
Mitotic Dysregulation at Tumor Initiation Creates a Therapeutic Vulnerability to Combination Anti-Mitotic and Pro-Apoptotic Agents for MYCN-Driven Neuroblastoma
 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Gene Expression Profiling of TH-MYCN+/+ Transgenic Mice Identifies Mitotic Gene Set Enrichment in Pre-Malignant Neuroblasts
2.2. Prophylactic Inhibition of Mitosis Impedes MYCN-Driven Tumor Initiation and Progression to Neuroblastoma
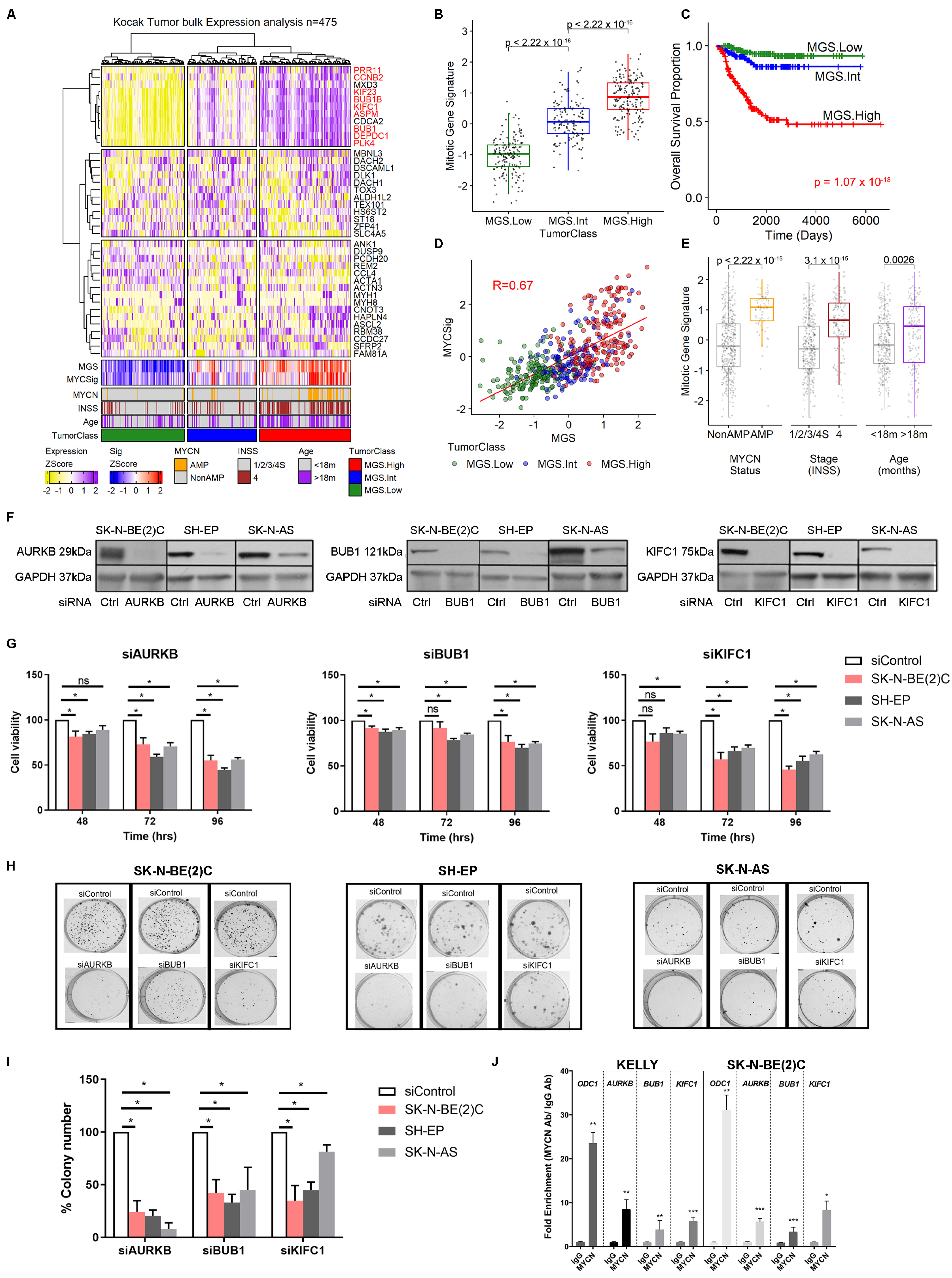
2.3. High Expression of Mitotic Genes Is Associated with Poor Prognosis in Neuroblastoma Patients
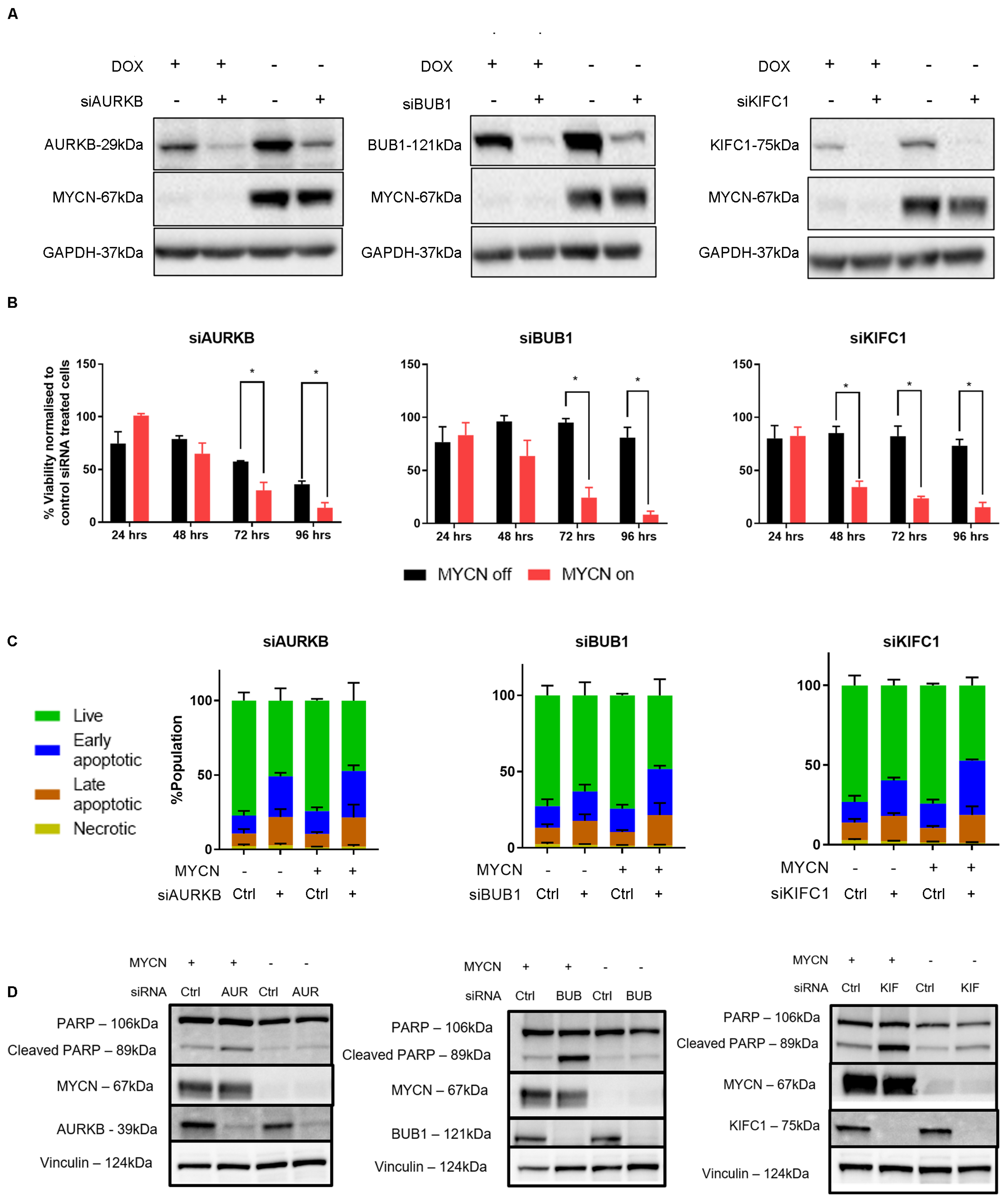
2.4. MYCN Overexpression Sensitizes Neuroblastoma Cells to Mitotic Gene Knockdown, by Induction of Apoptosis
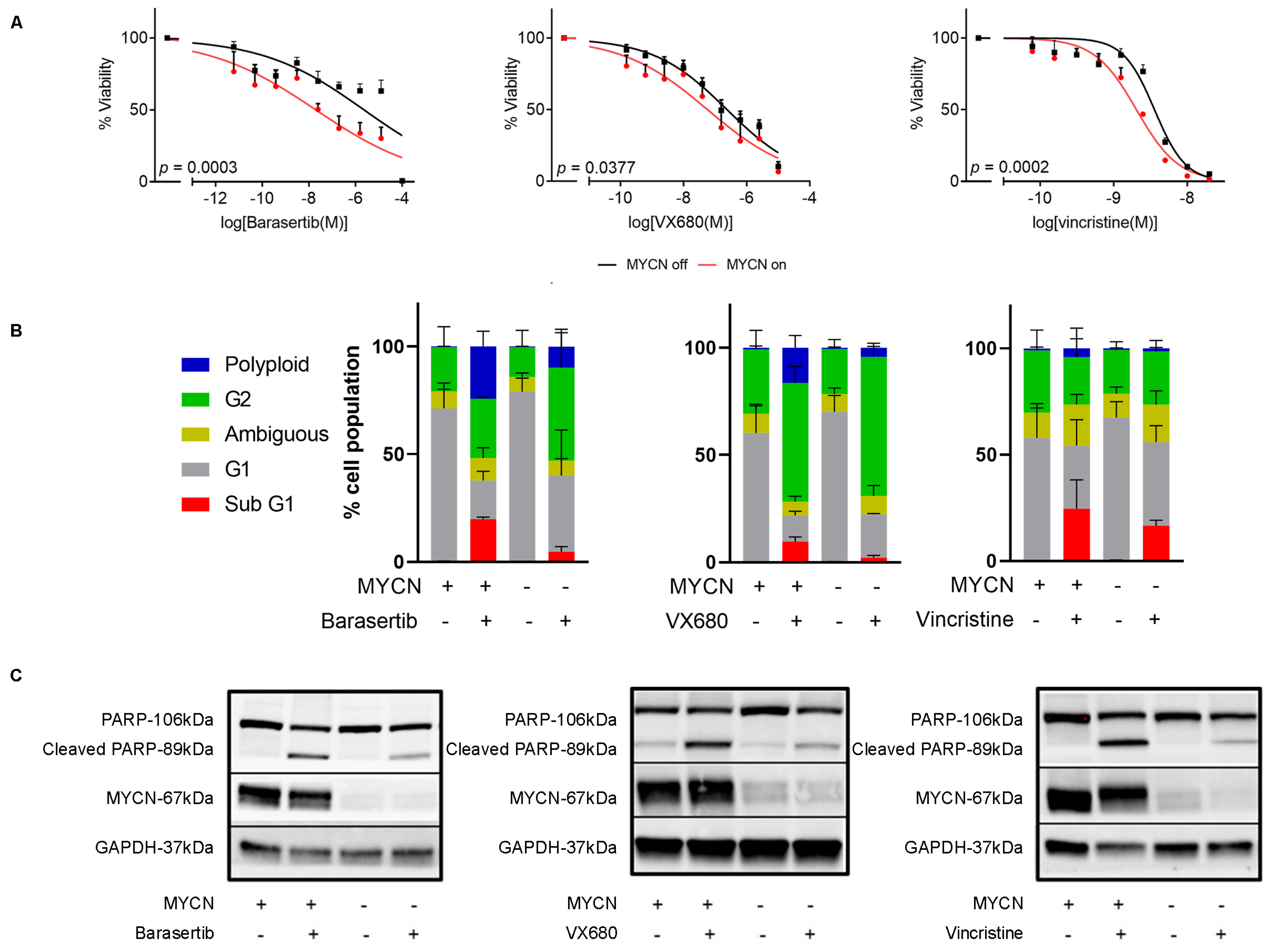
2.5. Chemical Inhibition of Mitosis Is Selectively Toxic to MYCN-Expressing Neuroblastoma
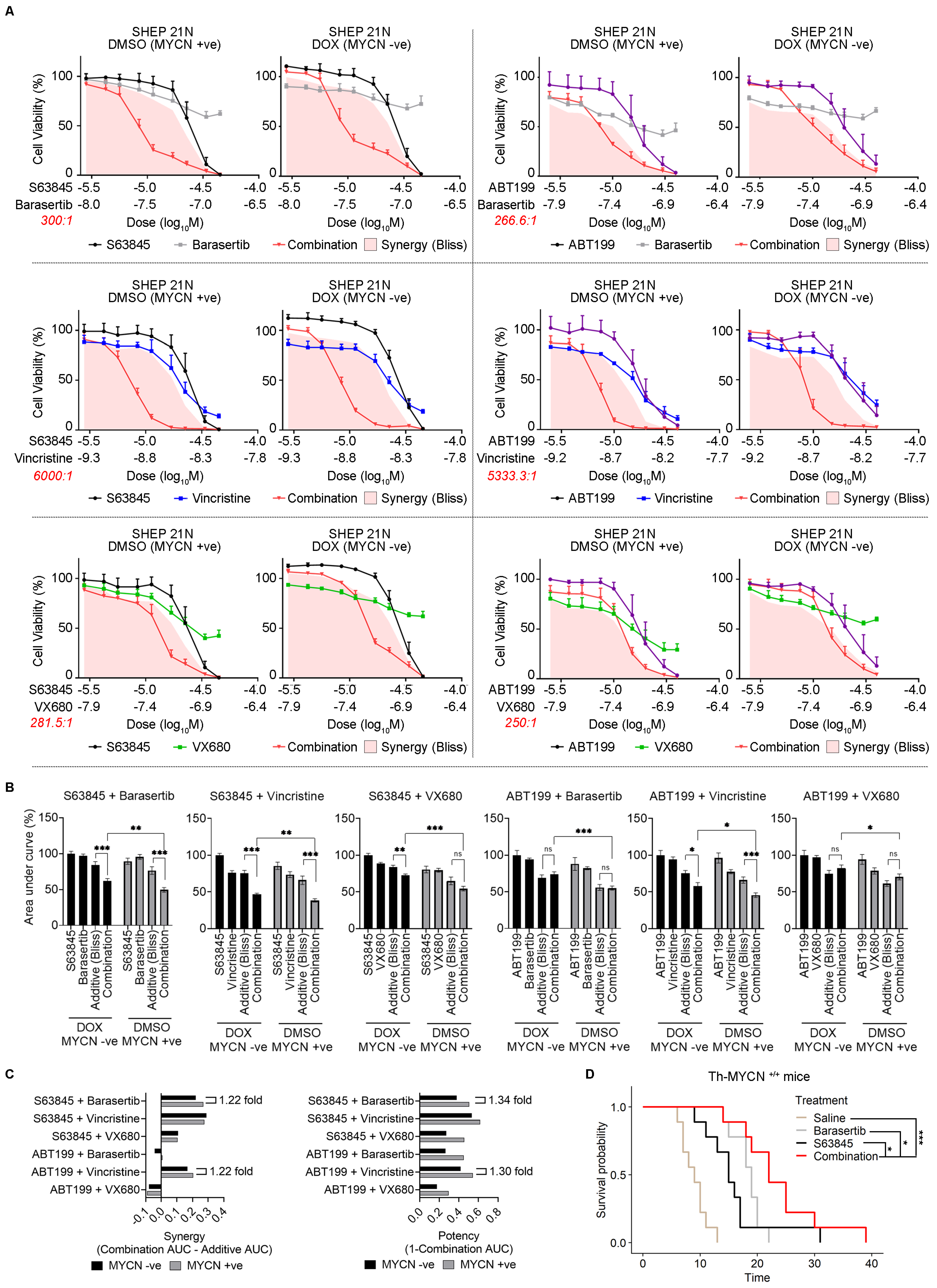
2.6. Combination Antimitotic/Pro-Apoptotic Therapy Is Efficacious in MYCN-Driven Neuroblastoma
3. Discussion
4. Materials and Methods
4.1. Neuroblastoma Cell Line Tissue Culture
4.2. siRNA Transfection
4.3. Cell Viability Assays
4.4. Flow Cytometry Assays
4.5. Colony Formation Assays
4.6. Western Blotting
4.7. Drug Treatments In Vitro
4.8. Combination Therapy In Vivo
4.9. Prophylactic Treatment in TH-MYCN Mice with Neuroblast Hyperplasia
4.10. Single-Cell Quantitative PCR
| Gene | Probe | Gene | Probe |
| Phox2b | Mm00435872_m1 | Bub1b | Mm00437811_m1 |
| hMYCN | Hs00232074_m1 | Kif23 | Mm00458527_m1 |
| Dlk1 | Mm00494477_m1 | Bub1 | Mm00660135_m1 |
| Gap43 | Mm00500404_m1 | Kifc1 | Mm00835842_g1 |
| Dbh | Mm00460472_m1 | Aspm | Mm00486659_m1 |
| Tubb3 | Mm00727586_s1 | Plk4 | Mm00550358_m1 |
| Sox10 | Mm01300162_m1 | Depdc1a | Mm01319324_g1 |
| Col1a2 | Mm00483888_m1 | Ccnb2 | Mm01171453_m1 |
| S100a1 | Mm01222827_m1 | Prr11 | Mm00723607_m1 |
4.11. Bioinformatics Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef]
- Marshall, G.M.; Carter, D.R.; Cheung, B.B.; Liu, T.; Mateos, M.K.; Meyerowitz, J.G.; Weiss, W.A. The prenatal origins of cancer. Nat. Rev. Cancer 2014, 14, 277–289. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E.; et al. Differential expression of myc family genes during murine development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. Embo J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Calao, M.; Sekyere, E.O.; Cui, H.J.; Cheung, B.B.; Thomas, W.D.; Keating, J.; Chen, J.B.; Raif, A.; Jankowski, K.; Davies, N.P.; et al. Direct effects of Bmi1 on p53 protein stability inactivates oncoprotein stress responses in embryonal cancer precursor cells at tumor initiation. Oncogene 2012, 32, 3616–3626. [Google Scholar] [CrossRef]
- Carter, D.R.; Murray, J.; Cheung, B.B.; Gamble, L.; Koach, J.; Tsang, J.; Sutton, S.; Kalla, H.; Syed, S.; Gifford, A.J.; et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci. Transl. Med. 2015, 7, 312ra176. [Google Scholar] [CrossRef] [PubMed]
- Hansford, L.M.; Thomas, W.D.; Keating, J.M.; Burkhart, C.A.; Peaston, A.E.; Norris, M.D.; Haber, M.; Armati, P.J.; Weiss, W.A.; Marshall, G.M. Mechanisms of embryonal tumor initiation: Distinct roles for MycN expression and MYCN amplification. Proc. Natl. Acad. Sci. USA 2004, 101, 12664–12669. [Google Scholar] [CrossRef]
- Cheung, N.K.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Beckers, A.; Van Peer, G.; Carter, D.R.; Gartlgruber, M.; Herrmann, C.; Agarwal, S.; Helsmoortel, H.H.; Althoff, K.; Molenaar, J.J.; Cheung, B.B.; et al. MYCN-driven regulatory mechanisms controlling LIN28B in neuroblastoma. Cancer Lett. 2015, 366, 123–132. [Google Scholar] [CrossRef]
- Beckers, A.; Van Peer, G.; Carter, D.R.; Mets, E.; Althoff, K.; Cheung, B.B.; Schulte, J.H.; Mestdagh, P.; Vandesompele, J.; Marshall, G.M.; et al. MYCN-targeting miRNAs are predominantly downregulated during MYCN-driven neuroblastoma tumor formation. Oncotarget 2015, 6, 5204–5216. [Google Scholar] [CrossRef]
- De Wyn, J.; Zimmerman, M.W.; Weichert-Leahey, N.; Nunes, C.; Cheung, B.B.; Abraham, B.J.; Beckers, A.; Volders, P.-J.; Decaesteker, B.; Carter, D.R.; et al. MEIS2 Is an Adrenergic Core Regulatory Transcription Factor Involved in Early Initiation of TH-MYCN-Driven Neuroblastoma Formation. Cancers 2021, 13, 4783. [Google Scholar] [CrossRef] [PubMed]
- Dorneburg, C.; Fischer, M.; Barth, T.F.; Mueller-Klieser, W.; Hero, B.; Gecht, J.; Carter, D.R.; de Preter, K.; Mayer, B.; Christner, L.; et al. LDHA in Neuroblastoma Is Associated with Poor Outcome and Its Depletion Decreases Neuroblastoma Growth Independent of Aerobic Glycolysis. Clin. Cancer Res. 2018, 24, 5772–5783. [Google Scholar] [CrossRef] [PubMed]
- Fabian, J.; Opitz, D.; Althoff, K.; Lodrini, M.; Hero, B.; Volland, R.; Beckers, A.; de Preter, K.; Decock, A.; Patil, N.; et al. MYCN and HDAC5 transcriptionally repress CD9 to trigger invasion and metastasis in neuroblastoma. Oncotarget 2016, 7, 66344–66359. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.Y.; Carter, D.R.; Liu, B.; Mayoh, C.; Beckers, A.; Lalwani, A.; Nagy, Z.; De Brouwer, S.; Decaesteker, B.; Hung, T.-T.; et al. Network Modeling of microRNA-mRNA Interactions in Neuroblastoma Tumorigenesis Identifies miR-204 as a Direct Inhibitor of MYCN. Cancer Res. 2018, 78, 3122–3134. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wu, G.; Zhan, X.; Nolan, A.; Koh, C.; De Marzo, A.; Doan, H.M.; Fan, J.; Cheadle, C.; Fallahi, M.; et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 2011, 6, e26057. [Google Scholar] [CrossRef]
- Bedoya-Reina, O.C.; Li, W.; Arceo, M.; Plescher, M.; Bullova, P.; Pui, H.; Kaucka, M.; Kharchenko, P.; Martinsson, T.; Holmberg, J.; et al. Single-nuclei transcriptomes from human adrenal gland reveal distinct cellular identities of low and high-risk neuroblastoma tumors. Nat. Commun. 2021, 12, 5309. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Yang, R.; Zhan, Y.; Lai, H.-D.; Ye, C.-J.; Yao, X.-Y.; Luo, W.-Q.; Cheng, X.-M.; Miao, J.-J.; Wang, J.-F.; et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell 2020, 38, 716–733. [Google Scholar] [CrossRef]
- Jansky, S.; Sharma, A.K.; Körber, V.; Quintero, A.; Toprak, U.H.; Wecht, E.M.; Gartlgruber, M.; Greco, A.; Chomsky, E.; Grünewald, T.G.P.; et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat. Genet. 2021, 53, 683–693. [Google Scholar] [CrossRef]
- Bogen, D.; Wei, J.S.; Azorsa, D.O.; Ormanoglu, P.; Buehler, E.; Guha, R.; Keller, J.M.; Griner, L.A.M.; Ferrer, M.; Song, Y.K.; et al. Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget 2015, 6, 35247–35262. [Google Scholar] [CrossRef]
- Ham, J.; Costa, C.; Sano, R.; Lochmann, T.L.; Sennott, E.M.; Patel, N.U.; Dastur, A.; Gomez-Caraballo, M.; Krytska, K.; Hata, A.N.; et al. Exploitation of the Apoptosis-Primed State of MYCN-Amplified Neuroblastoma to Develop a Potent and Specific Targeted Therapy Combination. Cancer Cell 2016, 29, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Topham, C.; Tighe, A.; Ly, P.; Bennett, A.; Sloss, O.; Nelson, L.; Ridgway, R.A.; Huels, D.; Littler, S.; Schandl, C.; et al. MYC Is a Major Determinant of Mitotic Cell Fate. Cancer Cell 2015, 28, 129–140. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Kocak, H.; Ackermann, S.; Hero, B.; Kahlert, Y.; Oberthuer, A.; Juraeva, D.; Roels, F.; Theissen, J.; Westermann, F.; Deubzer, H.; et al. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 2013, 4, e586. [Google Scholar] [CrossRef]
- Wang, C.; Gong, B.; Bushel, P.R.; Thierry-Mieg, J.; Thierry-Mieg, D.; Xu, J.; Fang, H.; Hong, H.; Shen, J.; Su, Z.; et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat. Biotechnol. 2014, 32, 926–932. [Google Scholar] [CrossRef]
- Yuan, X.; Seneviratne, J.A.; Du, S.; Xu, Y.; Chen, Y.; Jin, Q.; Jin, X.; Balachandran, A.; Huang, S.; Xu, Y.; et al. Single-cell profiling of peripheral neuroblastic tumors identifies an aggressive transitional state that bridges an adrenergic-mesenchymal trajectory. Cell. Rep. 2022, 41, 111455. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E.; et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 2018, 50, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Bliss, C. The toxicity of poisons applied jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Carter, D.R.; Sutton, S.K.; Pajic, M.; Murray, J.; Sekyere, E.O.; Fletcher, J.; Beckers, A.; De Preter, K.; Speleman, F.; George, R.E.; et al. Glutathione biosynthesis is upregulated at the initiation of MYCN-driven neuroblastoma tumorigenesis. Mol. Oncol. 2016, 10, 866–878. [Google Scholar] [CrossRef]
- Vanhauwaert, S.; Decaesteker, B.; De Brouwer, S.; Leonelli, C.; Durinck, K.; Mestdagh, P.; Vandesompele, J.; Sermon, K.; Denecker, G.; Van Neste, C.; et al. In silico discovery of a FOXM1 driven embryonal signaling pathway in therapy resistant neuroblastoma tumors. Sci. Rep. 2018, 8, 17468. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; Grabow, S.; Delbridge, A.R.; Adams, J.M.; Strasser, A. Prophylactic treatment with the BH3 mimetic ABT-737 impedes Myc-driven lymphomagenesis in mice. Cell. Death Differ. 2013, 20, 57–63. [Google Scholar] [CrossRef]
- Westermann, F.; Muth, D.; Benner, A.; Bauer, T.; Henrich, K.-O.; Oberthuer, A.; Brors, B.; Beissbarth, T.; Vandesompele, J.; Pattyn, F.; et al. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008, 9, R150. [Google Scholar] [CrossRef]
- Serrano-Del Valle, A.; Reina-Ortiz, C.; Benedi, A.; Anel, A.; Naval, J.; Marzo, I. Future prospects for mitosis-targeted antitumor therapies. Biochem. Pharmacol. 2021, 190, 114655. [Google Scholar] [CrossRef]
- DuBois, S.G.; Mosse, Y.P.; Fox, E.; Kudgus, R.A.; Reid, J.M.; McGovern, R.; Groshen, S.; Bagatell, R.; Maris, J.M.; Twist, C.J.; et al. Phase II Trial of Alisertib in Combination with Irinotecan and Temozolomide for Patients with Relapsed or Refractory Neuroblastoma. Clin. Cancer Res. 2018, 24, 6142–6149. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, R.; Wang, H.; Sun, M.; Lee, D.G.; Peng, J.; Thiele, C.J. Combining selinexor with alisertib to target the p53 pathway in neuroblastoma. Neoplasia 2022, 26, 100776. [Google Scholar] [CrossRef] [PubMed]
- Roeschert, I.; Poon, E.; Henssen, A.G.; Garcia, H.D.; Gatti, M.; Giansanti, C.; Jamin, Y.; Ade, C.P.; Gallant, P.; Schülein-Völk, C.; et al. Combined inhibition of Aurora-A and ATR kinase results in regression of MYCN-amplified neuroblastoma. Nat. Cancer 2021, 2, 312–326. [Google Scholar] [CrossRef]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- ggpubr: ‘ggplot2′ Based Publication Ready Plots. 2020. Available online: https://cran.r-project.org/web/packages/ggpubr/index.html (accessed on 20 September 2023).
- A Package for Survival Analysis in R_. 2022. Available online: https://cran.r-project.org/web/packages/survival/index.html (accessed on 20 September 2023).
- Survminer: Drawing Survival Curves Using ‘ggplot2’. 2020. Available online: https://cran.r-hub.io/web/packages/survminer/index.html (accessed on 20 September 2023).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, L.; Balachandran, A.; Larkin, R.; Seneviratne, J.A.; Chung, S.A.; Lalwani, A.; Tsubota, S.; Beck, D.; Kadomatsu, K.; Beckers, A.; et al. Mitotic Dysregulation at Tumor Initiation Creates a Therapeutic Vulnerability to Combination Anti-Mitotic and Pro-Apoptotic Agents for MYCN-Driven Neuroblastoma. Int. J. Mol. Sci. 2023, 24, 15571. https://doi.org/10.3390/ijms242115571
Zhai L, Balachandran A, Larkin R, Seneviratne JA, Chung SA, Lalwani A, Tsubota S, Beck D, Kadomatsu K, Beckers A, et al. Mitotic Dysregulation at Tumor Initiation Creates a Therapeutic Vulnerability to Combination Anti-Mitotic and Pro-Apoptotic Agents for MYCN-Driven Neuroblastoma. International Journal of Molecular Sciences. 2023; 24(21):15571. https://doi.org/10.3390/ijms242115571
Chicago/Turabian StyleZhai, Lei, Anushree Balachandran, Rebecca Larkin, Janith A. Seneviratne, Sylvia A. Chung, Amit Lalwani, Shoma Tsubota, Dominik Beck, Kenji Kadomatsu, Anneleen Beckers, and et al. 2023. "Mitotic Dysregulation at Tumor Initiation Creates a Therapeutic Vulnerability to Combination Anti-Mitotic and Pro-Apoptotic Agents for MYCN-Driven Neuroblastoma" International Journal of Molecular Sciences 24, no. 21: 15571. https://doi.org/10.3390/ijms242115571