
The BRCA1 c.4096+1G>A Is a Founder Variant Which Originated in Ancient Times
 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results
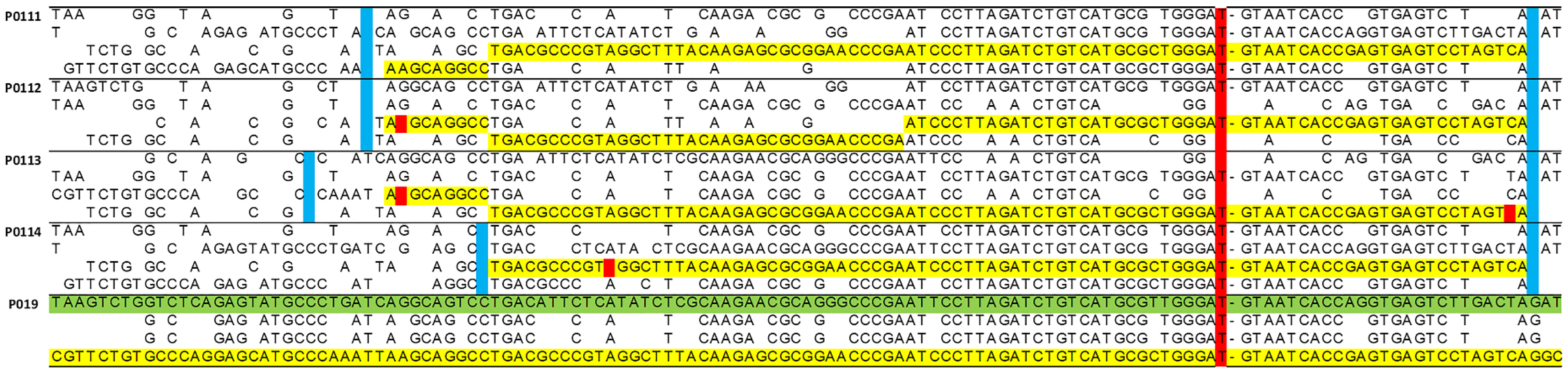
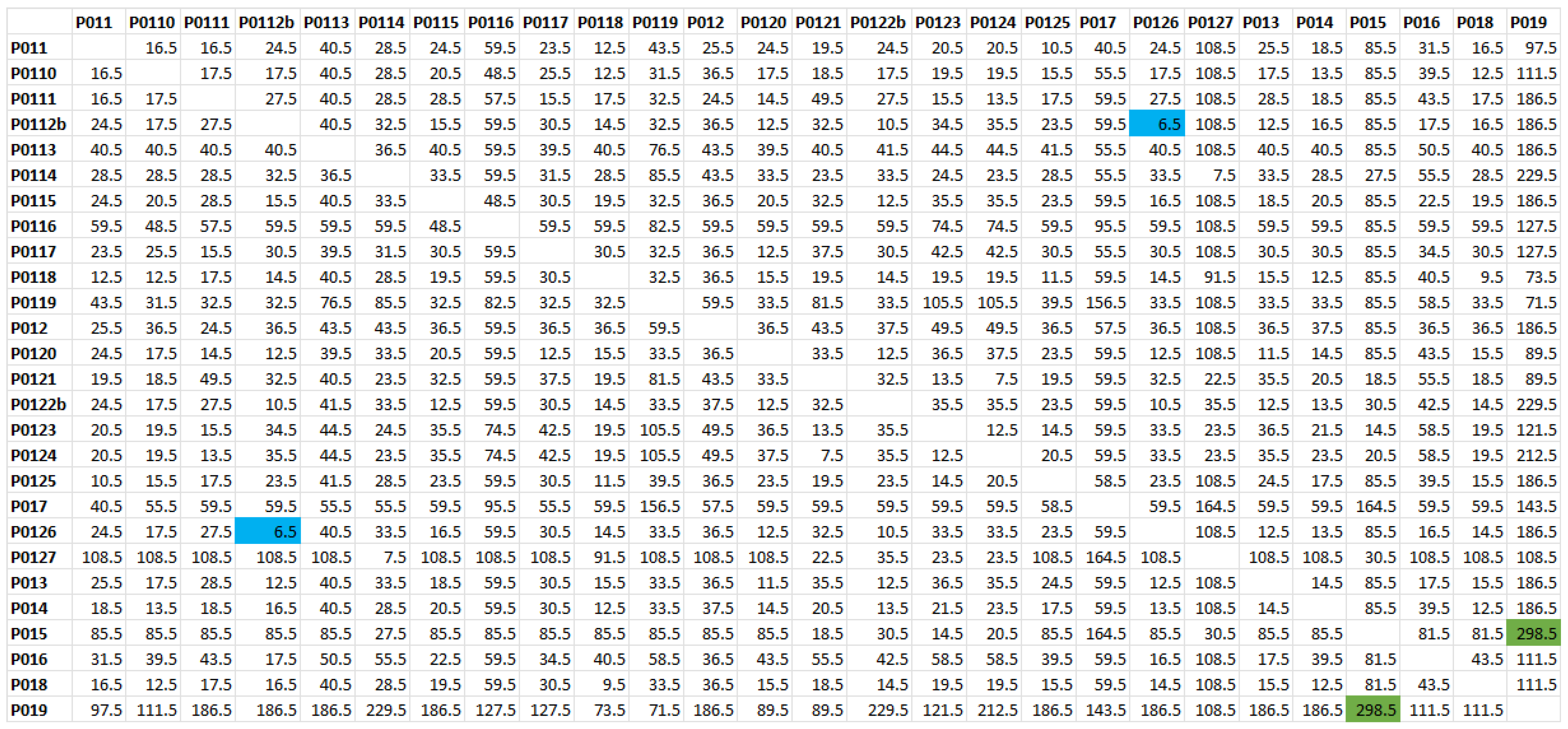
2.1. Haplotype Analysis
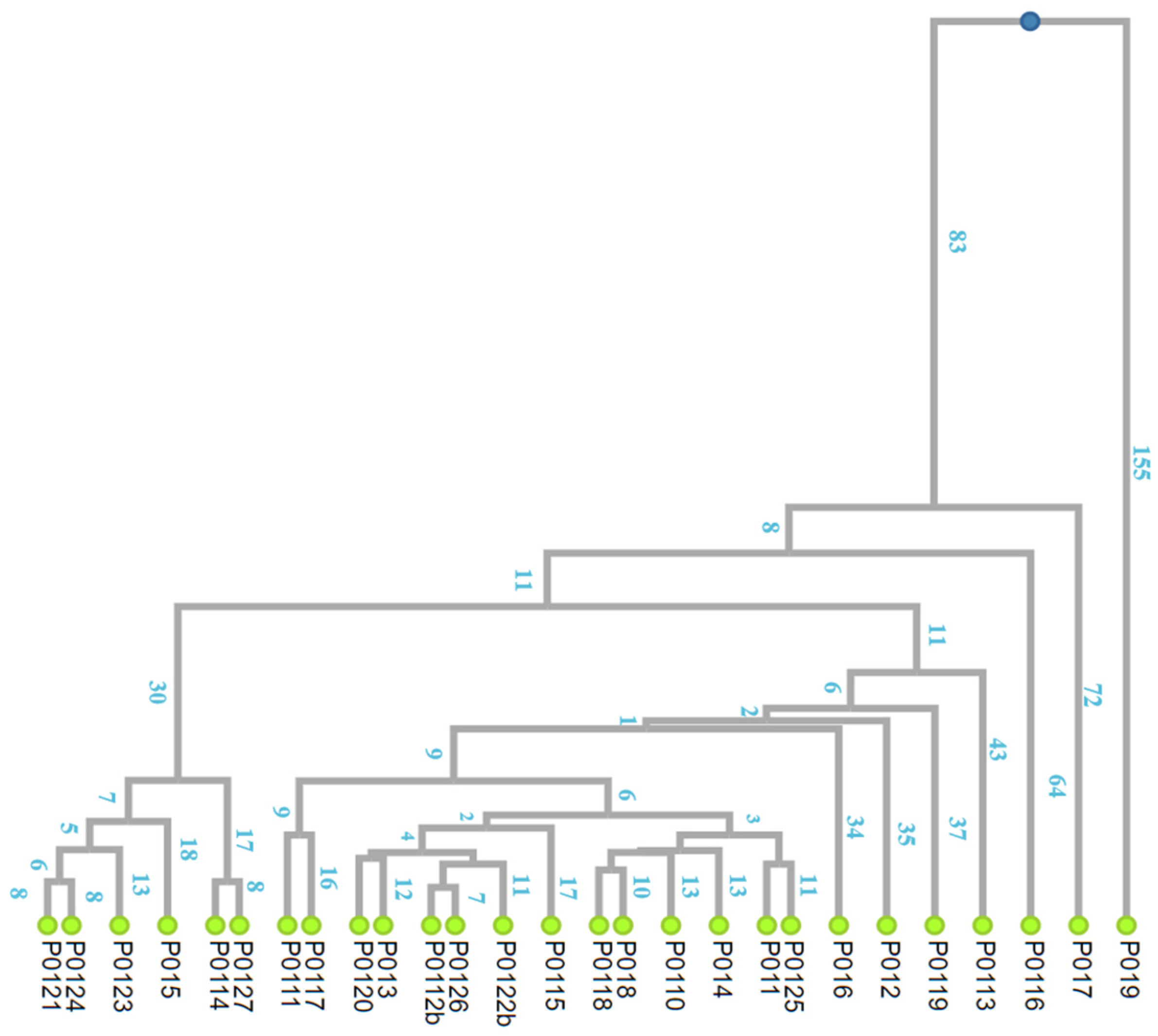
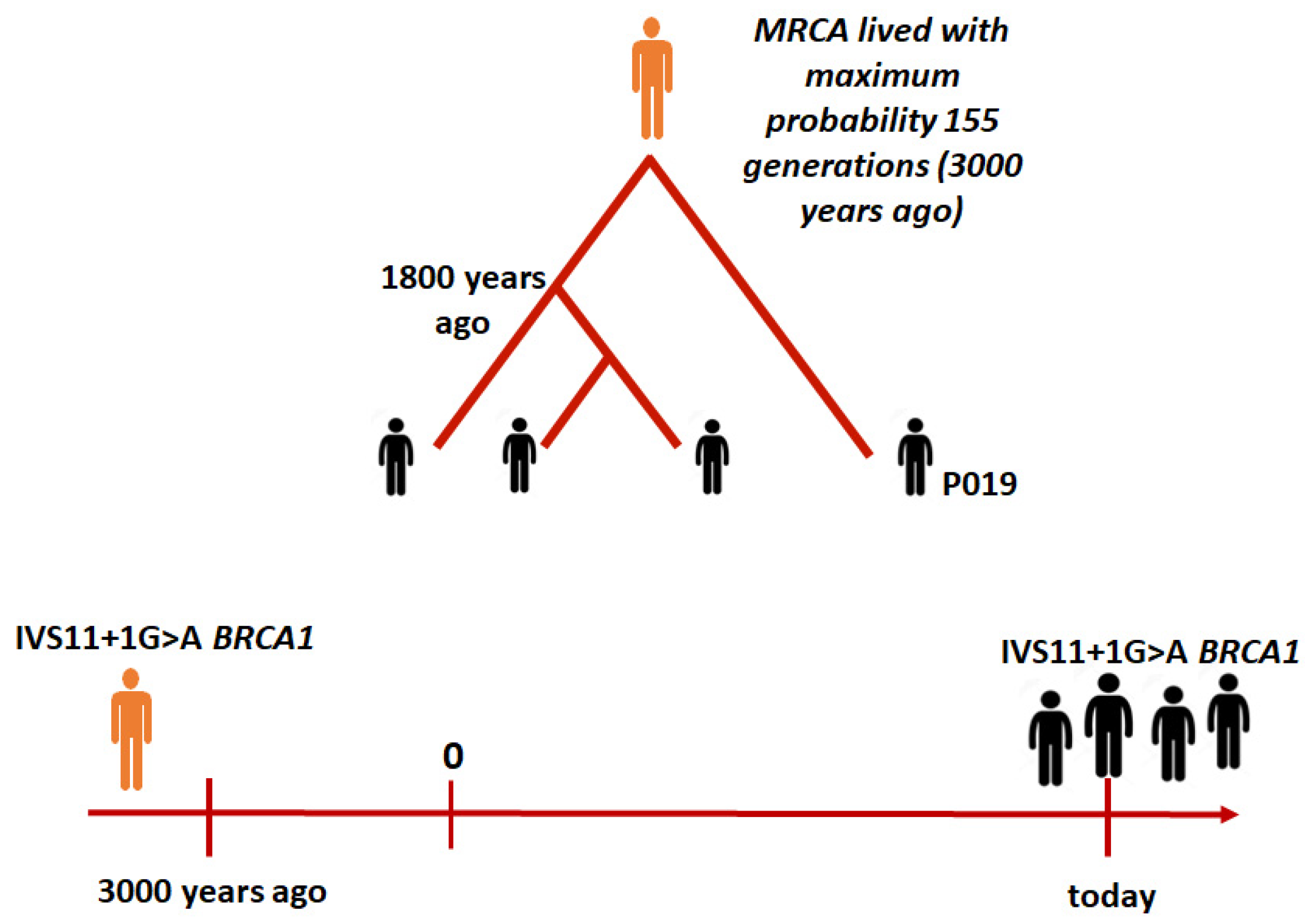
2.2. The Time to the MRCA
3. Discussion
4. Materials and Methods
4.1. Families and Genotyping
4.1.1. Description of Recruited Families
4.1.2. DNA Extraction and Genotyping
4.2. Haplotype Reconstruction
4.2.1. Determination of the Gametic Phase of the Markers
4.2.2. Inferring the Genealogy of the c.4096+1G>A Variant
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Angeli, D.; Salvi, S.; Tedaldi, G. Genetic Predisposition to Breast and Ovarian Cancers: How Many and Which Genes to Test? Int. J. Mol. Sci. 2020, 21, 1128. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Beitsch, P.D.; Whitworth, P.W.; Hughes, K.; Patel, R.; Rosen, B.; Compagnoni, G.; Baron, P.; Simmons, R.; Smith, L.A.; Grady, I.; et al. Underdiagnosis of Hereditary Breast Cancer: Are Genetic Testing Guidelines a Tool or an Obstacle? J. Clin. Oncol. 2019, 37, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.I.; King, M.C. Inherited breast and ovarian cancer. Hum. Mol. Genet. 1995, 4, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. How do mutations affecting the breast cancer genes BRCA1 and BRCA2 cause cancer susceptibility? DNA Repair. 2019, 81, 102668. [Google Scholar] [CrossRef]
- Brose, M.S.; Rebbeck, T.R.; Calzone, K.A.; Stopfer, J.E.; Nathanson, K.L.; Weber, B.L. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J. Natl. Cancer Inst. 2002, 94, 1365–1372. [Google Scholar] [CrossRef]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef]
- Chen, J.; Bae, E.; Zhang, L.; Hughes, K.; Parmigiani, G.; Braun, D.; Rebbeck, T.R. Penetrance of Breast and Ovarian Cancer in Women Who Carry a BRCA1/2 Mutation and Do Not Use Risk-Reducing Salpingo-Oophorectomy: An Updated Meta-Analysis. JNCI Cancer Spectr. 2020, 4, pkaa029. [Google Scholar] [CrossRef]
- Borde, J.; Ernst, C.; Wappenschmidt, B.; Niederacher, D.; Weber-Lassalle, K.; Schmidt, G.; Hauke, J.; Quante, A.S.; Weber-Lassalle, N.; Horvath, J.; et al. Performance of Breast Cancer Polygenic Risk Scores in 760 Female CHEK2 Germline Mutation Carriers. J. Natl. Cancer Inst. 2021, 113, 893–899. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Tonin, P.; Weber, B.; Offit, K.; Couch, F.; Rebbeck, T.R.; Neuhausen, S.; Godwin, A.K.; Daly, M.; Wagner-Costalos, J.; Berman, D.; et al. Frequency of recurrent BRCA1 and BRCA2 mutations in Ashkenazi Jewish breast cancer families. Nat. Med. 1996, 2, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Rafnar, T.; Benediktsdottir, K.R.; Eldon, B.J.; Gestsson, T.; Saemundsson, H.; Olafsson, K.; Salvarsdottir, A.; Steingrimsson, E.; Thorlacius, S. BRCA2, but not BRCA1, mutations account for familial ovarian cancer in Iceland: A population-based study. Eur. J. Cancer 2004, 40, 2788–2793. [Google Scholar] [CrossRef] [PubMed]
- Baudi, F.; Quaresima, B.; Grandinetti, C.; Cuda, G.; Faniello, C.; Tassone, P.; Barbieri, V.; Bisegna, R.; Ricevuto, E.; Conforti, S.; et al. Evidence of a founder mutation of BRCA1 in a highly homogeneous population from southern Italy with breast/ovarian cancer. Hum. Mutat. 2001, 18, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Calo, V.; Bruno, L.; Schiro, V.; Agnese, V.; Cascio, S.; Foddai, E.; Fanale, D.; Rizzo, S.; Di Gaudio, F.; et al. Is BRCA1-5083del19, identified in breast cancer patients of Sicilian origin, a Calabrian founder mutation? Breast Cancer Res. Treat. 2009, 113, 67–70. [Google Scholar] [CrossRef]
- Incorvaia, L.; Fanale, D.; Bono, M.; Calo, V.; Fiorino, A.; Brando, C.; Corsini, L.R.; Cutaia, S.; Cancelliere, D.; Pivetti, A.; et al. BRCA1/2 pathogenic variants in triple-negative versus luminal-like breast cancers: Genotype-phenotype correlation in a cohort of 531 patients. Ther. Adv. Med. Oncol. 2020, 12, 1758835920975326. [Google Scholar] [CrossRef] [PubMed]
- Incorvaia, L.; Fanale, D.; Badalamenti, G.; Bono, M.; Calo, V.; Cancelliere, D.; Castiglia, M.; Fiorino, A.; Pivetti, A.; Barraco, N.; et al. Hereditary Breast and Ovarian Cancer in Families from Southern Italy (Sicily)-Prevalence and Geographic Distribution of Pathogenic Variants in BRCA1/2 Genes. Cancers 2020, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Marroni, F.; Cipollini, G.; Peissel, B.; D’Andrea, E.; Pensabene, M.; Radice, P.; Caligo, M.A.; Presciuttini, S.; Bevilacqua, G. Reconstructing the genealogy of a BRCA1 founder mutation by phylogenetic analysis. Ann. Hum. Genet. 2008, 72, 310–318. [Google Scholar] [CrossRef]
- Holm, H.; Gudbjartsson, D.F.; Sulem, P.; Masson, G.; Helgadottir, H.T.; Zanon, C.; Magnusson, O.T.; Helgason, A.; Saemundsdottir, J.; Gylfason, A.; et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat. Genet. 2011, 43, 316–320. [Google Scholar] [CrossRef]
- Fu, Z.; Nakayama, T.; Sato, N.; Izumi, Y.; Kasamaki, Y.; Shindo, A.; Ohta, M.; Soma, M.; Aoi, N.; Sato, M.; et al. Haplotype-based case-control study of CYP4A11 gene and myocardial infarction. Hereditas 2012, 149, 91–98. [Google Scholar] [CrossRef]
- Slatkin, M.; Rannala, B. Estimating allele age. Annu. Rev. Genomics Hum. Genet. 2000, 1, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Bertorelle, G. Using linked markers to infer the age of a mutation. Hum. Mutat. 2001, 18, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.; Rauch, A.; Rotger, M.; Fellay, J.; Martinez, R.; Fux, C.; Thurnheer, C.; Gunthard, H.F.; Goldstein, D.B.; Furrer, H.; et al. The HCP5 single-nucleotide polymorphism: A simple screening tool for prediction of hypersensitivity reaction to abacavir. J. Infect. Dis. 2008, 198, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Bonatti, F.; Pepe, C.; Tancredi, M.; Lombardi, G.; Aretini, P.; Sensi, E.; Falaschi, E.; Cipollini, G.; Bevilacqua, G.; Caligo, M.A. RNA-based analysis of BRCA1 and BRCA2 gene alterations. Cancer Genet. Cytogenet. 2006, 170, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Rivera, D.; Paudice, M.; Gismondi, V.; Anselmi, G.; Vellone, V.G.; Varesco, L.; Ligurian, B.W.G. Implementing NGS-based BRCA tumour tissue testing in FFPE ovarian carcinoma specimens: Hints from a real-life experience within the framework of expert recommendations. J. Clin. Pathol. 2021, 74, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Santonocito, C.; Rizza, R.; Paris, I.; Marchis, L.; Paolillo, C.; Tiberi, G.; Scambia, G.; Capoluongo, E. Spectrum of Germline BRCA1 and BRCA2 Variants Identified in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome. Cancers 2020, 12, 1286. [Google Scholar] [CrossRef] [PubMed]
- Tsaousis, G.N.; Papadopoulou, E.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Kampouri, S.; Diamantopoulos, N.; Floros, T.; Iosifidou, R.; Katopodi, O.; et al. Analysis of hereditary cancer syndromes by using a panel of genes: Novel and multiple pathogenic mutations. BMC Cancer 2019, 19, 535. [Google Scholar] [CrossRef] [PubMed]
- Susswein, L.R.; Marshall, M.L.; Nusbaum, R.; Vogel Postula, K.J.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. 2016, 18, 823–832. [Google Scholar] [CrossRef]
- Palmero, E.I.; Carraro, D.M.; Alemar, B.; Moreira, M.A.M.; Ribeiro-Dos-Santos, A.; Abe-Sandes, K.; Galvao, H.C.R.; Reis, R.M.; de Padua Souza, C.; Campacci, N.; et al. The germline mutational landscape of BRCA1 and BRCA2 in Brazil. Sci. Rep. 2018, 8, 9188. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.H.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef]
- Tammaro, C.; Raponi, M.; Wilson, D.I.; Baralle, D. BRCA1 exon 11 alternative splicing, multiple functions and the association with cancer. Biochem. Soc. Trans. 2012, 40, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Raponi, M.; Smith, L.D.; Silipo, M.; Stuani, C.; Buratti, E.; Baralle, D. BRCA1 exon 11 a model of long exon splicing regulation. RNA Biol. 2014, 11, 351–359. [Google Scholar] [CrossRef]
- Gelli, E.; Colombo, M.; Pinto, A.M.; De Vecchi, G.; Foglia, C.; Amitrano, S.; Morbidoni, V.; Imperatore, V.; Manoukian, S.; Baldassarri, M.; et al. Usefulness and Limitations of Comprehensive Characterization of mRNA Splicing Profiles in the Definition of the Clinical Relevance of BRCA1/2 Variants of Uncertain Significance. Cancers 2019, 11, 295. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Garibay, G.; Fernandez-Garcia, I.; Mazoyer, S.; Leme de Calais, F.; Ameri, P.; Vijayakumar, S.; Martinez-Ruiz, H.; Damiola, F.; Barjhoux, L.; Thomassen, M.; et al. Altered regulation of BRCA1 exon 11 splicing is associated with breast cancer risk in carriers of BRCA1 pathogenic variants. Hum. Mutat. 2021, 42, 1488–1502. [Google Scholar] [CrossRef] [PubMed]
- Gandolfo, L.C.; Bahlo, M.; Speed, T.P. Dating rare mutations from small samples with dense marker data. Genetics 2014, 197, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Tuazon, A.M.A.; Lott, P.; Bohorquez, M.; Benavides, J.; Ramirez, C.; Criollo, A.; Estrada-Florez, A.; Mateus, G.; Velez, A.; Carmona, J.; et al. Haplotype analysis of the internationally distributed BRCA1 c.3331_3334delCAAG founder mutation reveals a common ancestral origin in Iberia. Breast Cancer Res. 2020, 22, 108. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, R.; Diop, J.P.D.; Bourdon-Huguenin, V.; Dem, A.; Diouf, D.; Dieng, M.M.; Diop, P.S.; Kane Gueye, S.M.; Ba, S.A.; Dia, Y.; et al. Evidence for an ancient BRCA1 pathogenic variant in inherited breast cancer patients from Senegal. NPJ Genom. Med. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.; Chan, S.H.; Teo, J.X.; Chong, S.T.; Li, S.T.; Courtney, E.; Ishak, D.; Sankar, H.; Ang, Z.L.T.; Chiang, J.; et al. Investigation into the origins of an ancient BRCA1 founder mutation identified among Chinese families in Singapore. Int. J. Cancer 2021, 148, 637–645. [Google Scholar] [CrossRef]
- Campos, B.; Diez, O.; Odefrey, F.; Domenech, M.; Moncoutier, V.; Martinez-Ferrandis, J.I.; Osorio, A.; Balmana, J.; Barroso, A.; Armengod, M.E.; et al. Haplotype analysis of the BRCA2 9254delATCAT recurrent mutation in breast/ovarian cancer families from Spain. Hum. Mutat. 2003, 21, 452. [Google Scholar] [CrossRef]
- Bergman, A.; Einbeigi, Z.; Olofsson, U.; Taib, Z.; Wallgren, A.; Karlsson, P.; Wahlstrom, J.; Martinsson, T.; Nordling, M. The western Swedish BRCA1 founder mutation 3171ins5; a 3.7 cM conserved haplotype of today is a reminiscence of a 1500-year-old mutation. Eur. J. Hum. Genet. 2001, 9, 787–793. [Google Scholar] [CrossRef]
- Francovich, L. Le Immigrazioni in Toscana: L’origine della Popolazione Locale dall’anno mille ad Oggi Attraverso una Rassegna Bibliografica; Edizioni Regione Toscana: Florence, Italy, 1999; pp. 7–29. Available online: https://www.regione.toscana.it/documents/10180/12662099/1ef68d378200c0f1f482eac197f971a5_1leimmigrazioniintoscana.pdf/ec247c28-8622-48f6-bf34-e75791edba41 (accessed on 1 October 2023).
- Amos, C.I.; Dennis, J.; Wang, Z.; Byun, J.; Schumacher, F.R.; Gayther, S.A.; Casey, G.; Hunter, D.J.; Sellers, T.A.; Gruber, S.B.; et al. The OncoArray Consortium: A Network for Understanding the Genetic Architecture of Common Cancers. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 126–135. [Google Scholar] [CrossRef]
- Hofmeister, R.J.; Ribeiro, D.M.; Rubinacci, S.; Delaneau, O. Accurate rare variant phasing of whole-genome and whole-exome sequencing data in the UK Biobank. Nat. Genet. 2023, 55, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Sneath, P.H.A.; Sokal, R.R. Numerical taxonomy. In The Principles and Practice of Numerical Classification; W.H. Freeman and Company: San Francisco, CA, USA, 1973; pp. 230–234. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Gender | Age of the Last Follow-Up or Age of Death | Relationship to the Probamd | c.4096+1G>A BRCA1 | Tumor | Age of Onset |
|---|---|---|---|---|---|---|
| P011 | F | 80 | Proband | Carrier | HGSOC | 52 |
| P012 | F | 76 | Proband | Carrier | Breast/HGSOC | 61/73 |
| P013 | F | 77 | Proband | Carrier | Breast/HGSOC | 62/67 |
| P013a | M | 75 | Brother | Wild-type | Healthy | |
| P014 | F | 51 | Proband | Carrier | Breast | 49 |
| P015 | F | 49 | Proband | Carrier | Breast | 39 |
| P016 | F | 59 | Proband | Carrier | Breast | 49 |
| P016a | F | 62 | Sister | Carrier | Healthy | |
| P016b | F | 26 | Nephew | Carrier | Healthy | |
| P016c | F | 80 | Mother | Wild-type | Breast | 79 |
| P017 | F | 33 | Proband | Carrier | Breast | 25 |
| P017a | M | 59 | Father | Carrier | Healthy | |
| P017b | F | 56 | Mother | Wild-type | Breast | 47 |
| P018 | F | 76 | Proband | Carrier | Breast | 60 |
| P018a | F | 56 | Nephew | Wild-type | Healthy | |
| P018b | F | 90 | Sister | Carrier | Breast | 86 |
| P019 | F | 43 | Proband | Carrier | Breast | 37 |
| P0110 | F | 63 | Proband | Carrier | Breast | 50 |
| P0110a | F | 38 | Daughter | Wild-type | Healthy | |
| P0111 | F | 89 | Proband | Carrier | Breast | 72 |
| P0111a | F | 65 | Daughter | Carrier | Healthy | |
| P0111b | M | 33 | Nephew | Carrier | Healthy | |
| P0111c | F | 28 | Nephew | Wild-type | Healthy | |
| P0112 | F | 44 | Proband | Carrier | Breast | 32 |
| P0112a | M | 71 | Father | Wild-type | Healthy | |
| P0112b | F | 71 | Mother | Carrier | Healthy | |
| P0113 | F | 39 | Proband | Carrier | Healthy | |
| P0114 | F | 54 | Proband | Carrier | Breast | 49 |
| P0115 | F | 69 | Proband | Carrier | Breast/Uterus | 55/65 |
| P0115a | F | 44 | Daughter | Carrier | Healthy | |
| P0116 | F | 79 | Proband | Carrier | Breast | 48 |
| P0116a | F | 58 | Daughter | Carrier | Healthy | |
| P0116b | F | 49 | Daughter | Wild-type | Healthy | |
| P0117 | F | 63 | Proband | Carrier | HGSOC | 59 |
| P0118 | F | 81 | Proband | Carrier | Breast/HGSOC | 77/72 |
| P0118a | F | 56 | Daughter | Wild-type | Healthy | |
| P0119 | F | 74 | Proband | Carrier | HGSOC | 61 |
| P0120 | F | 65 | Proband | Carrier | HGSOC | 62 |
| P0121 | F | 58 | Proband | Carrier | Breast | 54 |
| P0122 | F | 46 | Proband | Carrier | Breast | 44 |
| P0122a | M | 75 | Father | Wild-type | Healthy | |
| P0122b | F | 75 | Mother | Carrier | Healthy | |
| P0123 | F | 83 | Proband | Carrier | HGSOC | 78 |
| P0124 | F | 54 | Proband | Carrier | Breast | 52 |
| P0125 | F | 81 | Proband | Carrier | HGSOC | 78 |
| P0126 | F | 66 | Proband | Carrier | HGSOC | 58 |
| P0126a | F | 41 | Daughter | Carrier | Healthy | |
| P0127 | F | 47 | Proband | Carrier | Breast | 45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aretini, P.; Presciuttini, S.; Pastore, A.; Galli, A.; Panepinto, S.; Tancredi, M.; Ghilli, M.; Guglielmi, C.; Sidoti, D.; Congregati, C.; et al. The BRCA1 c.4096+1G>A Is a Founder Variant Which Originated in Ancient Times. Int. J. Mol. Sci. 2023, 24, 15507. https://doi.org/10.3390/ijms242115507
Aretini P, Presciuttini S, Pastore A, Galli A, Panepinto S, Tancredi M, Ghilli M, Guglielmi C, Sidoti D, Congregati C, et al. The BRCA1 c.4096+1G>A Is a Founder Variant Which Originated in Ancient Times. International Journal of Molecular Sciences. 2023; 24(21):15507. https://doi.org/10.3390/ijms242115507
Chicago/Turabian StyleAretini, Paolo, Silvano Presciuttini, Aldo Pastore, Alvaro Galli, Sara Panepinto, Mariella Tancredi, Matteo Ghilli, Chiara Guglielmi, Diletta Sidoti, Caterina Congregati, and et al. 2023. "The BRCA1 c.4096+1G>A Is a Founder Variant Which Originated in Ancient Times" International Journal of Molecular Sciences 24, no. 21: 15507. https://doi.org/10.3390/ijms242115507