

Genetic Code Expansion and a Photo-Cross-Linking Reaction Facilitate Ribosome Display Selections for Identifying a Wide Range of Affinity Peptides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
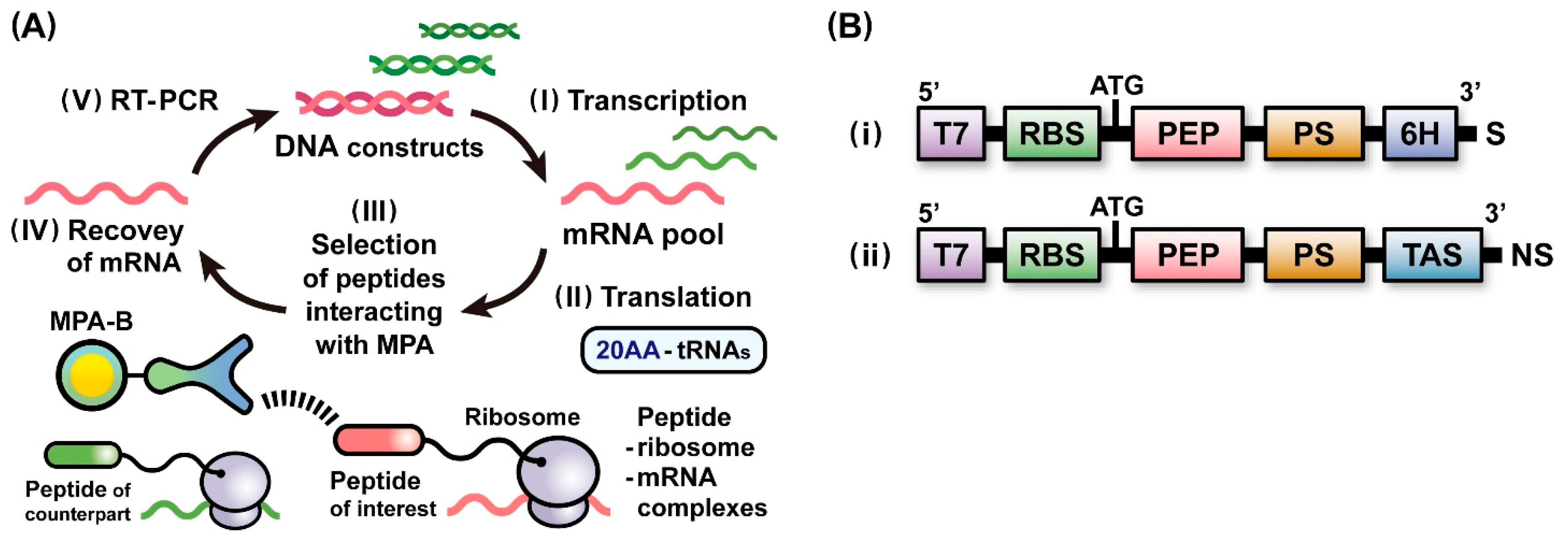
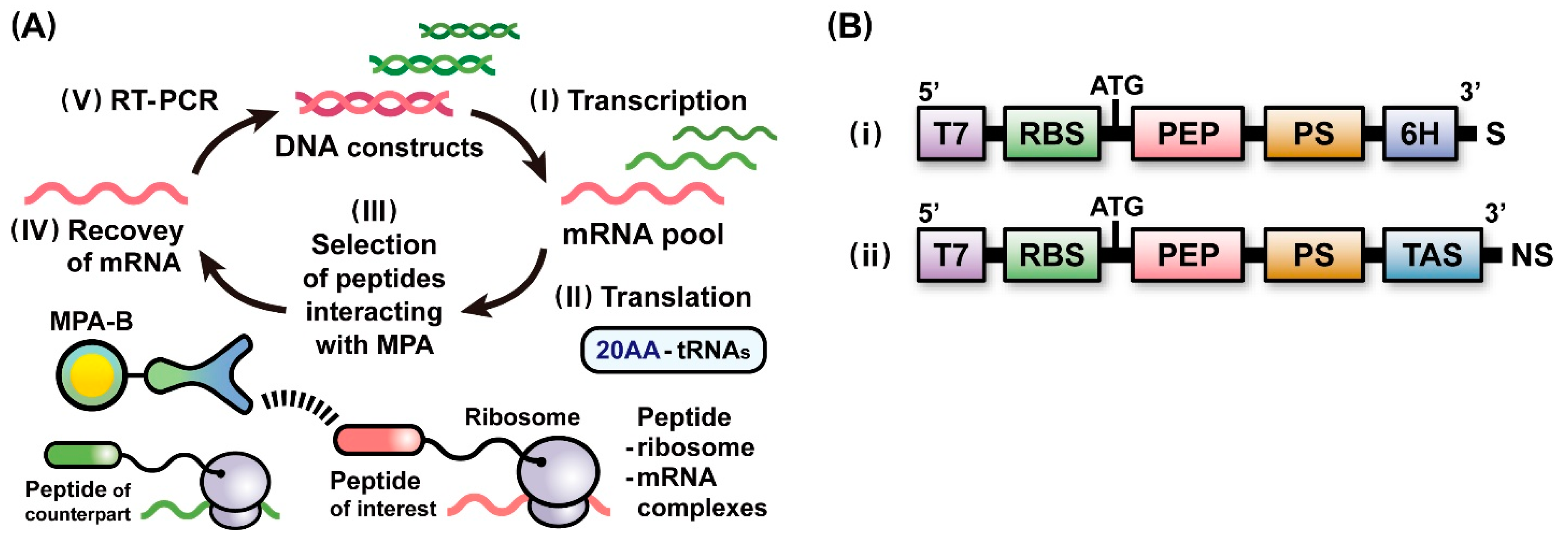
2.1. A Scheme of Ribosome Display Selection for Identifying Affinity Peptides
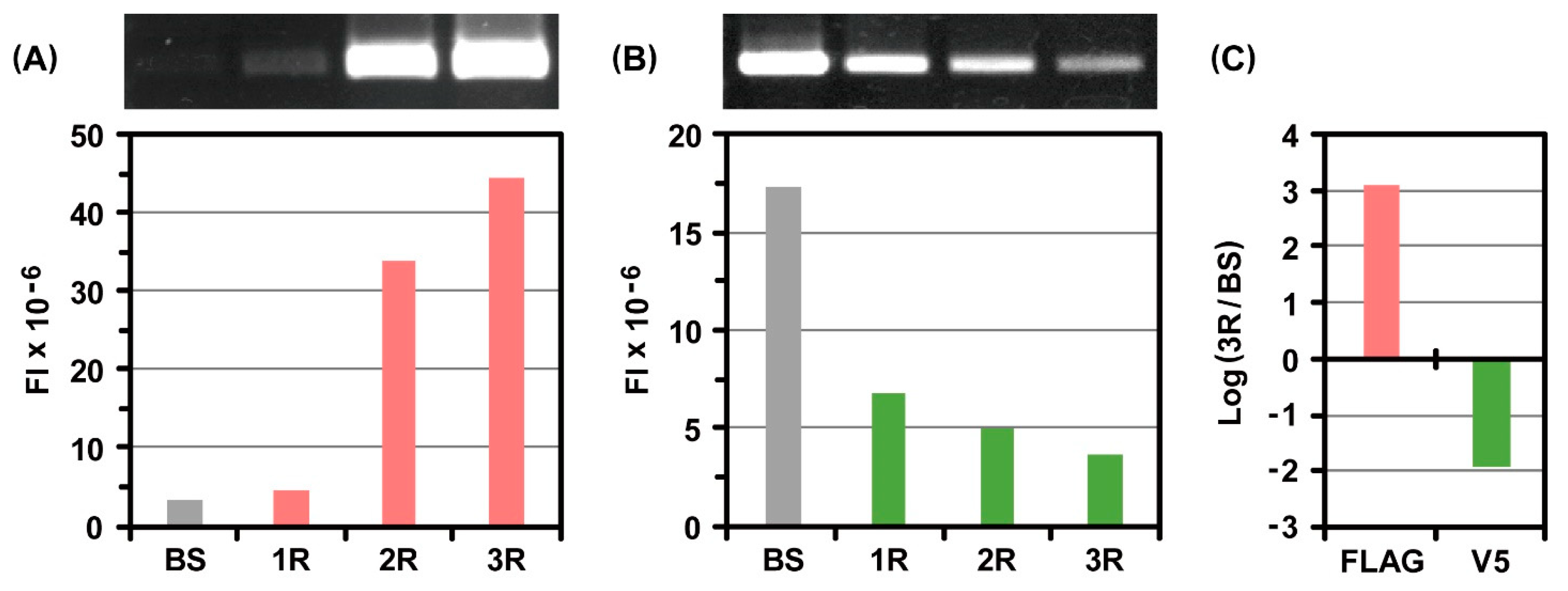
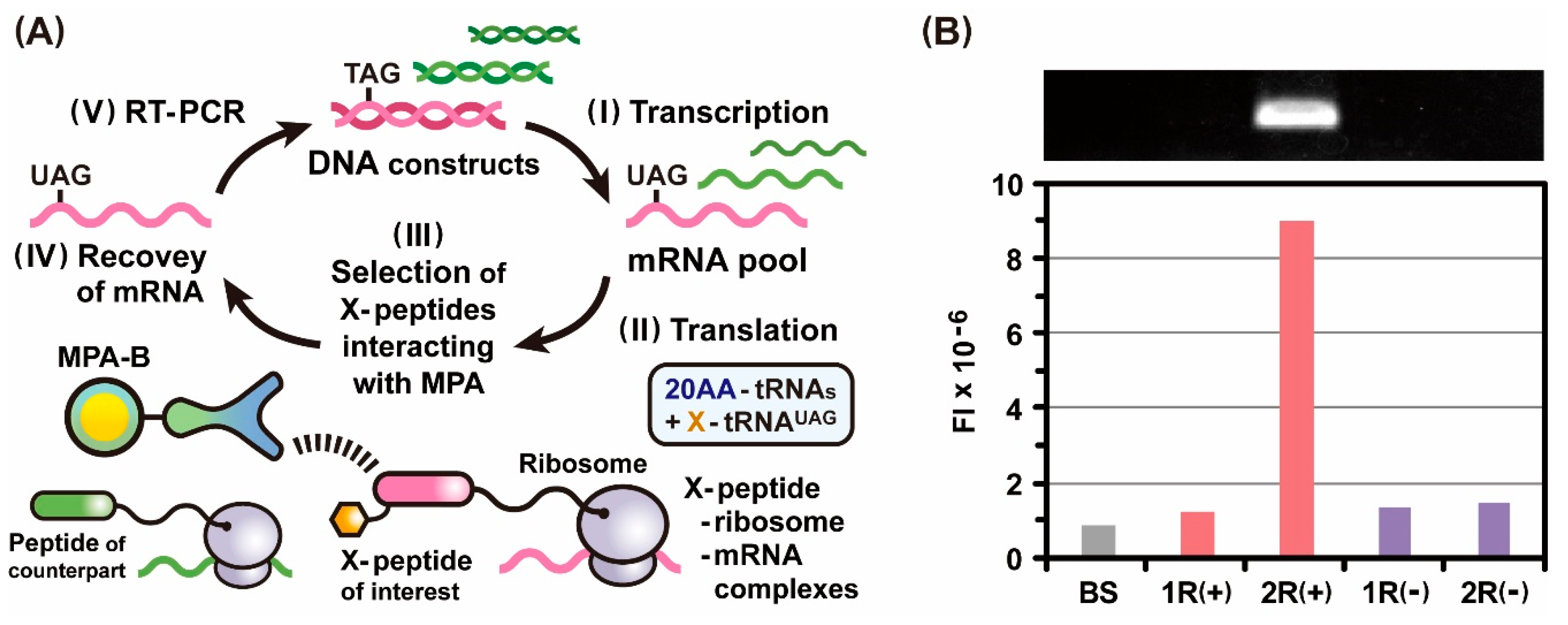
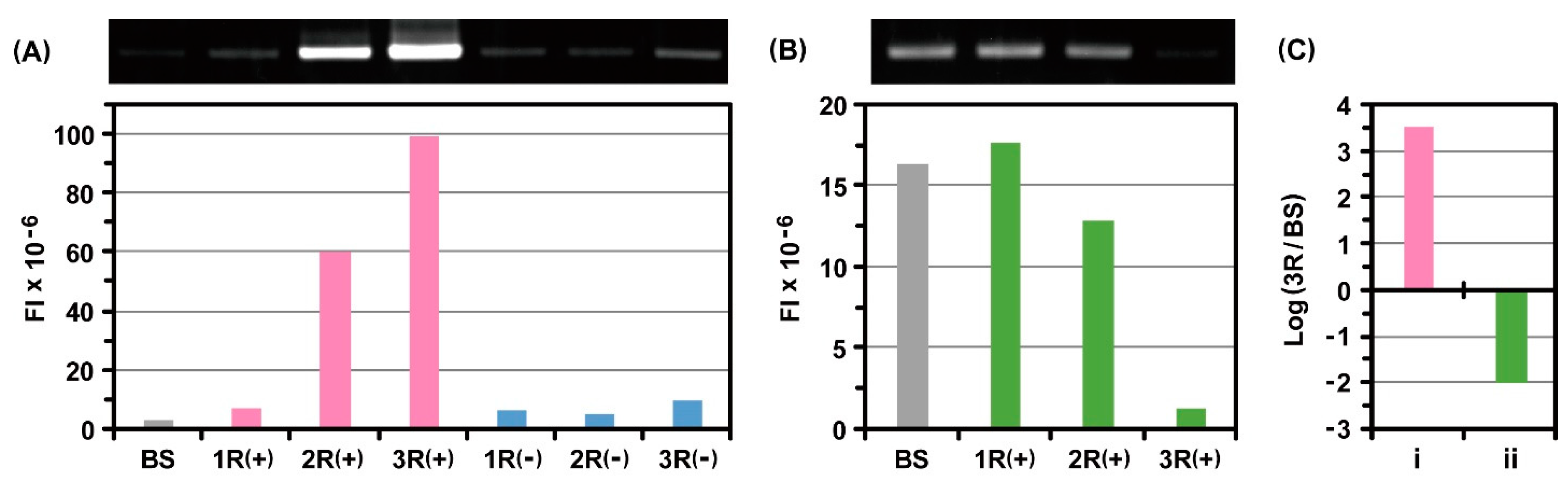
2.2. Incorporation of Genetic Code Expansion into the Ribosome Display Selection
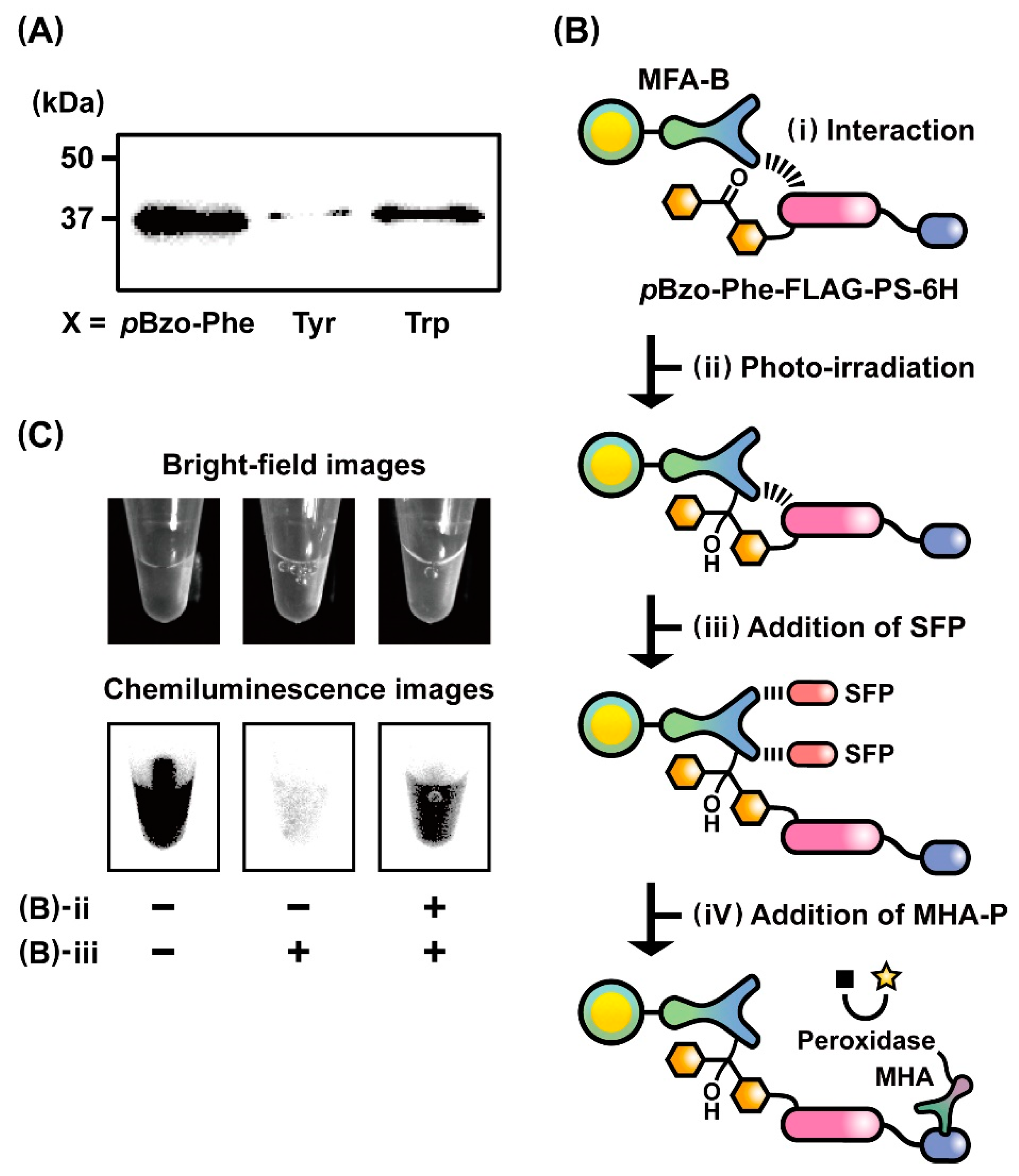
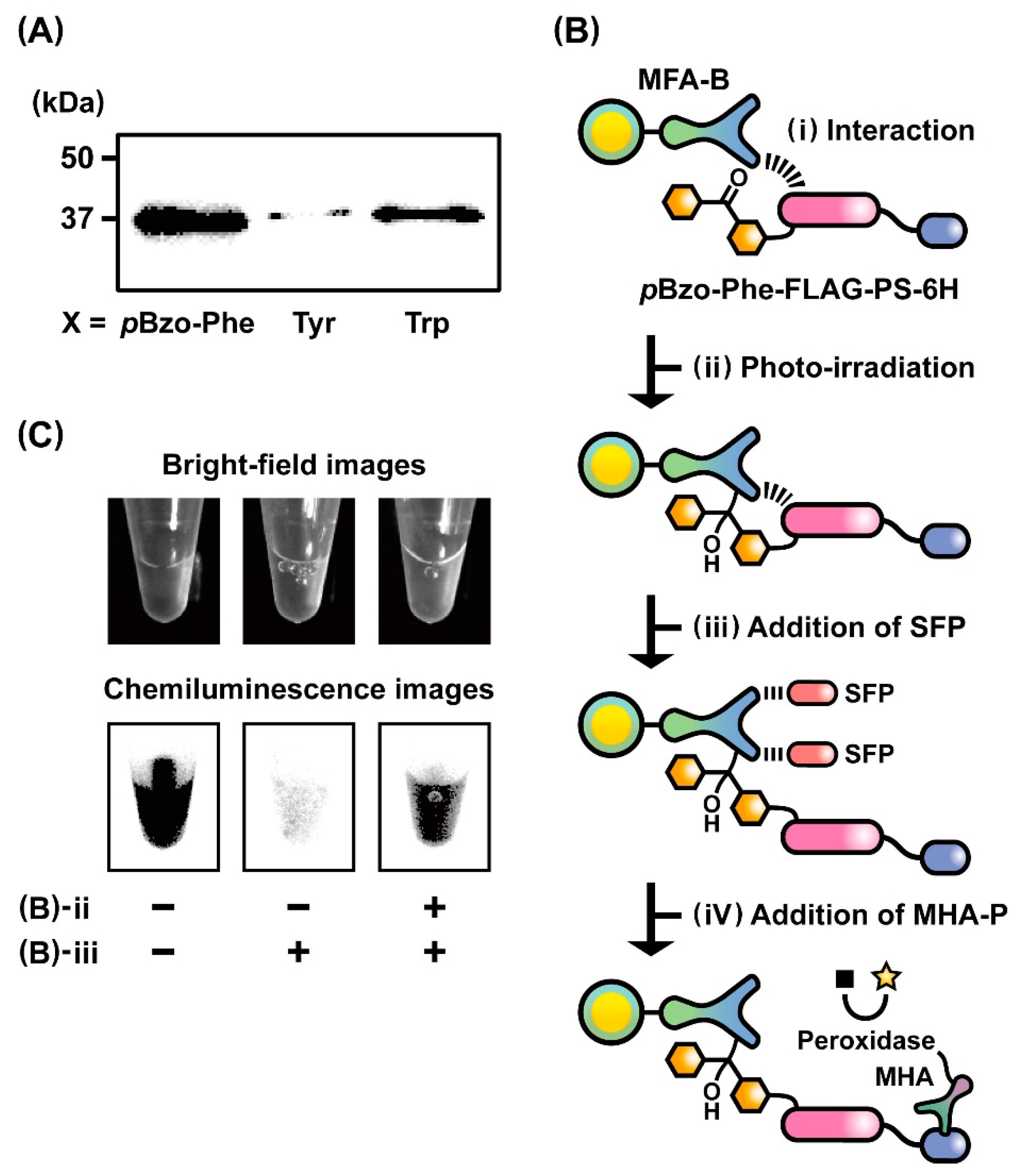
2.3. Evaluation of the Photo-Cross-Linking Reactions of pBzo-Phe-Incorporated Peptides
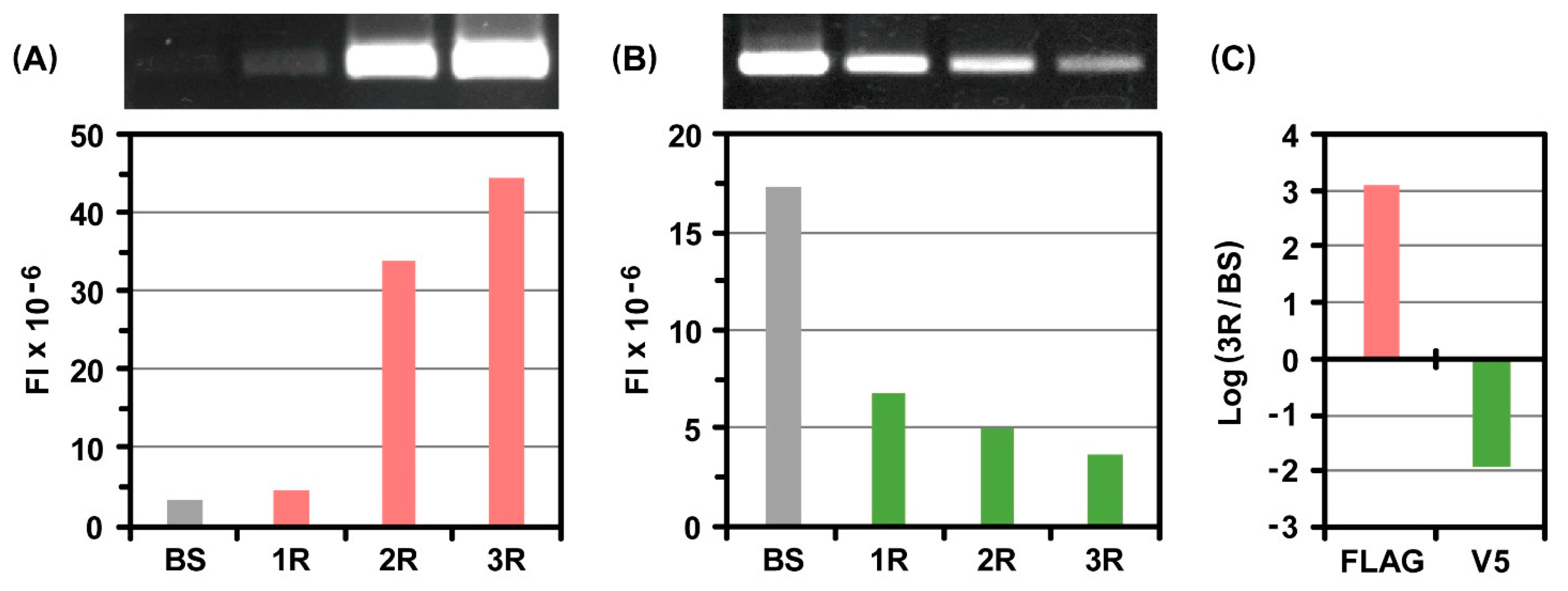
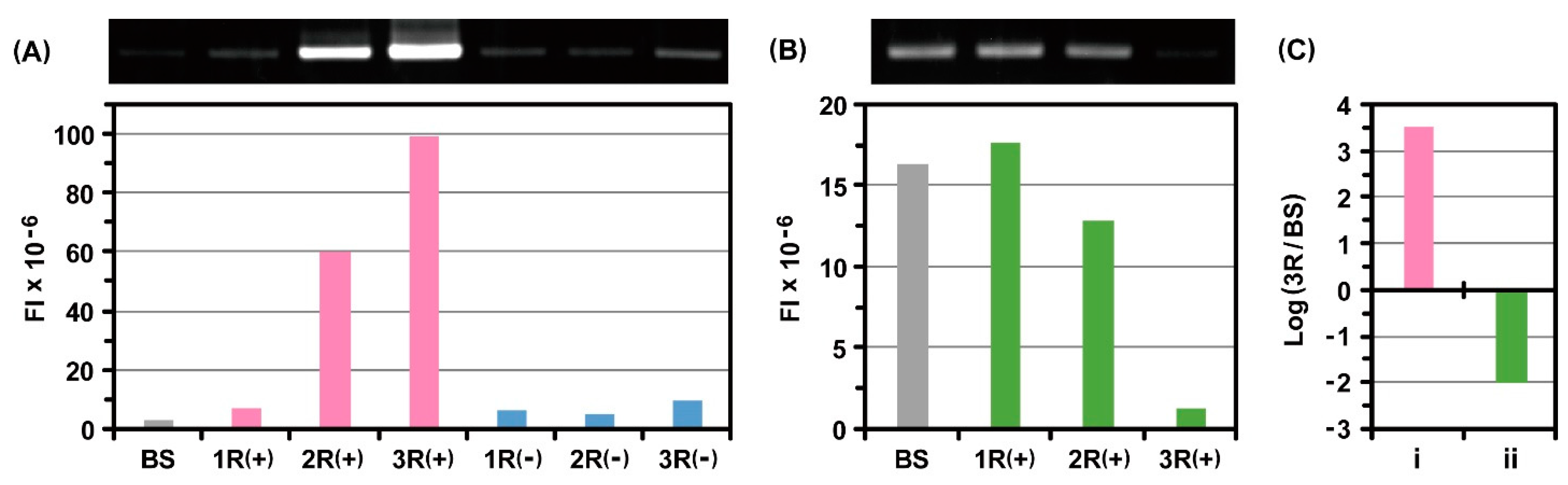
2.4. Photo-Cross-Linkable Ribosome Display Selection for Identifying Low-Affinity Peptides
3. Materials and Methods
3.1. Construction of Plasmid and Linear DNAs
3.2. Western Blot Analysis of Protein Expression
3.3. Ribosome Display Selections with or without Genetic Code Expansion
3.4. RT-PCR and Electrophoretic Gel Analysis after Ribosome Display Selections
3.5. Chemiluminescent Image Analysis of Photo-Cross-Linking Reactions
3.6. Photo-Cross-Linkable Ribosome Display Selection with Genetic Code Expansion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wimberly, B.T.; Brodersen, D.E.; Clemons, W.M., Jr.; Morgan-Warren, R.J.; Carter, A.P.; Vonrhein, C.; Hartsch, T.; Ramakrishnan, V. Structure of the 30S ribosomal subunit. Nature 2000, 407, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Schluenzen, F.; Tocilj, A.; Zarivach, R.; Harms, J.; Gluehmann, M.; Janell, D.; Bashan, A.; Bartels, H.; Agmon, I.; Franceschi, F.; et al. Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell 2000, 102, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Opron, K.; Burton, Z.F. Ribosome structure, function, and early evolution. Int. J. Mol. Sci. 2019, 20, 40. [Google Scholar] [CrossRef]
- D'Orazio, K.N.; Green, R. Ribosome states signal RNA quality control. Mol. Cell 2021, 81, 1372–1383. [Google Scholar] [CrossRef]
- Korostelev, A.A. The structural dynamics of translation. Annu. Rev. Biochem. 2022, 91, 245–267. [Google Scholar] [CrossRef]
- Madin, K.; Sawasaki, T.; Ogasawara, T.; Endo, Y. A high efficient and robust cell-free protein synthesis system prepared from wheat embryos: Plants apparently contain a suicide system directed at ribosomes. Proc. Natl. Acad. Sci. USA 2000, 97, 559–564. [Google Scholar] [CrossRef]
- Rifo, R.S.; Ricci, E.P.; Décimo, D.; Moncorgé, O.; Ohlmann, T. Back to basics: The untreated rabbit reticulocyte lysate as a competitive system to recapitulate cap/poly(A) synergy and the selective advantage of IRES-driven translation. Nucleic Acids Res. 2007, 35, e121. [Google Scholar] [CrossRef]
- Hino, M.; Kataoka, M.; Kajimoto, K.; Yamamoto, T.; Kido, J.; Shinohara, Y.; Baba, Y. Efficiency of cell-free protein synthesis based on a crude cell extract from Escherichia coli, wheat germ, and rabbit reticulocytes. J. Biotechnol. 2008, 133, 183–189. [Google Scholar] [CrossRef]
- Stech, M.; Quast, R.B.; Sachse, R.; Schulze, C.; Wüstenhagen, D.A.; SKubick, S. A continuous-exchange cell-free protein synthesis system based on extracts from cultured insect cells. PLoS ONE 2014, 9, e96635. [Google Scholar] [CrossRef]
- Hibi, K.; Amikura, K.; Sugiura, N.; Masuda, K.; Ohno, S.; Yokogawa, T.; Ueda, T.; Shimizu, Y. Reconstituted cell-free protein synthesis using in vitro transcribed tRNAs. Commun. Biol. 2020, 3, 350. [Google Scholar] [CrossRef] [PubMed]
- Tinafar, A.; Jaenes, K.; Pardee, K. Synthetic biology goes cell-free. BMC Biol. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Brookwell, A.; Oza, J.P.; Caschera, F. Biotechnology applications of cell-free expression systems. Life 2021, 11, 1367. [Google Scholar] [CrossRef] [PubMed]
- Yue, K.; Chen, J.; Li, Y.; Kai, L. Advancing synthetic biology through cell-free protein synthesis. Comput. Struct. Biotechnol. J. 2023, 21, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Wada, A. Development of next-generation peptide binders using in vitro display technologies and their potential applications. Front. Immunol. 2013, 4, 224. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wiedmann, M.M.; Suga, H. RNA display methods for the discovery of bioactive macrocycles. Chem. Rev. 2019, 119, 10360–10391. [Google Scholar] [CrossRef]
- Jaroszewicz, W.; Morcinek-Orłowska, J.; Pierzynowska, K.; Gaffke, L.; Węgrzyn, G. Phage display and other peptide display technologies. FEMS Microbiol. Rev. 2022, 46, fuab052. [Google Scholar] [CrossRef]
- Mattheakis, L.C.; Bhatt, R.R.; Dower, W.J. An in-vitro polysome display system for identifying ligands from very large peptide libraries. Proc. Natl. Acad. Sci. USA 1994, 91, 9022–9026. [Google Scholar] [CrossRef]
- Zahnd, C.; Amstutz, P.; Plückthun, A. Ribosome display: Selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 2007, 4, 269–279. [Google Scholar] [CrossRef]
- Wada, A.; Hara, S.; Osada, H. Ribosome display and photo-cross-linking techniques for in vitro identification of target proteins of bioactive small molecules. Anal. Chem. 2014, 86, 6768–6773. [Google Scholar] [CrossRef]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.J.; Kuhn, B.T.; Bräuer, P.; Newstead, S.; Dawson, R.J.P.; Geertsma, E.R.; Seeger, M.A. Generation of synthetic nanobodies against delicate proteins. Nat. Protoc. 2020, 15, 1707–1741. [Google Scholar] [CrossRef]
- Nemoto, N.; Miyamoto-Sato, E.; Husimi, Y.; Yanagawa, H. In vitro virus: Bonding of mRNA bearing puromycin at the 3’-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett. 1997, 414, 405–408. [Google Scholar] [PubMed]
- Roberts, R.W.; Szostak, J.W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar] [CrossRef] [PubMed]
- Kamalinia, G.; Grindel, B.J.; Takahashi, T.T.; Millward, S.W.; Roberts, R.W. Directing evolution of novel ligands by mRNA display. Chem. Soc. Rev. 2021, 50, 9055–9103. [Google Scholar] [CrossRef] [PubMed]
- Smith, G. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Hoess, R.H. Protein design and phage display. Chem. Rev. 2001, 101, 3205–3218. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Song, E.-W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef]
- Chin, J.W. Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. [Google Scholar] [CrossRef]
- Martin, R.W.; Des Soye, B.J.; Kwon, Y.-C.; Kay, J.; Davis, R.G.; Thomas, P.M.; Majewska, N.I.; Chen, C.X.; Marcum, R.D.; Weiss, M.G.; et al. Cell-free protein synthesis from genomically recoded bacteria enables multisite incorporation of noncanonical amino acids. Nat. Commun. 2018, 9, 1203. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Kuratani, T.; Seki, E.; Hino, N.; Sakamoto, K.; Yokoyama, S. Structural basis for genetic-code expansion with bulky lysine derivatives by an engineered pyrrolysyl-tRNA synthetase. Cell Chem. Biol. 2019, 26, 936–949. [Google Scholar] [CrossRef]
- Adachi, J.; Katsura, K.; Seki, E.; Takemoto, C.; Shirouzu, M.; Terada, T.; Mukai, T.; Sakamoto, K.; Yokoyama, S. Cell-free protein synthesis using S30 extracts from Escherichia coli RFzero strains for efficient incorporation of non-natural amino acids into proteins. Int. J. Mol. Sci. 2019, 20, 492. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Johnston, W.A.; Alexandrov, K. Cell-Free Approach for Non-canonical amino acids incorporation into polypeptides. Front. Bioeng. Biotechnol. 2020, 8, 1031. [Google Scholar] [CrossRef]
- Katoh, T.; Suga, H. In vitro genetic code reprogramming for the expansion of usable noncanonical amino acids. Annu. Rev. Biochem. 2022, 21, 221–243. [Google Scholar] [CrossRef] [PubMed]
- Kita, A.; Hino, N.; Higashi, S.; Hirota, K.; Narumi, R.; Adachi, J.; Takafuji, K.; Ishimoto, K.; Okada, Y.; Sakamoto, K.; et al. Adenovirus vector-based incorporation of a photo-cross-linkable amino acid into proteins in human primary cells and cancerous cell lines. Sci. Rep. 2016, 6, 36946. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Matsuda, T.; Ohtake, K.; Yanagisawa, T.; Yokoyama, S.; Fujiwara, Y.; Watanabe, T.; Hohsaka, H.; Sakamoto, K. Incorporation of a doubly functionalized synthetic amino acid into proteins for creating chemical and light-induced conjugates. Bioconjug. Chem. 2016, 27, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Klompus, S.; Leviatan, S.; Kalka, I.N.; Weinberger, A.; Wijmenga, C.; Fu, J.; Zhernakova, A.; Weersma, R.K.; Segal, E. Population-wide diversity and stability of serum antibody epitope repertoires against human microbiota. Nat. Med. 2021, 27, 1442–1450. [Google Scholar] [CrossRef]
- Ledsgaard, L.; Ljungars, A.; Rimbault, C.; Sørensen, C.V.; Tulika, T.; Wade, J.; Wouters, Y.; McCafferty, J.; Laustsen, A.H. Advances in antibody phage display technology. Drug Discov. Today 2022, 27, 2151–2169. [Google Scholar] [CrossRef]
- Henson, S.N.; Elko, E.A.; Swiderski, P.M.; Liang, Y.; Engelbrektson, A.L.; Piña, A.; Boyle, A.S.; Fink, Z.; Facista, S.J.; Martinez, V.; et al. PepSeq: A fully in vitro platform for highly multiplexed serology using customizable DNA-barcoded peptide libraries. Nat. Protoc. 2023, 18, 396–423. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Ito, K. The ribosomal exit tunnel functions as a discriminating gate. Cell 2002, 108, 629–636. [Google Scholar] [CrossRef]
- Evans, M.S.; Ugrinov, K.G.; Frese, M.-A.; Clark, P.L. Homogeneous stalled ribosome nascent chain complexes produced in vivo or in vitro. Nat. Methods 2005, 2, 757–762. [Google Scholar] [CrossRef]
- Ranawakage, D.C.; Takada, T.; Kamachi, Y. HiBiT-qIP, HiBiT-based quantitative immunoprecipitation, facilitates the determination of antibody affinity under immunoprecipitation conditions. Sci. Rep. 2019, 9, 6895. [Google Scholar] [CrossRef] [PubMed]
- Taira, H.; Matsushita, Y.; Kojima, K.; Shiraga, K.; Hohsaka, T. Comprehensive screening of amber suppressor tRNAs suitable for incorporation of non-natural amino acids in a cell-free translation system. Biochem. Biophys Res. Commun. 2008, 374, 304–308. [Google Scholar] [CrossRef]
- Abe, R.; Ohashi, H.; Iijima, I.; Ihara, M.; Takagi, H.; Hohsaka, T.; Ueda, H. “Quenchbodies”: Quench-based antibody probes that show antigen-dependent fluorescence. J. Am. Chem. Soc. 2011, 133, 17386–17394. [Google Scholar] [CrossRef]
- Chadani, Y.; Sugata, N.; Niwa, T.; Ito, Y.; Iwasaki, S.; Taguchi, H. Nascent polypeptide within the exit tunnel stabilizes the ribosome to counteract risky translation. EMBO J. 2021, 40, e108299. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. 4'-(1-Azi-2,2,2-trifluoroethyl)phenylalanine, a photolabile carbene-generating analog of phenylalanine. J. Am. Chem. Soc. 1984, 106, 7540–7545. [Google Scholar] [CrossRef]
- Smith, D.P.; Anderson, J.; Plante, J.; Ashcroft, A.E.; Radford, S.E.; Wilson, A.J.; Parker, M.J. Trifluoromethyldiazirine: An effective photo-induced cross-linking probe for exploring amyloid formation. Chem. Commun. 2008, 44, 5728–5730. [Google Scholar] [CrossRef]
- L'abbe, G. Decomposition and addition reactions of organic azides. Chem. Rev. 1969, 69, 345–363. [Google Scholar] [CrossRef]
- Pendergrast, P.S.; Chen, Y.; Ebright, Y.W.; Ebright, R.H. Determination of the orientation of a DNA binding motif in a protein-DNA complex by photocrosslinking. Proc. Natl. Acad. Sci. USA 1992, 89, 10287–10291. [Google Scholar] [CrossRef]
- Dormán, G.; Nakamura, H.; Pulsipher, A.; Prestwich, G.D. The life of pi star: Exploring the exciting and forbidden worlds of the benzophenone photophore. Chem. Rev. 2016, 116, 15284–15398. [Google Scholar] [CrossRef]
- Wegner, G.J.; Lee, H.J.; Corn, R.M. Characterization and optimization of peptide arrays for the study of epitope-antibody interactions using surface plasmon resonance imaging. Anal. Chem. 2002, 74, 5161–5168. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furuhashi, T.; Sakamoto, K.; Wada, A. Genetic Code Expansion and a Photo-Cross-Linking Reaction Facilitate Ribosome Display Selections for Identifying a Wide Range of Affinity Peptides. Int. J. Mol. Sci. 2023, 24, 15661. https://doi.org/10.3390/ijms242115661
Furuhashi T, Sakamoto K, Wada A. Genetic Code Expansion and a Photo-Cross-Linking Reaction Facilitate Ribosome Display Selections for Identifying a Wide Range of Affinity Peptides. International Journal of Molecular Sciences. 2023; 24(21):15661. https://doi.org/10.3390/ijms242115661
Chicago/Turabian StyleFuruhashi, Takuto, Kensaku Sakamoto, and Akira Wada. 2023. "Genetic Code Expansion and a Photo-Cross-Linking Reaction Facilitate Ribosome Display Selections for Identifying a Wide Range of Affinity Peptides" International Journal of Molecular Sciences 24, no. 21: 15661. https://doi.org/10.3390/ijms242115661
APA StyleFuruhashi, T., Sakamoto, K., & Wada, A. (2023). Genetic Code Expansion and a Photo-Cross-Linking Reaction Facilitate Ribosome Display Selections for Identifying a Wide Range of Affinity Peptides. International Journal of Molecular Sciences, 24(21), 15661. https://doi.org/10.3390/ijms242115661