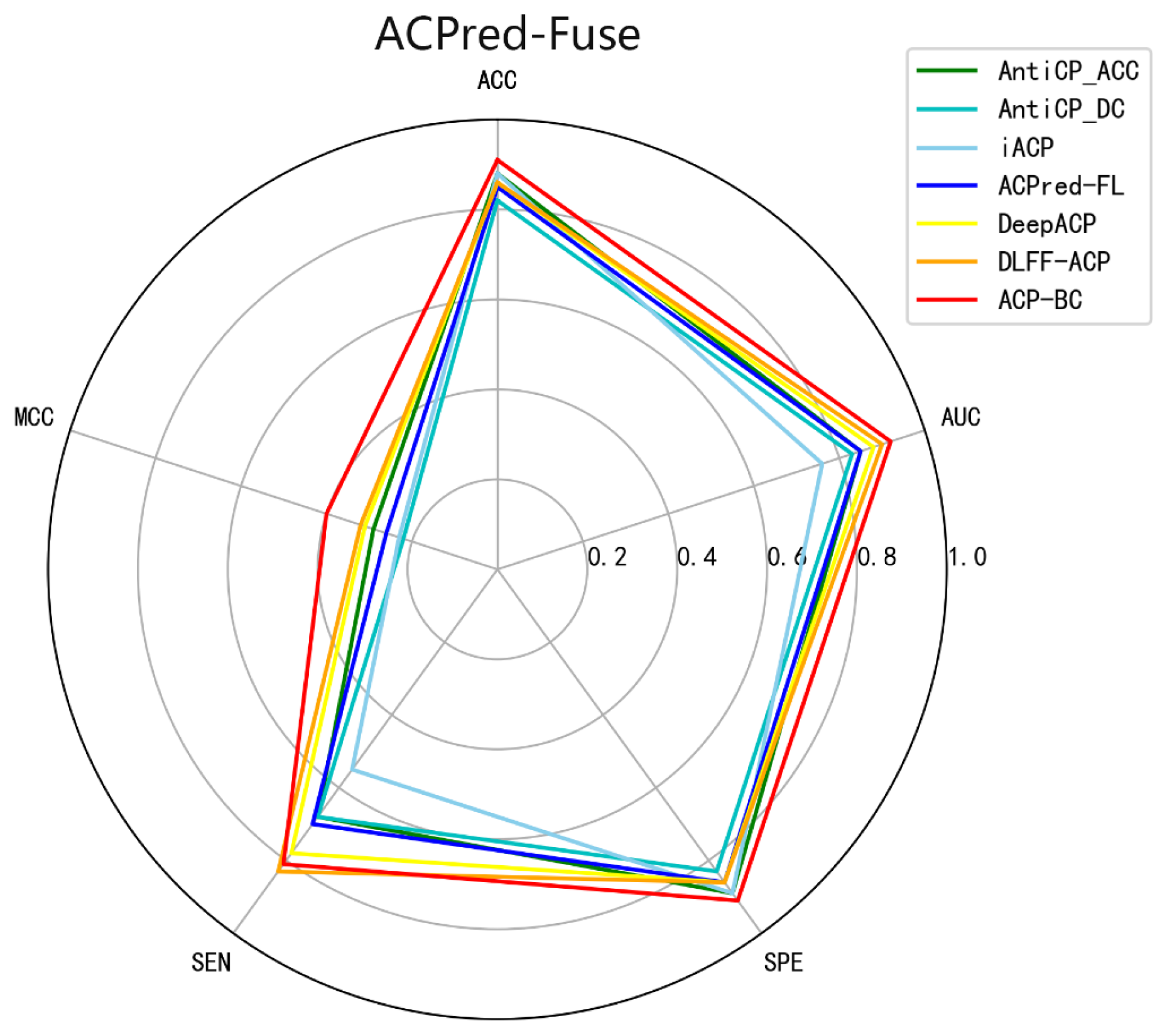
Cancer is a prevalent and deadly disease, and its treatment has always been a long-standing challenge. Anticancer peptides have demonstrated potent anticancer activity, and distinguishing between anticancer and non-anticancer peptides is a crucial step in anticancer peptide research. In this study, we propose a novel anticancer peptide identification model called ACP-BC, which integrates multiple features including sequence information and chemical information. Extensive experiments have shown that our method achieves high accuracy and robustness, making it suitable for anticancer peptide identification. In the following sections, we analyze the reasons behind the improved performance of our model.
Firstly, we employ an enhanced data augmentation method to preprocess the dataset, randomly replacing, shuffling, reversing, or subsampling each amino acid residue in each sequence with a probability of p. Experimental results demonstrate that using p = 0.01 to augment the entire ACPs dataset effectively enhances the model’s performance and generalization by doubling the amount of data.
For feature extraction from protein sequence information, we utilize BI-LSTM, BERT, and manually selected features as three channels to effectively capture different hierarchical features of amino acid sequences. In the first channel, the entire protein sequence is encoded through an embedding layer, which is trained together with the entire model. The resulting embedded representation of the original sequence is then input into a three-layer bidirectional LSTM, and the output of the Bi-LSTM serves as the information for the first channel. Experimental findings suggest that setting the embedding layer and LSTM’s hidden neuron counts to 256 and 512, respectively, yields optimal results. In the second channel, we introduce the structural information of amino acids in an innovative way. We employ a BERT model pre-trained on SMILES sequences to extract deep abstract features. Initially, the original sequence is converted into a molecular structure representation, and then SMILES, a structured symbolic language, is utilized to simplify chemical molecular formulas. Subsequently, the obtained SMILES-formatted data is input into the BERT model for fine-tuning, resulting in chemical molecular formula features related to protein sequences. In the process of selecting detailed parameters for the BERT model, a series of experiments are conducted, ultimately choosing ChemBERTa with a SMILES tokenizer as the feature extractor. In the third channel, we optimize the feature engineering methods used in other studies. We combine BPF, DPC, and PAAC features as manually selected features, which extract positional information, compositional information, and local information of the protein sequences, respectively. Through a series of ablation experiments, we demonstrate the effectiveness of these manually selected features in capturing diverse aspects of the sequences. The three channels of feature information complement each other, and their fusion enables better extraction of various hidden layers of information in protein sequences. To better fine-tune the BERT model, we employ multiple fully connected layers to integrate, abstract, and predict the extracted features. In the entire model, we assign a smaller learning rate to the BERT model than other layers by an order of magnitude, which is more suitable for fine-tuning training. In evaluating the effectiveness of our approach, we train and test our model on ACP740 and ACP240 datasets, and validate it on an independent ACP20 dataset [
19,
45,
62]. Experimental results demonstrate that our designed model achieves excellent performance, surpassing other models in multiple metrics such as ACC and MCC. It also performs exceptionally well on the independent dataset. In summary, our model exhibits great effectiveness, robustness, and generalization ability, and it can be readily applied to the identification of ACPs.
5. Conclusions
In this study, we proposed a model aimed at accurately identifying anticancer peptides by Bi-LSTM and chemical information, called ACP-BC, which is a two-class classification problem. To find useful features, we compare the performance of an autoencoder feature, a newly proposed chemical molecular feature, and four commonly used feature combinations on two benchmark datasets. The experimental results demonstrate that the combination of these six features plays a positive role in ACPs and non-ACPs identification. Finally, we employ a fully connected network to handle the feature combinations for ACPs recognition.
Comparing with six existing state-of-the-art methods, ACP-BC shows improvements in various performance metrics, including ACC, MCC, SE, SP, and AUC. ACP-BC also exhibits improved performance metrics on the ACP740 dataset. When tested on an independent dataset ACP20, ACP-BC accurately predicts all 10 ACPs samples. Through a series of experiments, we demonstrate the effective and accurate identification of ACPs and non-ACPs by ACP-BC. However, our proposed method still has limitations and we are still unable to accurately identify certain anticancer peptides. There are many reasons for this phenomenon. On the one hand, this may be a problem with the dataset. Some samples of anticancer peptides are relatively unique and have significant distribution differences compared to other samples, making it difficult for the model to identify them based on experience. On the other hand, it may be that our model itself needs improvement and fails to accurately extract more effective and recognizable features, resulting in the model being unable to accurately recognize anticancer peptides. In following research work, more datasets can be introduced and combined with new machine learning methods, such as contrastive learning, to reduce the impact of non real negative samples on model performance. At the same time, we try to gain a deeper understanding of anticancer peptides and use methods that are more suitable for extracting anticancer peptides features, in order to identify anticancer peptides more accurately. In future work, we plan to incorporate more complex and effective features and deploy them on a network to develop an intelligent system for accurate ACPs identification.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}