Abstract
High myopia is the most severe and pathological form of myopia. It occurs when the spherical refractive error exceeds –6.00 spherical diopters (SDs) or the axial length (AL) of the eye is greater than 26 mm. This article focuses on early-onset high myopia, an increasingly common condition that affects children under 10 years of age and can lead to other serious ocular pathologies. Through the genetic analysis of 21 families with early-onset high myopia, this study seeks to contribute to a better understanding of the role of genetics in this disease and to propose candidate genes. Whole-exome sequencing studies with a panel of genes known to be involved in the pathology were performed in families with inconclusive results: 3% of the variants found were classified as pathogenic, 6% were likely pathogenic and the remaining 91% were variants of uncertain significance. Most of the families in this study were found to have alterations in several of the proposed genes. This suggests a polygenic inheritance of the pathology due to the cumulative effect of the alterations. Further studies are needed to validate and confirm the role of these alterations in the development of early-onset high myopia and its polygenic inheritance.
1. Introduction
Myopia is the most common eye disorder in the world [1]. From a physiological point of view, it involves a refractive error where the light rays entering the eye focus on a point in front of the retina, leading to decreased visual acuity. This refractive error is due to a postnatal axial elongation of the eye in the process of emmetropization [2,3]. Although myopia is usually considered a benign condition that can be corrected with the use of glasses, contact lenses or refractive surgery, it is becoming a public health concern due to its increasing prevalence in younger populations and its progression to its most severe and pathological form, high myopia (HM) (Figure 1). HM is defined as a spherical refractive error exceeding –6.00 spherical diopters (SD) or an axial length (AL) greater than 26 mm [4,5].
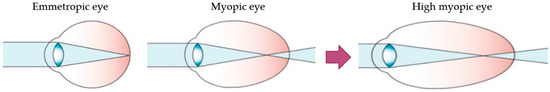
Figure 1.
Schematic optics of the eye. In emmetropic eyes, the parallel rays of a distant object are focused precisely on the photoreceptors located on the retina. Myopic eyes have longer axial length, and the image of a distant object falls in front of the photoreceptors and cannot be brought into focus by accommodation. The myopic eye can become highly myopic, accentuating this defocus considerably. Modified from Morgan et al. [5].
This study focuses on early-onset HM (EoHM), which occurs in children under 10 years of age [3]. Through an examination of the disease from a genetic perspective, this analysis seeks to contribute to a better understanding of the role of genetics in its development.
HM increases the susceptibility to other ocular complications that have a significant impact on quality of life and can lead to irreversible vision loss such as cataracts, glaucoma, retinal detachment and macular degeneration. It is therefore important to diagnose myopia and its progression to HM for early prevention and intervention before the onset of more severe pathological manifestations. In addition, HM can appear as one of the first clinical manifestations of conditions such as Marfan or Stickler syndromes, retinal disorders or congenital stationary night blindness (CSNB), allowing for the early diagnosis and treatment of these diseases [4].
The prevalence of myopia is highest in East Asia, affecting 90% of high school graduates. The reported prevalence of myopia among Caucasians in Europe is 30.6% and that of HM is 2.7%. Based on current trends, the study by Holden et al., estimates that the number of people affected will significantly increase worldwide by 2050, as may be observed in Figure 2 [4,6].
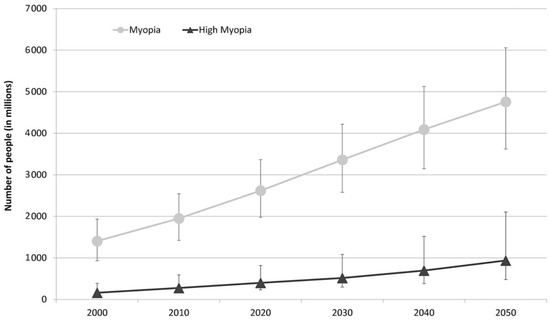
Figure 2.
Graph representing the number of people estimated to develop myopia and HM for each decade from 2000 to 2050 [6].
The etiology of myopia and HM is heterogeneous and multifactorial, involving a combination of environmental (external), microenvironmental (internal) and genetic factors [4].
Currently, the most studied environmental factor believed to protect individuals from developing myopia is the time spent outdoors [4]. Conversely, several studies have linked the progression of myopia to the time spent in near work and exposure to artificial light [3,5]. In the case of EoHM, however, given the young age of the patients, environmental factors are not considered to have a significant influence on the development of myopia. This suggests that there is a greater genetic burden involved in EoHM, supporting a genetic approach to the disease [3].
Microenvironmental factors, such as oxidative stress, oxidation and angiogenesis, can induce or accentuate EoHM. Given its high oxygen demand and direct exposure to natural light, the retina is particularly vulnerable to oxidative stress. In addition, patients with EoHM have an imbalance in the oxidative/antioxidative status of the retina, suggesting that oxidative stress plays a direct role in the development of this pathology [3].
Genetic factors, as explained above, play a critical and major role in the development of EoHM. Genetic information initiates and regulates the processes involved in emmetropization, including the regulation of microenvironmental factors and visual feedback:
- Regulation of microenvironmental factors such as the level of angiogenic growth factors (VEGF, MCP1 and IL5) or proinflammatory cytokines (IL6, IFN-γ, IP-10, eotaxin and MIP-1α) in the aqueous and vitreous humors [3].
- Visual feedback driven by optical defocus. The defocus signal is detected by the retina and triggers a multilayered signaling cascade involving a large number of coding and regulatory genes, sequentially affecting the retina, retinal pigment epithelium (RPE), choroid, sclera and its extracellular matrix (ECM). In this last step of the cascade, changes occur in the composition of the ECM and are reflected in axial elongation (Figure 3). Alterations in these genes and their function can disrupt emmetropization and result in excessive axial length [2].
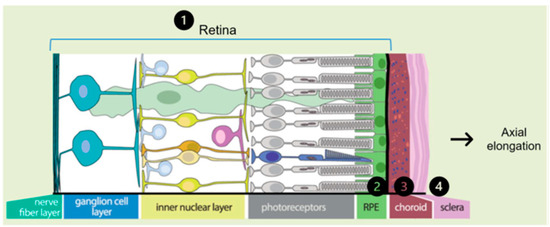 Figure 3. Representation of the multilayer signaling cascade that enables axial elongation. The signaling cascade order is represented by the numbers 1 to 4 Modified from Tedja et al. [7].
Figure 3. Representation of the multilayer signaling cascade that enables axial elongation. The signaling cascade order is represented by the numbers 1 to 4 Modified from Tedja et al. [7].
The classification of genetic alterations affecting visual feedback may be divided into two main groups: those involved in the retina, affecting the function of photoreceptors, RPEs, and ON and OFF bipolar neurons; and those that occur in the sclera, affecting the composition and metabolism of the ECM [5].
The role of genetic factors in myopia is supported by several studies. It has been observed that children with a family history of myopia have longer axial lengths and a higher risk of developing myopia during childhood compared to the average population [4]. Due to differences in study design and methods of analysis, heritability estimates in the literature range from 15% to 98%; however, the true heritability of myopia is probably between 60% and 80%.
Genome-wide association studies (GWASs) and segregation analyses have identified more than 26 loci and 400 genes involved in refractive error and the development of common myopia [4,7]. Fewer genes have been associated with EoHM: 11 genes with autosomal dominant inheritance have been identified for its non-syndromic form (ZNF644, SCO2, SLC39A5, CCDC111, P4HA2, BSG, CPSF1, NDUFAF7, TNFRSF21, XYLT and DZIP1); four genes with autosomal recessive inheritance (LRPAP1, CTSH, LEPREL1 and LOXL3); and two X-linked genes (ARR3 and OPN1LW). Other studies have associated EoHM with loci involved in the development of common myopia and refractive error, as well as with other genes such as CTNND2, JOANA, CACNA1F, RPGR, PRSS56, BMP3, KCNQ5, LAMA2, TOX, TJP2, RDH5, ZIC2, RASGRF1, GJD2, RBFOX1, SHISA6, FAM150B-ACP1, LINC00340, FBN1, DIS3L-MAP2K1, ARID2-SNAT1 and SLC14A2 [3].
Most of the variants studied, especially in common myopia, carry a low risk and can be prevalent in the general population, contributing to a small extent and cumulatively to the overall risk. Therefore, it is postulated that the inheritance of myopia and high myopia follows a polygenic pattern, in which several genes together contribute to the manifestation of the disease [8].
In this study, next-generation sequencing (NGS) was used to sequence the whole exome of 21 families diagnosed with EoHM. Whole-exome sequencing makes it possible to study alterations and their involvement in a larger number of genes, thus allowing for the diagnosis and proposal of new candidate causal genes for EoHM in patients.
2. Results and Discussion
The results of this study were obtained from an ongoing project seeking to identify new genes responsible for EoHM in a sample of families from a tertiary hospital in Spain. This project also aims to evaluate the implementation of NGS and its relevance to the management of patients with EoHM. A total of 21 probands (33.33% female [7/21] and 66.66% male [14/21]) and nine affected relatives with EoHM from 21 unrelated families were recruited based on their phenotype (Table 1) and the inclusion criteria indicated in the Materials and Methods.

Table 1.
Clinical evaluation of probands.
The ages of the probands range from 6 to 80, yet they all share the common characteristic of exhibiting HM before the age of 10, thus constituting a unified group for genetic analysis. It is worth noting that 62% of the subjects in this study have not yet reached the age of 10 (Figure 4).
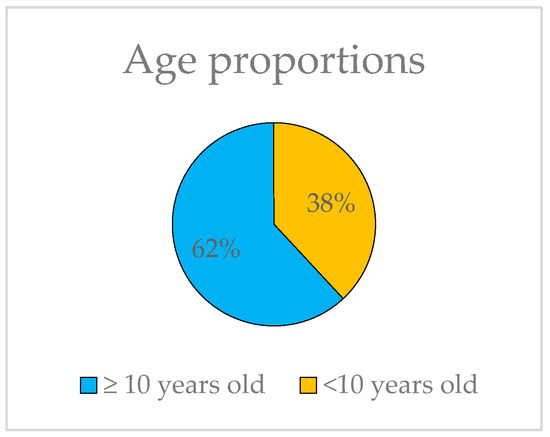
Figure 4.
Representation of the proportions of probands younger and older than 10 years old.
The average spherical equivalent for the probands is −12.044 diopters in the right eye and −11.499 diopters in the left eye. Table 2 shows the descriptive statistics, including the mean and standard deviation of the refractive outcomes in this cohort.
Table 2.
Refractive results.
Figure 5 depicts the clinical characteristics of the EoHM patients based on fundoscopic examination. 33% of them had not developed yet any features (normal). The most prevalent phenotypic feature is diffuse chorioretinal atrophy, present in 19% of the cases alone and adding a total of 38% accompanied by other features, followed by a tesselated fundus, with 9%.
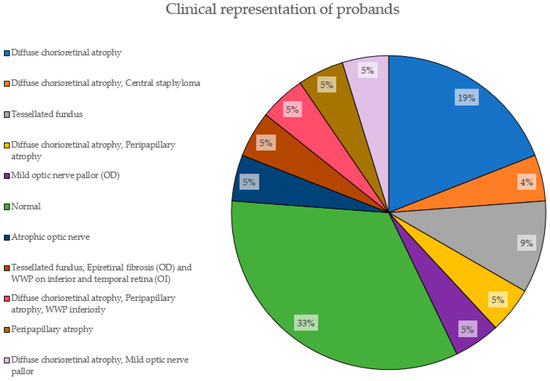
Figure 5.
Clinical representation of probands. The proportions of features presented by the probands in this study are shown here, with their corresponding percentage.
For the gender-based difference analysis, the patients were divided into two groups: more severe EoHM and less severe EoHM, obtaining the following frequencies (Table 3).
Table 3.
Frequencies.
Since we have frequencies lower than 5, a Fisher’s exact test was performed, resulting in a p-value of 0.6557. Given that this p-value is greater than 0.05, there is insufficient evidence to reject the null hypothesis (it can be found in Materials and Methods section). Thus, there is no association between severity and gender; they are independent variables.
The results obtained were evaluated considering the limitations of exome sequencing such as the low capture efficiency in the sequencing of certain regions, the inability to analyze regulatory regions far from the exons, the lack of previous scientific studies of yet unknown genes and the inability to conduct functional studies of all the variants found.
As one of the objectives of this work was to identify new genes responsible for EoHM and exome sequencing implies the possibility of finding variants in genes not previously related to ocular pathology, we considered variants classified as pathogenic (P), likely pathogenic (LP) and variants of uncertain significance (VUS) not previously associated with HM, but that are involved in the regulation of emmetropization.
We found 83 variants that may be involved in the development of EoHM. Two (3%) were classified as P, five (6%) was LP and 74 (91%) was VUS (Figure 6). The American College of Medical Genetics (ACMG) criteria for classifying pathogenic variants may be observed in Table A1.
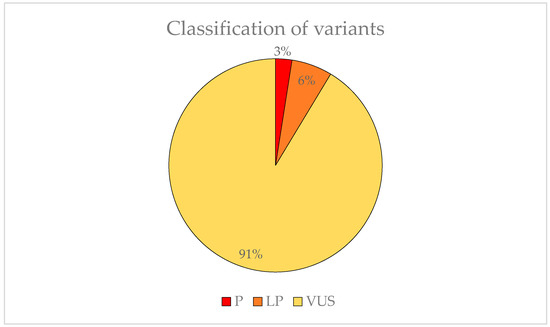
Figure 6.
Representation of the variant pathogenicity found in possible candidate genes for EoHM.
The alterations found in the families included in the study and their relationship to EoHM are detailed below.
2.1. OFT-00074 Family
Candidate genes on Table 4. One of the proposed causal alterations for EoHM in this family was classified as pathogenic and affects the splicing of the TRPM1 gene. Its associated pathology is night blindness, congenital stationary (complete), 1C and autosomal recessive [9]. It encodes a permeable calcium cation channel, mainly expressed in retinal bipolar cells. Because CSNB and EoHM are associated and cases of EoHM have been observed in CSNB patients with altered TRPM1, the involvement of this gene in its development has been proposed, although the cause of this association is not yet known [10,11,12]. It has been hypothesized that TRPM1 plays a role in embryonic development by influencing synaptic activity, optic nerve formation, photoreceptor arrangement and ON bipolar cell function [13]. In the Gene Ontology Resource database, this gene is associated with visual perception and the cellular response to light stimulus (Table A2) [14].
Other VUS alterations detected in this family included that of ARHGEF18, which codes for a guanine nucleotide exchange factor (GEF) that directly controls the activation of Rho GTPases and is involved in retinal development and degeneration [15]. This gene is a key regulator of RhoA-Rock2 signaling, which is crucial for the maintenance of polarity in the vertebrate retinal epithelium. In addition, ARHGEF18 is required to maintain apico-basal polarity, tight junction localization and cortical actin, thus shaping the morphology of these cells [16]. This gene is associated with retinitis pigmentosa 78 [9].
KDM6B encodes a lysine-specific demethylase that demethylates histone H3 (epigenetic control) and other non-histone proteins. It is involved in processes of organogenesis and retinal development at the amacrine, horizontal and ganglion cell level, allowing the focusing or defocusing signal to correctly reach the brain from the retina [17].
The selected candidate genes did not have AD inheritance and were not found in homozygosity, suggesting the possibility of a cumulative effect of the proposed alterations as a cause of EoHM in the affected individuals.
Table 4.
Candidate genes of the OFT-00074 family.
Table 4.
Candidate genes of the OFT-00074 family.
 | ACMG Criteria | ACMG Result | Variant | Zygosity | Inheritance | Total Families | Model of Inheritance | Gene Reported by |
|---|---|---|---|---|---|---|---|---|
| ARHGEF18 | PM2 | VUS | NM_015318.3:c.2167C>T:p.(Arg723Cys) | Het | Unknown | 1 | AR | [16] |
| KDM6B | PM2, BP4 | VUS | NM_001080424.1:c.1582C>T:p.(Pro528Ser) | Het | Unknown | 2 | Unknown | [17] |
| TRPM1 | PVS1, PM2, PP5 | P | NM_002420.5:c.1023+1G>A | Het | Unknown | 2 | AR | [10,11,12,13] |
Het: heterozygous; AR: autosomal recessive.
2.2. OFT-00097 Family
Candidate genes on Table 5. The proband in this family had a variant inherited from his mother of the most altered gene identified in this study, HSPG2. This is a gene with incomplete penetrance [18], encoding perlecan, a large multidomain proteoglycan. It binds and cross-links to ECM components and molecules on the cell surface, allowing it to interact with laminin, prolargin, type IV collagen, FGFBP1, FBLN2, FGF7 and transthyretin and maintain the endothelial barrier function in the vascular ECM. HSPG2 also promotes the activity of growth factors such as FGF2, thus stimulating endothelial growth and fibroblast regeneration [15]. The study by Wan et al., includes it as one of the new candidate genes for EoHM [19].
Other altered genes affecting the structure of the sclera were also observed, such as COL9A2, a collagen associated with Stickler syndrome, which, being a heterozygous variant, may have contributed to HM in conjunction with the other alterations without developing the full syndrome [20]. Fibulin1 (FBLN1) is a glycoprotein incorporated into a fibrillar ECM [15], which is expressed in fibroblasts of the sclera, enabling cell–matrix interactions, and is involved in the regulation of ocular growth [21].
CACNA1F presents a pathogenic alteration in homozygosis that adds a stop codon. The protein expressed is a multi-pass transmembrane that functions as an alpha-1 subunit of the voltage-dependent calcium channel and mediates the entry of calcium ions into the cell. CACNA1F is associated with several X-linked eye disorders including CSNB type 2A and Aland Island eye disease [15]. The gene is involved in the cone and rod response and was identified in a study of patients with CSNB as predominant in those who also had EoHM [22].
CSMD1, another of the most altered genes found in this study, is located at the MYP10 locus and is mainly expressed in the peripheral retina and in the area surrounding the macula. This gene plays a critical role in cone growth, including signal transduction and matrix adhesion, and has been proposed in multiple studies as a candidate gene for the development of EoHM [19,23].
Finally, an alteration in ADAMTSL1 is proposed as possibly contributing to the development of EoHM by encoding a thrombospondin motif metalloproteinase (ADAMTS). This protein may play important roles in the ECM, thus establishing a link to myopia [15].
In the case of this family, the most relevant alterations that may have influenced the EoHM of the probands are those present in the HSPG2, FBLN1 and CACNA1F genes, due to their inheritance pattern, although the other genes mentioned may also have contributed. Specifically, CSMD1 has been observed and proposed in this study and other related studies [19,23].
Table 5.
Candidate genes of the OFT-00097 family.
Table 5.
Candidate genes of the OFT-00097 family.
 | ACMG Criteria | ACMG Result | Variant | Zygosity | Inheritance | Total Families | Model of Inheritance | Gene Reported by |
|---|---|---|---|---|---|---|---|---|
| ADAMTSL1 | PM2 | VUS | NM_001040272.5:c.1819G>A:p.(Glu607Lys) | Het | Unknown | 1 | Unknown | [15] |
| CACNA1F | PVS1, PM2, PP5 | P | NM_005183.3:c.4504C>T:p.(Arg1502 *) | Hemi | Maternal | 2 | XL | [22,24] |
| COL9A2 | PM2 | VUS | NM_001852.3:c.1652C>T:p.(Ala551Val) | Het | Unknown | 1 | AR | [20] |
| CSMD1 | PM2 | VUS | NM_033225.5:c.1712A>G:p.(Asn571Ser) | Het | Maternal | 4 | Unknown | [19,23] |
| FBLN1 | PM2 | VUS | NM_006486.2:c.1157C>T:p.(Thr386Met) | Het | Unknown | 1 | AD | [21] |
| HSPG2 | PM2 | VUS | NM_005529.6:c.12691G>A:p.(Glu4231Lys) | Het | Maternal | 6 | AD/AR | [19] |
Het: heterozygous; Hemi: hemizygous; AD: autosomal dominant; AR: autosomal recessive; XL: X-linked. * Gain of a stop codon, does not substitute for any amino acid.
2.3. OFT-00155 Family
Candidate genes on Table 6. In this family, we searched for alterations common to the two affected individuals, as well as including other genes, also shared with the unaffected sibling, reported in studies of EoHM or myopia. All the altered genes are involved in the maintenance of the sclera, with the exception of CACNA1F, as explained above, and COL9A3, a collagen described in Stickler syndrome, in which the most involved ocular structure is the choroid [15]. The role of HSPG2 in the development of EoHM has been detailed above.
Table 6.
Candidate genes of the OFT-00155 family.
The LAMA1 gene is at the MYP2 locus and belongs to a family of structural glycoproteins in the ECM of the sclera and lens [15,25]. It is one of the genes most associated with the development of EoHM, supported by several studies [25,26,27]. Because it has an AR inheritance and is heterozygous, it is likely not the only altered gene causing EoHM in this family. There could be a cumulative effect enhanced by the other alterations indicated in Table 4, as most of these affect the sclera. For example, an alteration was observed in LAMA5, another glycoprotein in basement membranes with biological functions similar to LAMA1 [15].
Alterations were also found in the THBS2 and THBS1 genes, homotrimeric-disulfide-linked glycoproteins that mediate cell–cell and cell–matrix interactions. THBS1 can bind to fibrinogen, fibronectin, laminin, type-V collagen and alpha-V/beta-1 integrins [15]. In a study of patterns of messenger RNA (mRNA) in sclera remodeling during the development of lens-induced myopia in Soricidae, changes were observed in the SPARC, THBS1, THBS2, TNC and SPP1 genes, suggesting that these may play a role in increasing scleral sliding velocity [28].
2.4. OFT-00175 Family
Candidate genes on Table 7. All the alterations found in the OFT-00175 family were classified as VUS. The proband had only one de novo alteration in HSPG2; the other alterations were inherited from his father.
The altered genes affecting the retina are PCDH15 and TRPM1. PCDH15 belongs to the cadherin superfamily, which encodes integral membrane proteins responsible for mediating calcium-dependent cell–cell adhesion. It plays an essential role in the maintenance of normal retinal and cochlear function [15]. PCDH15 was proposed in the WES study by Wan et al., as a candidate gene related to EoHM [19], with an AR or digenic inheritance model. Digenic inheritance is that in which a mutation in two genes is necessary to cause a given phenotype. In this case, there were alterations in four genes.
Finally, BMPR2 is a member of the bone morphogenetic protein (BMP) family of transmembrane serine/threonine kinase receptors. Ligands for this receptor are members of the transforming growth factor (TGF)-β superfamily [15]. This gene is also expressed in the retina but may have some relation to the development of EoHM given its association with the function of scleral fibroblasts in early myopia and incomplete penetrance [29].
Table 7.
Candidate genes of the OFT-00175 family.
Table 7.
Candidate genes of the OFT-00175 family.
 | ACMG Criteria | ACMG Result | Variant | Zygosity | Inheritance | Total Families | Model of Inheritance | Gene Reported by |
|---|---|---|---|---|---|---|---|---|
| BMPR2 | PM2 | VUS | NM_001204.6:c.1931A>G:p.(Asn644Ser) | Het | Paternal | 1 | AD | [29] |
| HSPG2 | PM2 | VUS | NM_005529.6:c.10481G>A:p.(Arg3494Gln) | Het | De novo | 6 | AD/AR | [19] |
| PCDH15 | PM2 | VUS | NM_001142769.1:c.4396A>G:p.(Ser1466Gly) | Het | Paternal | 2 | AR/Digenic | [19] |
| TRPM1 | BP4 | VUS | NM_002420.5:c.4433C>T:p.(Thr1478Met) | Het | Paternal | 2 | AR | [10,11,12,13] |
Het: heterozygous; AD: autosomal dominant; AR: autosomal recessive.
2.5. OFT-00178 Family
Candidate genes on Table 8. In this family, variants inherited from the mother and father were found affecting both scleral and retinal development, which could cumulatively cause EoHM.
Genes involved in the identified retinal development included PCDH15 and CSMD1, discussed above. LRP2, another gene in this group, encodes a multi-ligand endocytic receptor, has a role in cell signaling and has been associated with Donnai–Barrow and Stickler syndromes, both of which are characterized by EoHM [15]. This gene was found to have a necessary function for normal eye growth through inactivation of Lrp2 in the mouse forebrain (including the neural retina and retinal and ciliary pigment epithelia), resulting in a 40% greater axial elongation compared to controls. Bipolar, photoreceptor and retinal ganglion cells were also affected [30].
The proband had two heterozygous alterations in the MAP3K1 gene, one inherited from his mother and the other from his father, known as double heterozygous or compound heterozygous. This gene is known to be involved in eye development, with high expression in the retina. Its low expression in the mouse retina affects vascularization, RPE, photoreceptor loss and early degeneration [31,32].
LAMA4 is another gene associated with alterations affecting the sclera. Expression changes in the HIF-1α/miR-150-5p/LAMA4/p38 MAPK axis have been observed in ECM degradation of scleral fibroblasts under hypoxic conditions, leading to the pathological progression of HM. An increased expression of LAMA4 has been observed in patients with HM [33].
At the scleral level, excessive PLG expression has been reported in HM patients. Here, plasmin (in its active form) can degrade fibrin and convert potential matrix metalloproteinases (pro-MMPs) into active MMPs, capable of destroying the ECM, thus reducing scleral stiffness and making it unable to maintain its necessary stiffness, strength and elasticity. Plasmin also participates in other processes involved in the pathogenesis of HM such as tissue remodeling and angiogenesis [34].
VASH1 enables actin-binding and metallocarboxypeptidase activity. It is involved in the negative regulation of angiogenesis, the migration of blood vessel endothelial cells and proteolysis [15] and is expressed in the neural retina, sclera, choroid and RPE [35].
Several of these alterations may play a significant role in EoHM.
Table 8.
Candidate genes of the OFT-00178 family.
Table 8.
Candidate genes of the OFT-00178 family.
 | ACMG Criteria | ACMG Result | Variant | Zygosity | Inheritance | Total Families | Model of Inheritance | Gene Reported by |
|---|---|---|---|---|---|---|---|---|
| CSMD1 | PM2 | VUS | NM_033225.5:c.8042G>A:p.(Gly2681Asp) | Het | Paternal | 4 | Unknown | [19,23] |
| CSMD1 | PM2, BP4 | VUS | NM_033225.5:c.4375G>A:p.(Ala1459Thr) | Het | Paternal | 4 | Unknown | [19,23] |
| LAMA4 | PM2 | VUS | NM_001105207.2:c.673G>A:p.(Ala225Thr) | Het | Paternal | 1 | AD | [33] |
| LRP2 | PM2, PP3 | VUS | NM_004525.2:c.10202C>G:p.(Thr3401Arg) | Het | Maternal | 1 | AR | [30] |
| MAP3K1 | PM2, BP4 | VUS | NM_005921.1:c.299G>A:p.(Gly100Glu) | Het | Maternal | 1 | AD | [31,32] |
| MAP3K1 | PM2, PM4 | VUS | NM_005921.1:c.3646_3648delATC:p.(Ile1216del) | Het | Paternal | 1 | AD | [31,32] |
| PCDH15 | PM2, BP4 | VUS | NM_001142769.1:c.1519G>A:p.(Val507Ile) | Het | Paternal | 2 | AR/Digenic | [19] |
| PLG | PM2, PP3 | VUS | NM_000301.3:c.598A>G:p.(Thr200Ala) | Het | Paternal | 1 | AR | [34] |
| VASH1 | PM2 | VUS | NM_014909.4:c.953G>A:p.(Arg318Gln) | Het | Maternal | 1 | Unknown | [35] |
Het: heterozygous; AD: autosomal dominant; AR: autosomal recessive.
2.6. OFT-00191 Family
Candidate genes on Table 9. The PER3 gene belongs to the period gene family, which encodes components of the circadian rhythms of locomotor activity, metabolism and behavior [15]. It has a role in the negative regulation of transcription by RNA polymerase II [14]. Circadian rhythm genes are associated with refractive error. PER3 is located near the MYP14 locus and is expressed in ON and OFF bipolar cells [36].
COL11A1, also associated with Stickler and Marshall syndromes, may only present a phenotype in the eye, such as EoHM [15,37]. As for FRMPD1, the protein it encodes directly interacts with Gpsm2 (G-protein signaling modulator 2) and is necessary for the optimization of the rod-to-bipolar synaptic transmission when Gαt is present at the synapse [38].
In the two affected individuals of this family, the effect of the alteration in COL11A1 predominates, making it the primary factor in the pathogenesis of the disease given its inheritance pattern, classification, extensive evidence in the literature and association with Stickler and Marshall syndromes.
Table 9.
Candidate genes of the OFT-00191 family.
Table 9.
Candidate genes of the OFT-00191 family.
 | ACMG Criteria | ACMG Result | Variant | Zygosity | Inheritance | Total Families | Model of Inheritance | Gene Reported by |
|---|---|---|---|---|---|---|---|---|
| COL11A1 | PP3, PM2 | LP | NM_001854.3:c.2900G>T:p.(Gly967Val) | Het | Paternal | 1 | AD/AR | [15,24] |
| FRMPD1 | PM2, BP4 | VUS | NM_014907.2:c.2469C>A:p.(Ser823Arg) | Het | Paternal | 2 | Unknown | [38] |
| PER3 | PM2, BP4 | VUS | NM_016831.2:c.3502A>G:p.(Thr1168Ala) | Het | Paternal | 1 | AD | [36] |
Het: heterozygous; AD: autosomal dominant; AR: autosomal recessive.
2.7. OFT-00209 Family
Candidate genes on Table 10. The candidate genes for EoHM in this family include GLB1. The protein it encodes (galactosidase beta 1) is present in human choroidal endothelial cells and acts as a receptor for elastin-derived peptides (EDPs). Elevated levels of circulating EDP do not affect retinal function in mice but increase the expression and deposition of collagen IV in the RPE/choroid complex [39]. COL9A1 is associated with Stickler syndrome [15]; KDM6B has been discussed above.
Table 10.
Candidate genes of the OFT-00209 family.
2.8. OFT-00217 Family
Candidate genes on Table 11. The proband in family OFT-00217 inherited alterations from both her mother and father. AGRN is located at the MYP14 locus, encoding agrin, a large proteoglycan with multiple isoforms. It contains several laminin G domains, a Kazal-like serine protease inhibitor and epidermal growth factor. The study by Zheng et al. [40] found that AGRN is involved in baseline refractive development, demonstrating that this gene interacts with EGR1, which is implicated in refractive development and regulates synaptic physiology in the retina [15,40]. Several studies of different populations have associated rare or infrequent heterozygous alterations in AGRN with the development of HM [40].
The GRM6 gene is involved in ON synaptic transmission in bipolar cells, controlling the release of dopamine, which has been suggested as a factor in ocular growth. Mutations in this gene cause CSNB type 1B. HM is often seen in CSNB type 1B patients with altered GRM6. A link between GRM6 and susceptibility to HM has been suggested in a number of studies [41].
Alterations in the CNTN6 gene may be involved in the development of EoHM, as contactin-6 is responsible for the correct development and maintenance of the central nervous system, particularly in axonal projection, branching and synapses. Meguro et al., associated an altered expression of this gene affecting GABA receptor levels or synaptogenesis with HM [42]. FRMD4B is also involved at the retinal level, through direct interaction of the protein it encodes with cytohesin-3, and functions as a scaffolding protein. Cytohesin-3 plays an important role in insulin, epidermal and nerve growth factor signaling, with a recent study [42] suggesting that the FRMD4B-cytohesin-3 complex affects cell junction dynamics in the retina and contributes to the regulation of photoreceptor cell growth and development [42].
LRP1 affects the sclera, encoding protein 1 of the low-density lipoprotein receptor family of cytokines and growth factors, and is involved in several cellular processes, even the negative regulation of gene expression [14]. LRP1 deficiency leads to the disruption of TGF-β and may result in abnormal remodeling of the ECM of the developing eye, making this gene a candidate for HM [43].
Further studies are needed to demonstrate the direct involvement of these genes with EoHM for an accurate diagnosis, but for now, they are good candidates.
Table 11.
Candidate genes of the OFT-00217 family.
Table 11.
Candidate genes of the OFT-00217 family.
 | ACMG Criteria | ACMG Result | Variant | Zygosity | Inheritance | Total Families | Model of Inheritance | Gene Reported by |
|---|---|---|---|---|---|---|---|---|
| AGRN | PM2 | VUS | NM_198576.3:c.4799C>T:p.(Ala1600Val) | Het | Paternal | 2 | AR | [40] |
| CNTN6 | PM2, BP4 | VUS | NM_014461.3:c.260A>G:p.(Asn87Ser) | Het | Paternal | 1 | Unknown | [42] |
| CNTN6 | PM2, BP4 | VUS | NM_014461.3:c.2553G>C:p.(Met851Ile) | Het | Paternal | 1 | Unknown | [42] |
| FRMD4B | PM2, PP3 | VUS | NM_015123.2:c.554T>C:p.(Leu185Ser) | Het | Maternal | 1 | Unknown | [42] |
| GRM6 | PM2, PVS1, PP5 | LP | NM_000843.3:c.3G>T:p.(Met1?) | Het | Paternal | 1 | AR | [24,41] |
| LRP1 | PM2, PP2 | VUS | NM_002332.2:c.1415G>A:p.(Arg472Gln) | Het | Paternal | 2 | AD/AR | [43] |
Het: heterozygous; AD: autosomal dominant; AR: autosomal recessive.
2.9. OFT-00223 Family
Candidate genes on Table 12. In the analysis of this family, we studied the alterations present in the two affected individuals. The protein encoded by USH2A is abundantly expressed in the macular and peripheral retina, mainly by photoreceptors. Alterations in this gene have been associated with Usher syndrome type IIa and retinitis pigmentosa. In a WES study of 20 patients with EoHM, USH2A was identified as a candidate gene, with pathogenic variants in four participants [19]. ALKBH5 enables mRNA N6-methyladenosine dioxygenase activity, is involved in the response to hypoxia [15], and a decrease in its expression has been observed in individuals with EoHM [44].
Table 12.
Candidate genes of the OFT-00223 family.
This study proposes the involvement of both alterations in the development of EoHM.
2.10. OFT-00253 Family
Candidate genes on Table 13. In this family, we detected four genes that had not been observed in the other families studied. CEP290 encodes the centrosomal protein 290, which is highly expressed in the retina of individuals with HM, being predominant in the retinal photoreceptors. Wan et al., proposed this gene as a candidate for causing EoHM [19].
CPSF1 is known to play a role in mRNA processing; however, the relationship between CPSF1 and human eye diseases, including myopia, remains unknown. Results from several studies suggest that mutations in CPSF1 may be a new cause of EoHM [40,45,46]. OPN4 encodes melanopsin, a photoreceptor opsin expressed in retinal ganglion and amacrine cells. These are necessary for correct refractive development and protection from myopia progression, as they are involved in the eye’s response to myopigenic stimuli, acting in part through dopaminergic mechanisms [47].
The proband was double heterozygous for the MYOM1 gene, which is located at the MYP2 locus and is a structural constituent of the cytoskeleton thought to integrate the thin and thick filaments and confer elasticity to the M-band of the sarcomere in striated muscle. Alterations in this gene have been associated with the development of HM [48].
Table 13.
Candidate genes of the OFT-00253 family.
Table 13.
Candidate genes of the OFT-00253 family.
 | ACMG Criteria | ACMG Result | Variant | Zygosity | Inheritance | Total Families | Model of Inheritance | Gene Reported by |
|---|---|---|---|---|---|---|---|---|
| CEP290 | PM2, BP4 | VUS | NM_025114.3:c.6791A>G:p.(Lys2264Arg) | Het | Maternal | 1 | AR | [19] |
| CPSF1 | PM2 | VUS | NM_013291.2:c.3128C>T:p.(Pro1043Leu) | Het | Maternal | 2 | Unknown | [40,45,46] |
| HSPG2 | PM2 | VUS | NM_005529.6:c.3346G>A:p.(Gly1116Ser) | Het | Paternal | 6 | AD/AR | [19] |
| MYOM1 | PM2, PP3 | VUS | NM_003803.3:c.4580G>T:p.(Gly1527Val) | Het | Maternal | 1 | Unknown | [48] |
| MYOM1 | PM2, PP3 | VUS | NM_003803.3:c.3032T>C:p.(Val1011Ala) | Het | Paternal | 1 | Unknown | [48] |
| OPN4 | BP6 | VUS | NM_033282.3:c.1411_1412insT:p.(Ser473fs) | Het | Paternal | 1 | Unknown | [47] |
| THBS2 | PM2 | VUS | NM_003247.3:c.799G>A:p.(Glu267Lys) | Het | Paternal | 2 | Unknown | [28] |
Het: heterozygous; AD: autosomal dominant; AR: autosomal recessive.
2.11. OFT-00268 Family
Candidate genes on Table 14. The proband, his mother (supposedly unaffected) and grandmother (affected) presented two alterations in the CSMD1 gene that compromise its function and could be the cause of EoHM.
Table 14.
Candidate gene of the OFT-00268 family.
2.12. OFT-00332 Family
Candidate genes on Table 15. Most of the genes proposed in this family may have AD inheritance and affected individuals at different levels: retina (ZNF644 and ARHGEF15), choroid (CFH) and sclera (HSPG2 and LRP1), as well as CPSF1, which is known to be involved in the pathology although its exact cause is unknown. ZNF644 encodes a zinc finger transcription factor in the RPE, regulating genes involved in ocular development. An alteration in this gene may therefore impact the structure of the eye, leading to the progression of EoHM. It has been related to the non-syndromic form of EoHM in several studies with an AD model of inheritance [15,49,50]. ARHGEF15, a guanine nucleotide exchange factor specific for RhoA, has been found to activate VEGF-induced Cdc42, promoting retinal angiogenesis [51].
Table 15.
Candidate genes of the OFT-00332 family.
CFH has antioxidant effects and regulates caspase-dependent apoptosis in retinal pigment epithelial cells under oxidative stress. This gene also blocks the pro-inflammatory effects of malondialdehyde, a major product of lipid peroxidation, and protects against oxidative stress in vivo in mice. Statistically significant higher levels of CFH have been observed in HM patients versus mild myopia and control groups, being greater in eyes with choroidal atrophy and its neovascularization, suggesting that it plays a key role in the development of myopia [52].
2.13. OFT-00403 Family
Candidate genes on Table 16. All three family members shared a single candidate variant, TSG101, which has a role in cell growth and differentiation and may act as a negative growth regulator. In the study by Le et al., the loss of TSG101 severely altered the polarity of the RPE, forming irregular aggregates with a non-polarized distribution of cell adhesion proteins and the activation of epidermal growth factor receptor signaling [53]. In this particular case, the variant has a low allele frequency in the overall population and is synonymous; in an in silico splicing analysis using Alamut Visual 2.15 software, it was found unaltered by the variant. TSG101 could therefore be a candidate for EoHM; however, further studies of this gene and its variants with other genes are necessary to determine the cause of the pathology.
Table 16.
Candidate genes of the OFT-00403 family.
Table 16.
Candidate genes of the OFT-00403 family.
 | ACMG Criteria | ACMG Result | Variant | Zygosity | Inheritance | Total Families | Model of Inheritance | Gene Reported by |
|---|---|---|---|---|---|---|---|---|
| TSG101 | PM2 | VUS | NM_006292.3:c.942C>T:p.(Ile314Ile) | Het | Maternal | 3 | Unknown | [53] |
Het: heterozygous.
2.14. OFT-00429 Family
Candidate genes on Table 17. The proband in this family had two deletions in the HSPG2 gene found in previous families. As the alterations are close to each other, it may be observed that they are in cis, which means they were inherited from the same parent.
This individual also had an altered LRPAP1 gene, which encodes a chaperone of LRP1, inhibits its degradation and influences the activity of TGF-β. An LRP1 deficiency and up-regulation of TGF-β have been observed in individuals with EoHM. TGF-β plays an important role in scleral ECM remodeling in myopia, and LRPAP1 has been linked to EoHM in several studies [43,54].
MMP9 is another gene involved in scleral ECM composition, implicated in the breakdown of type IV and V collagens [15,55]. Higher levels of MMP9 have been found to reduce scleral elastin, making it more prone to deformation under intraocular pressure, linking it to the development of myopia and HM [55,56].
Finally, affecting the retina, we observed alterations in the FLRT3 gene, which is involved in various signaling pathways and has been related to the development of the central nervous system and the eye due to its expression during ocular development in mouse embryos. It has also been associated with the development of HM in Central European families [57].
Table 17.
Candidate genes of the OFT-00429 family.
Table 17.
Candidate genes of the OFT-00429 family.
 | ACMG Criteria | ACMG Result | Variant | Zygosity | Inheritance | Total Families | Model of Inheritance | Gene Reported by |
|---|---|---|---|---|---|---|---|---|
| FLRT3 | PM2, PP3 | VUS | NM_013281.3:c.325T>G:p.(Leu109Val) | Het | Unknown | 2 | AD/Digenic/Multigenic | [57] |
| HSPG2 | PM2, PM4 | VUS | NM_005529.6:c.742_744delCTT:p.(Leu248del) | Het | Unknown | 6 | AD/AR | [19] |
| HSPG2 | PVS1, PM2 | LP | NM_005529.6:c.738delT:p.(Leu247fs) | Het | Unknown | 6 | AD/AR | [19] |
| LRPAP1 | PM2 | VUS | NM_002337.3:c.298G>A:p.(Gly100Ser) | Het | Unknown | 1 | AR | [43,54] |
| MMP9 | PM2, BP4 | VUS | NM_004994.2:c.1270C>A:p.(Arg424Ser) | Het | Unknown | 1 | AR | [55,56] |
Het: heterozygous; AD: autosomal dominant; AR: autosomal recessive.
2.15. OFT-00474 Family
Candidate genes on Table 18. All the alterations selected as candidate genes in this proband were inherited from his father. The importance of LAMA1 and FRMPD1 in the development of EoHM has been discussed above. The only novel gene is PRIMPOL, a DNA primase-polymerase that facilitates DNA damage tolerance by mediating uninterrupted fork progression after UV irradiation and reinitiating DNA synthesis [15]. PRIMPOL mRNA, which is expressed in many tissues including scleral fibroblasts, retinal epithelial and Müller cells, has been associated with the development of HM in several studies [58,59,60].
Table 18.
Candidate genes of the OFT-00474 family.
2.16. OFT-00477 Family
Candidate genes on Table 19. The only candidate gene for the development of EoHM in this family was TSG101. This gene and its synonymous variant are the same as those observed in Family OFT-00403. Further study could be interesting to confirm or rule out its role in the pathology.
Table 19.
Candidate gene of the OFT-00477 family.
2.17. OFT-00506 Family
Candidate genes on Table 20. In this family, we identified a single candidate alteration in the COL9A3 gene. This gene was also altered in family OFT-00155 and has an AD inheritance pattern, making it a good candidate.
Table 20.
Candidate gene of the OFT-00506 family.
2.18. OFT-00546 Family
Candidate genes on Table 21. The results for this family, like others in this study, suggest the cumulative effect of several altered genes involved in the development of EoHM inherited from both parents, although it includes genes that had not been observed in previous families. ABCA4 is expressed in the outer segments of the cone and rod photoreceptors of the retina and in the sclera, mediating the transport of an essential molecule, all-trans-retinal aldehyde, across the photoreceptor cell membrane upon activation following phototransduction. Mutations in ABCA4 can lead to multiple vision-related phenotypes, including retinitis pigmentosa, fundus flavimaculatus, cone-rod dystrophy and Stargardt disease. A variant in this gene responsible for myopia has recently been discovered, and has been proposed in the study by Wan et al., as a candidate gene for the development of EoHM [19].
Table 21.
Candidate genes of the OFT-00546 family.
LAMA2 encodes the alpha-2 subunit of laminin, a major component of the basement membrane, with an important role in connecting the collagen fibers of the sclera. This gene has been linked to refractive error and myopia in different ethnic populations [61,62]. It may be that altered LAMA2 and LAMA5 have a greater effect on the development of EoHM than if they were isolated events, as observed in family OFT-00155t with LAMA1 and LAMA5.
Alterations were also found in the LTBP2 gene. This gene belongs to the family of latent TGF-β binding proteins, which are ECM proteins with a multidomain structure. It has been associated with Weill–Marchesani 3 and Stickler syndromes [15].
2.19. OFT-00586 Family
Candidate genes on Table 22. The alterations observed in this proband and her son affecting the development of EoHM primarily involve the retina. In addition to LTBP2 and TSG101, which were also altered in previous families, this family presented altered BICC1. BICC1, located at the MYP15 locus [63], encodes an RNA-binding protein that regulates expression [15] and has been associated with the development of HM in several studies among different populations [63,64]. Alterations were also found in CNTN4, which encodes contactin-4, a glycosylphosphatidylinositol-anchored neuronal membrane protein that may play a role in the formation of axonal connections and arborization in the developing nervous system. CNTN4 has been associated with HM in the literature [42].
Table 22.
Candidate genes of the OFT-00586 family.
2.20. OFT-00601 Family
Candidate genes on Table 23. Both affected members of this family had altered CSMD1, which is the candidate gene involved in the development of EoHM in this and other families included in our study.
Table 23.
Candidate gene of the OFT-00601 family.
2.21. OFT-00710 Family
Candidate genes on Table 24. In the last family, all the variants selected as candidates for the development of EoHM were maternally inherited. In addition to alterations in the USH2A and LTBP2 genes, an alteration was also observed in FBN1, a member of the fibrillin family of proteins. Mutations in this gene are associated with Marfan syndrome (with HM as one of its clinical manifestations) and the MASS phenotype [15]. We propose all three genes as candidates for the progression of EoHM.
Table 24.
Candidate genes of the OFT-00710 family.
In our analysis of families with EoHM, we found that most of the alterations identified were classified as VUS, reflecting the need for further studies to determine the pathogenicity of these variants and to confirm the involvement of the proposed genes in the pathology. Table A2 shows a summary of the different variants found in each family, its classification, functional annotation and frequencies in the gnomAD global population. We also added the main pathways related to each gene.
The 74 VUS identified are located within a total of 47 genes. Out of these 47 genes, 19.15% are associated with pathologies that may manifest as HM without other clinical characteristics [37]. This percentage includes genes linked to myopia (CPSF1, ZNF644, LRPAP1 and PRIMPOL, 8.51%), Stickler syndrome (COL9A1, COL9A2 and COL9A3, 6.38%), Marfan syndrome (FBN1, 2.13%) and CSNB (TRPM1, 2.13%).
The remaining 80.85% of genes are associated with various other pathologies. However, this does not imply that they cannot contribute to EoHM, as previously discussed, since they play a role in the development of the eye and ocular structures and have been considered candidates in prior studies.
Five of the proposed genes codify transcription factors, which are KDM6B, PER3, LRP1, ZNF644 and PRIMPOL.
Figure 7 illustrates that most of the identified variants are missense mutations, accounting for 83% of them. This can explain the substantial proportion of variants classified as VUS observed previously. Missense variants can have a variable impact on the function of the protein encoded by the gene by altering a single amino acid and, moreover, not all options are covered. In contrast, stop-gained variants are far more likely to result in a complete loss of protein function, so they are rarely classified as VUS.
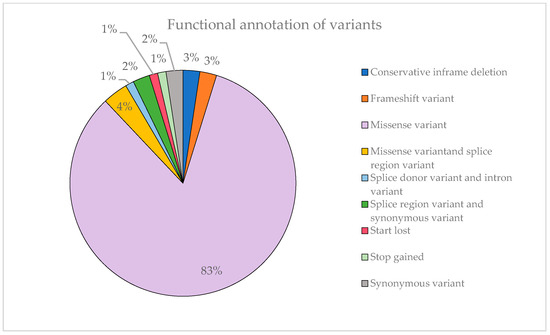
Figure 7.
Representation of the functional annotation of variants and their proportion.
Notably, most alterations in the probands were primarily concentrated in the retina or sclera (Figure 8), an observation that coincides with the literature.
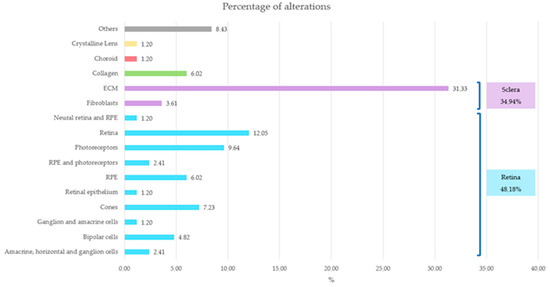
Figure 8.
Representation of the affected ocular structures and their corresponding percentage. Retina structures are represented in blue, sclera structures in purple, choroid in red, crystalline lens in yellow, collagen in green and others in gray.
The main optical function of the crystalline lens is to transmit light, focusing it on the retina [65]. The retina is a layer of photoreceptor cells and glial cells that captures incoming photons from the lens, it can discern whether the perceived image is blurred or not, and it transmits the photons along neuronal pathways as both electrical and chemical signals for the brain to perceive a visual picture [66]. The choroid supplies the outer retina with nutrients and maintains the temperature and volume of the eye [67]. And the sclera influences eye size, facilitating the excessive axial elongation that occurs during myopigenesis [68]. If the variants impact genes that are expressed and have a function in ocular structures, their function may become compromised, leading to failures in processes such as visual acuity, light detection and regulation of ocular axial growth, resulting in a larger axial length in most of them. Among these, axial growth is particularly critical in the pathology under consideration.
Several genes were found to be altered in more than one family, suggesting a stronger association with the shared pathology. Genes with alterations in two or more families were TRPM1, KDM6B, HSPG2, CACNA1F, CSMD1, LAMA1, COL9A3, LAMA5, THBS2, PCDH15, FRMPD1, AGRN, LRP1, USH2A, CPSF1, TSG101, FLRT3 and LTBP2. Specifically, HSPG2 was altered in six of the twenty-one families, while CSMD1 was altered in four, being the only candidate gene in several of them.
Although EoHM does not depend exclusively on the presence of alterations in these genes, it is influenced by the degree of penetrance, which was incomplete in some cases and unknown in others. Development of this pathology is also determined by the interaction and cumulative effect of these genes, with a genetic background affecting various levels of the multilayer signaling cascade (Figure 3 and Figure 8). Some genetic alterations may not have a significant impact in isolation in healthy individuals, but may have a more pronounced effect in the case of probands when combined with other alterations, thereby contributing to the development of the disease.
In addition to their biological function, most of the genes we propose have also been identified as candidates for EoHM in independent studies, supporting their involvement here.
3. Materials and Methods
A combined ophthalmological and genetic study was performed by the Multidisciplinary Unit of Ophthalmogenetics (UMOG) of the La Paz University Hospital, in accordance with the principles of the Declaration of Helsinki, and was approved by the ethics committee.
The inclusion criteria in our study were the following: (1) bilateral myopia with a refractive error of ≤−6 diopters in at least one eye with onset before the age of 10 years; (2) inconclusive result in the massive sequencing study implementing a panel (OFT-v3-1) of 419 genes related to ophthalmological disorders with suspected genetic cause, 93 genes and regions related to the pathogenesis of EoHM or within loci related to EoHM; (3) absence of syndromic phenotype; and (4) absence of corneal disease or other ophthalmologic diseases leading to secondary high myopia. In addition, whenever possible, parents and other relatives of the patients were included in the study.
A total of 63 individuals from 21 unrelated families were recruited in accordance with the inclusion criteria, including 30 affected individuals aged between 6 and 80 years old. Once informed written consent was obtained from the probands and their parents or guardians, they entered the study and followed the workflow represented in Figure 9. The clinical ophthalmologic evaluation of patients and first-degree relatives who signed the consent form was performed by the Ophthalmology unit of Hospital La Paz (HULP-3576).
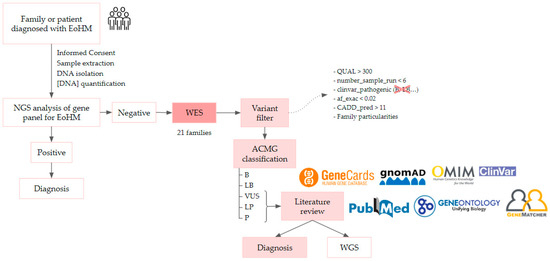
Figure 9.
EoHM diagnostic workflow, it is described in more detail below. ACMG: American College of Medical Genetics; WES, whole-exome sequencing; WGS, whole-genome sequencing.
The method followed for the statistical analysis was a Fisher’s exact test to determine whether severity is dependent on gender, with the following hypotheses:
Null Hypothesis (H0): Severity and gender are independent variables.
Alternative Hypothesis (H1): Severity and gender are dependent variables.
The Fisher’s exact test was conducted using the ‘fisher.test()’ function in RStudio.
Participants first underwent a complete ophthalmological evaluation, including best-corrected visual acuity, refraction before and after cycloplegia, funduscopic examination, LA measurement, retinography and OCT imaging.
A genetic study was then performed on genomic DNA obtained from leukocytes isolated from a peripheral venous blood sample in the pre-analytical area of our institute using the Chemagic Magnetic Separation Module I (Chemagen, PerkinElmer, Waltham, MA, USA). Concentrations of the isolated DNA were quantified using a NanoDrop ND-1000 spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA). Library preparation was carried out using Nextera DNA Exome (Illumina DNA Prep with Enrichment) and IDT for Illumina DNA/RNA UD Indexes Set A, B, C or D, Tagmentation. Sequencing was performed on high-quality sequencers, HiSeq4000 and NovaSeq6000, capturing 19,433 genes using xGen™ Exome Research Panel v2 IDT.
The Clinical Bioinformatics team at the Institute of Medical and Molecular Genetics (INGEMM) then performed the first analysis of the sequences obtained using an analytical algorithm developed to identify single nucleotide polymorphisms (SNPs) and insertions and deletions of small DNA fragments (indels) within the exome capture regions. This process comprises a sample pre-processing step, alignment of reads to a reference genome, identification and functional annotation of variants, and filtering of those variants. All these steps employ open tools widely used in the scientific community as well as proprietary tools. In addition, the steps are robustly designed and include process parameters and adequate quality controls to deliver a reliable report on the variants in question.
After sequencing, basecall file conversion (BCL) to FASTQ files was performed using Illumina’s demultiplexing software (bcl2fastq2 v2.20.0). The resulting DNA sequence reads were aligned and mapped to the human genome reference sequence (GRCh37/hg19) using bowtie2-align v2.0.6, after pre-processing and trimming using Trimmomatic v0.36 software. Realignment and recalibration of the reads were performed using the Genome Analysis Toolkit (GATK v3.3.0), and PCR duplicates were removed using Picard-tools v1.141. In addition, samtools v1.3.1 and BEDtools v2.26 software tools were used for the bioinformatics statistical analysis. SNPs and indels were detected using GATK v3.3.0, and the algorithms used for CNV detection were LACONv (unpublished in-house-developed algorithm) and eXome-Hidden Markov Model (XHMM v1.1). The variants were annotated in the Variant Call File (VCF) with predicted functional effect using SnpEff v4.3s. Furthermore, the following databases were also used for annotation: dbNSFP v3.5, dbSNP v151, ClinVar date 20180930, ExAC-1, SIFT ensembl 66, Polyphen-2 v2.2.2, MutationAssessor v3, FATHMM v2.3, CADD v1.4 and dbscSNV1.1.
The second analysis consisted of assessing the clinical significance of the variants found in the patients, relating them to their phenotype. To do so, we first filtered the data from bioinformatics using the criteria indicated in Table 25.
Table 25.
Filters used after the bioinformatic analysis.
The variants were then classified according to ACMG prioritization standards with Franklin by Genoox, and doing so, we can know if the classification of the variant is B, LB, VUS, LP or P. Finally, we studied the clinical pathogenic significance of the VUS, LP and P variants and their relationship to the pathology under study; it was assessed by consulting several databases. The main databases consulted were Pubmed (pubmed.ncbi.nlm.nih.gov/), GeneCards (genecards.org/), gnomAD (gnomad.broadinstitute.org/), OMIM (omim.org/), Clinvar (ncbi.nlm.nih.gov/clinvar/) and GeneOntologyResource (geneontology.org/), as well as contacting different scientists through GeneMatcher (genematcher.org/).
The criteria followed to identify the candidate genes causing the pathology were scientific articles that (1) include the described gene involved in EoHM; (2) report the gene as a candidate implicated in EoHM, HM or syndromes that include EoHM among their manifestations; or (3) involve the development, homeostasis or correct functioning of the main tissues affected in EoHM (retina, choroid and sclera).
Candidate variants in affected subjects and relatives with indications of a causal relationship with EoHM should be validated using Sanger sequencing and functional studies. If no candidate variants are found, it would be necessary to extend the study with other techniques such as whole-genome sequencing (WGS).
4. Conclusions
Using WES, this study proposes 51 candidate genes that may cause EoHM. The genes identified are TRPM1, ARHGEF18, KDM6B, HSPG2, COL9A2, FBLN1, CACNA1F, CSMD1, ADAMTSL1, LAMA1, COL9A3, LAMA5, THBS2, THBS1, PCDH15, BMPR2, LRP2, MAP3K1, LAMA4, PLG, VASH1, PER3, COL11A1, FRMPD1, GLB1, COL9A1, AGRN, GRM6, LRP1, CNTN6, FRMD4B, USH2A, ALKBH5, CEP290, CPSF1, OPN4, MYOM1, ZNF644, CFH, ARHGEF15, TSG101, LRPAP1, FLRT3, MMP9, PRIMPOL, ABCA4, LAMA2, LTBP2, BICC1, CNTN4 and FBN1.
The presence of several alterations associated with EoHM in the same patient may indicate the existence of incomplete penetrance or polygenic inheritance of the disease, suggesting a cumulative pathogenic effect of different VUSs. Further studies of the proposed candidate genes are needed to learn more about their actual involvement in EoHM and their cumulative effect. Such studies could include WES with different cohorts, WGS, transcriptome analysis, DNA methylation or long-read sequencing of these genes.
Sharing the results obtained in this study in conjunction with other previously published reports in the literature will contribute to a more accurate diagnosis of patients with EoHM in the future.
Our study also underscores the importance of the coordinated work of multidisciplinary teams to enhance patient care and diagnosis.
Author Contributions
Conceptualization, E.V. and S.N.; methodology, E.S.-C. and C.G.-A.; software, Á.D.P., L.D.D.-B. and C.R.-A.; validation, E.S.-C., C.G.-A., C.R.-J., R.M., N.A., V.E.F.M. and M.V.G.-P.; formal analysis, A.L.-V., M.N.-M., M.G.-C. and J.F.M.; investigation, E.S.-C., C.G.-A., O.D.M. and J.C.-R.; resources, J.C.A. and J.B.; data curation, M.d.L.Á.G.-C. and C.C.; writing—original draft preparation, E.S.-C. and C.G.-A.; writing—review and editing, E.V. and E.G.-I.; visualization, S.N.; supervision, E.V. and P.R.-S.; project administration, M.d.L.Á.G.-C. and L.D.-M.; funding acquisition, E.V. All authors have read and agreed to the published version of the manuscript.
Funding
This study has been funded by Instituto de Salud Carlos III (ISCIII) through the project “18/1234” and co-funded by ERDF, “A way to make Europe” and by ONCE grant number 2020/0197782.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of La Paz University Hospital of Madrid (protocol code PI-4016 approved on the 3 February 2020, HULP-3576).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to the patients and their families.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACMG | American College of Medical Genetics |
| AD | Autosomal dominant |
| ADAMTS | A disintegrin and metalloproteinase with thrombospondin motif |
| AL | Axial length |
| AR | Autosomal recessive |
| Astig | Astigmatism |
| BCVA | Best-corrected visual acuity |
| BMP | Bone morphogenetic protein |
| CSNB | Congenital stationary night blindness |
| ECM | Extracellular matrix |
| EDP | Elastin-derived peptides |
| EoHM | Early-onset High Myopia |
| Fx | Fixation |
| GEF | Guanine nucleotide exchange factor |
| Gpsm2 | G-protein signaling modulator 2 |
| GWAS | Genome-wide association study |
| Hemi | Hemizygous |
| Het | Heterozygous |
| HM | High myopia |
| LP | Likely pathogenic |
| NA | Not available |
| NFx | Non-fixation |
| NGS | Next-Generation Sequencing |
| NLP | No light perception |
| OD | Right eye |
| OS | Left eye |
| P | Pathogenic |
| RD | Retinal detachment |
| RPE | Retinal pigment epithelium |
| SD | Spherical diopters |
| SE | Spherical equivalent |
| SPcc | Sphere with cycloplegia |
| UMOG | Multidisciplinary Unit of Ophthalmogenetics |
| VUS | Variant of uncertain significance |
| WES | Whole-exome sequencing |
| WGS | Whole-genome sequencing |
| WWP | White without pressure |
| XL | X-linked |
Appendix A
Table A1.
American College of Medical Genetics and Genomics criteria for classifying pathogenic variants.
Table A1.
American College of Medical Genetics and Genomics criteria for classifying pathogenic variants.
| Code | Description |
| PVS1 | Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where loss of function is a known mechanism of disease. |
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. |
| PS2 | De novo (both maternity and paternity confirmed) in a patient with the disease and no family history. |
| PS3 | Well-established in vitro or in vivo functional studies supporting a damaging effect on the gene or gene product. |
| BS1 | Allele frequency is greater than expected for the disorder. |
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain (e.g., active site of an enzyme) with no benign variation. |
| PM2 | Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, and Exome Aggregation Consortium. |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant. |
| PM4 | Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants. |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been previously observed. |
| PP1 | Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease. |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease. |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.) |
| PP5 | Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation. |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.) |
| BP6 | Reputable source recently reports variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation. |
Table A2.
Summary of the different variants found in each family.
Table A2.
Summary of the different variants found in each family.
| Family | Variant | Classification | Functional Annotation | GnomAD Allele Frequency | Gene | Gene Ontology Main Pathways Associated |
| OFT-00074 | NM_015318.3:c.2167C>T:p.(Arg723Cys) | VUS | missense_variant | 0 | ARHGEF18 | Small-GTPase-mediated signal transduction, negative regulation of stress fiber assembly, extracellular exosome in part of neural stem cell, regulation of Rho protein signal transduction, plasma membrane |
| NM_001080424.1:c.1582C>T:p.(Pro528Ser) | VUS | missense_variant | 0.00001577 | KDM6B | Beta-catenin binding, histone demethylase activity, metal ion binding, histone h3k27me2/h3k27me3 demethylase activity, inflammatory response to antigenic stimulus, chromatin remodeling, positive regulation of transcription by RNA polymerase II, regulation of gene expression | |
| NM_002420.5:c.1023+1G>A | P | splice_donor_variant&intron_variant | 0.00002005 | TRPM1 | Calcium channel activity, G-protein-coupled glutamate receptor signaling pathway, visual perception, cellular response to light stimulus, axon | |
| OFT-00097 | NM_001040272.5:c.1819G>A:p.(Glu607Lys) | VUS | missense_variant | 0.0001279 | ADAMTSL1 | Hydrolase activity, extracellular matrix organization, extracellular region |
| NM_005183.3:c.4504C>T:p.(Arg1502*) | P | stop_gained | 0 | CACNA1F | Voltage-gated calcium channel activity, metal ion binding, visual perception, detection of light stimulus involved in visual perception, photoreceptor outer segment | |
| NM_001852.3:c.1652C>T:p.(Ala551Val) | VUS | missense_variant | 0.00005996 | COL9A2 | Extracellular matrix structural constituent conferring tensile strength, protein homodimerization activity, skeletal system development, extracellular region, endoplasmic reticulum lumen, collagen-containing extracellular matrix, collagen type IX trimer, extracellular space, extracellular matrix organization, | |
| NM_033225.5:c.1712A>G:p.(Asn571Ser) | VUS | missense_variant | 0.00001450 | CSMD1 | Male gonad development, female gonad development, oviduct epithelium development | |
| NM_006486.2:c.1157C>T:p.(Thr386Met) | VUS | missense_variant | 0.0001520 | FBLN1 | Collagen-containing extracellular matrix, fibronectin binding, integrin binding, extracellular matrix structural constituent, calcium ion binding, peptidase activator activity, fibrinogen binding, extracellular matrix organization, extracellular region, extracellular space, extracellular matrix, extracellular exosome | |
| NM_005529.6:c.12691G>A:p.(Glu4231Lys) | VUS | missense_variant | 0.00003196 | HSPG2 | Collagen-containing extracellular matrix, amyloid-beta binding, calcium ion binding, extracellular matrix structural constituent conferring compression resistance, collagen V binding, angiogenesis, response to hypoxia, positive regulation of endothelial cell proliferation, inflammatory response, negative regulation of angiogenesis, extracellular region, extracellular space, extracellular exosome | |
| OFT-00155 | NM_005183.3:c.2924G>A:p.(Arg975Gln) | LP | missense_variant | 0 | CACNA1F | Voltage-gated calcium channel activity, metal ion binding, visual perception, detection of light stimulus involved in visual perception, photoreceptor outer segment |
| NM_001853.3:c.1258C>G:p.(Gln420Glu) | VUS | missense_variant | 0.0002642 | COL9A3 | Extracellular matrix structural constituent conferring tensile strength, protein homodimerization activity, extracellular region, collagen-containing extracellular matrix, collagen type IX trimer, extracellular matrix organization, extracellular space | |
| NM_005529.6:c.4493C>T:p.(Ser1498Phe) | VUS | missense_variant | 0.00004378 | HSPG2 | Collagen-containing extracellular matrix, amyloid-beta binding, calcium ion binding, extracellular matrix structural constituent conferring compression resistance, collagen V binding, angiogenesis, response to hypoxia, positive regulation of endothelial cell proliferation, inflammatory response, negative regulation of angiogenesis, extracellular region, extracellular space, extracellular exosome | |
| NM_005559.3:c.3958A>G:p.(Ile1320Val) | VUS | missense_variant | 0.000003977 | LAMA1 | Collagen-containing extracellular matrix, extracellular matrix structural constituent, glycosphingolipid binding, morphogenesis of an epithelial sheet, neuron projection development, establishment of epithelial cell apical/basal polarity, retinal blood vessel morphogenesis, positive regulation of integrin-mediated signaling pathway, extracellular region, extracellular space, extracellular matrix, collagen-containing extracellular matrix, protein complex involved in cell–matrix adhesion, laminin-1 complex, laminin-3 complex | |
| NM_005560.4:c.1744C>T:p.(Pro582Ser) | VUS | missense_variant | 0.000004338 | LAMA5 | Collagen-containing extracellular matrix, integrin binding, extracellular matrix structural constituent, morphogenesis of a polarized epithelium, integrin-mediated signaling pathway, morphogenesis of embryonic epithelium, regulation of epithelial cell proliferation, postsynapse organization, extracellular region, synaptic cleft, collagen-containing extracellular matrix, extracellular exosome, extracellular matrix of synaptic cleft, laminin-5 complex, laminin-10 complex, laminin-11 complex | |
| NM_003246.3:c.1122C>T:p.(Pro374Pro) | VUS | splice_region_variant&synonymous_variant | 0.00006059 | THBS1 | Positive regulation of MAP kinase activity, negative regulation of endothelial cell proliferation, negative regulation of cell–matrix adhesion, negative regulation of angiogenesis, collagen-containing extracellular matrix, fibronectin binding, integrin binding, extracellular matrix structural constituent, calcium ion binding, fibroblast growth factor binding, laminin binding, proteoglycan binding, transforming growth factor beta binding, fibrinogen binding, collagen V binding, response to hypoxia, sprouting angiogenesis, chronic inflammatory response, apoptotic process, inflammatory response, positive regulation of fibroblast migration, negative regulation of fibroblast growth factor receptor signaling pathway, positive regulation of extrinsic apoptotic signaling pathway via death domain receptors, positive regulation of reactive oxygen species metabolic process, extracellular region, extracellular space, extracellular matrix, fibrinogen complex | |
| NM_003247.3:c.1019C>T:p.(Thr340Met) | VUS | missense_variant | 0.00008847 | THBS2 | Extracellular matrix structural constituent, calcium ion binding, protein binding, negative regulation of angiogenesis, positive regulation of synapse assembly, extracellular region, collagen-containing extracellular matrix | |
| OFT-00175 | NM_001204.6:c.1931A>G:p.(Asn644Ser) | VUS | missense_variant | 0 | BMPR2 | Positive regulation of gene expression, positive regulation of SMAD protein signal transduction, positive regulation of epithelial cell migration, proteoglycan biosynthetic process, positive regulation of transcription by RNA polymerase II, retina vasculature development in camera-type eye, cellular response to BMP stimulus, endothelial cell apoptotic process, extracellular space, nucleoplasm, plasma membrane, transforming growth factor beta receptor activity |
| NM_005529.6:c.10481G>A:p.(Arg3494Gln) | VUS | missense_variant | 0.00001199 | HSPG2 | Collagen-containing extracellular matrix, amyloid-beta binding, calcium ion binding, extracellular matrix structural constituent conferring compression resistance, collagen V binding, angiogenesis, response to hypoxia, positive regulation of endothelial cell proliferation, inflammatory response, negative regulation of angiogenesis, extracellular region, extracellular space, extracellular exosome | |
| NM_001142769.1:c.4396A>G:p.(Ser1466Gly) | VUS | missense_variant | 0.000004036 | PCDH15 | Sensory perception of sound, calcium ion binding, photoreceptor cell maintenance, sensory perception of light stimulus, photoreceptor outer segment, extracellular region | |
| NM_002420.5:c.4433C>T:p.(Thr1478Met) | VUS | missense_variant | 0.0005196 | TRPM1 | Calcium channel activity, G-protein-coupled glutamate receptor signaling pathway, visual perception, cellular response to light stimulus, axon | |
| OFT-00178 | NM_033225.5:c.8042G>A:p.(Gly2681Asp) | VUS | missense_variant | 0.0003290 | CSMD1 | Male gonad development, female gonad development, oviduct epithelium development |
| NM_033225.5:c.4375G>A:p.(Ala1459Thr) | VUS | missense_variant | 0.00006418 | CSMD1 | Male gonad development, female gonad development, oviduct epithelium development | |
| NM_001105207.2:c.673G>A:p.(Ala225Thr) | VUS | missense_variant | 0.00001994 | LAMA4 | Collagen-containing extracellular matrix, extracellular matrix structural constituent, extracellular region | |
| NM_004525.2:c.10202C>G:p.(Thr3401Arg) | VUS | missense_variant | 0.00004599 | LRP2 | Negative regulation of apoptotic process, vitamin D metabolic process, phosphatidylinositol 3-kinase/protein kinase B signal transduction, cellular response to growth factor stimulus, neuron projection arborization, extracellular exosome | |
| NM_005921.1:c.299G>A:p.(Gly100Glu) | VUS | missense_variant | 0 | MAP3K1 | Protein serine/threonine kinase activity, MAP kinase activity, MAPK cascade, protein phosphorylation, Fc-epsilon receptor signaling pathway | |
| NM_005921.1:c.3646_3648delATC:p.(Ile1216del) | VUS | conservative_inframe_deletion | 0.000008190 | MAP3K1 | Protein serine/threonine kinase activity, MAP kinase activity, MAPK cascade, protein phosphorylation, Fc-epsilon receptor signaling pathway | |
| NM_001142769.1:c.1519G>A:p.(Val507Ile) | VUS | missense_variant | 0.00009902 | PCDH15 | Sensory perception of sound, calcium ion binding, photoreceptor cell maintenance, sensory perception of light stimulus, photoreceptor outer segment, extracellular region | |
| NM_000301.3:c.598A>G:p.(Thr200Ala) | VUS | missense_variant | 0.0007181 | PLG | Endopeptidase activity, serine-type endopeptidase activity, extracellular matrix disassembly, modulating synaptic transmission, negative regulation of cell–cell adhesion mediated by cadherin, extracellular region, extracellular space, collagen-containing extracellular matrix, extracellular exosome | |
| NM_014909.4:c.953G>A:p.(Arg318Gln) | VUS | missense_variant | 0 | VASH1 | Angiogenesis, negative regulation of endothelial cell proliferation, negative regulation of angiogenesis, extracellular space | |
| OFT-00191 | NM_001854.3:c.2900G>T:p.(Gly967Val) | LP | missense_variant | 0 | COL11A1 | Extracellular matrix structural constituent, extracellular matrix structural constituent conferring tensile strength, extracellular matrix binding, proteoglycan metabolic process, visual perception, extracellular matrix organization, collagen fibril organization, collagen type XI trimer, endoplasmic reticulum lumen, collagen-containing extracellular matrix, collagen type XI trime |
| NM_014907.2:c.2469C>A:p.(Ser823Arg) | VUS | missense_variant | 0.0002050 | FRMPD1 | Regulation of G-protein-coupled receptor signaling pathway | |
| NM_016831.2:c.3502A>G:p.(Thr1168Ala) | VUS | missense_variant | 0.0005746 | PER3 | Negative regulation of transcription by RNA polymerase II, regulation of circadian sleep/wake cycle, sleep, protein stabilization, circadian regulation of gene expression, transcription cis-regulatory region binding | |
| OFT-00209 | NM_001851.4:c.6G>T:p.(Lys2Asn) | VUS | missense_variant | 0.00004374 | COL9A1 | Extracellular matrix structural constituent conferring tensile strength, extracellular region, collagen-containing extracellular matrix, collagen type IX trimer, extracellular matrix organization, extracellular space |
| NM_000404.3:c.1498A>G:p.(Thr500Ala) | P | missense_variant | 0.00001216 | GLB1 | Beta-galactosidase activity, glycosphingolipid metabolic process, galactose catabolic process, heparan sulfate proteoglycan catabolic process | |
| NM_001080424.1:c.3221C>G:p.(Ala1074Gly) | VUS | missense_variant | 0 | KDM6B | Beta-catenin binding, histone demethylase activity, metal ion binding, histone h3k27me2/h3k27me3 demethylase activity, inflammatory response to antigenic stimulus, chromatin remodeling, positive regulation of transcription by RNA polymerase II, regulation of gene expression | |
| OFT-00217 | NM_198576.3:c.4799C>T:p.(Ala1600Val) | VUS | missense_variant | 0.00005405 | AGRN | Collagen-containing extracellular matrix, extracellular matrix structural constituent, laminin binding, heparan sulfate proteoglycan binding, clustering of voltage-gated sodium channels, positive regulation of transcription by RNA polymerase II, extracellular region, collagen-containing extracellular matrix, extracellular exosome, neuromuscular junction development |
| NM_014461.3:c.260A>G:p.(Asn87Ser) | VUS | missense_variant | 0.00002830 | CNTN6 | Notch signaling pathway, central nervous system development, axon guidance | |
| NM_014461.3:c.2553G>C:p.(Met851Ile) | VUS | missense_variant | 0.00001194 | CNTN6 | Notch signaling pathway, central nervous system development, axon guidance | |
| NM_015123.2:c.554T>C:p.(Leu185Ser) | VUS | missense_variant | 0.00005346 | FRMD4B | Establishment of epithelial cell polarity, ruffle, extracellular space | |
| NM_000843.3:c.3G>T:p.(Met1?) | LP | start_lost | 0.00003368 | GRM6 | Glutamate receptor activity detection of visible light, detection of light stimulus involved in visual perception, retina development in camera-type eye, positive regulation of calcium ion import across plasma membrane | |
| NM_002332.2:c.1415G>A:p.(Arg472Gln) | VUS | missense_variant&splice_region_variant | 0.0001351 | LRP1 | Negative regulation of metallopeptidase activity, RNA binding, heparan sulfate proteoglycan binding, negative regulation of gene expression, regulation of extracellular matrix disassembly, negative regulation of Wnt signaling pathway | |
| OFT-00223 | NM_017758.3:c.952C>A:p.(Pro318Thr) | VUS | missense_variant | 0.00001068 | ALKBH5 | mRNA N6-methyladenosine dioxygenase activity, response to hypoxia, mRNA processing, mRNA export from nucleus, regulation of translation, oxidative single-stranded RNA demethylation, mRNA destabilization, oxidative RNA demethylase activity |
| NM_206933.2:c.15172T>C:p.(Phe5058Leu) | VUS | missense_variant | 0.00003185 | USH2A | Photoreceptor inner segment, photoreceptor connecting cilium, neuronal cell body, collagen binding, visual perception, photoreceptor cell maintenance, sensory perception of light stimulus, extracellular region, USH2 complex | |
| OFT-00253 | NM_025114.3:c.6791A>G:p.(Lys2264Arg) | VUS | missense_variant | 0.00006424 | CEP290 | Protein transport, eye photoreceptor cell development, positive regulation of DNA-templated transcription, extracellular region, photoreceptor connecting cilium, camera-type eye development, non-motile cilium assembly |
| NM_013291.2:c.3128C>T:p.(Pro1043Leu) | VUS | missense_variant | 0.00001200 | CPSF1 | mRNA 3’-UTR AU-rich region binding, mRNA polyadenylation, co-transcriptional RNA 3’-end processing, cleavage and polyadenylation pathway, mRNA cleavage and polyadenylation specificity factor complex, nucleus, mRNA polyadenylation | |
| NM_005529.6:c.3346G>A:p.(Gly1116Ser) | VUS | missense_variant | 0.00005501 | HSPG2 | Collagen-containing extracellular matrix, amyloid-beta binding, calcium ion binding, extracellular matrix structural constituent conferring compression resistance, collagen V binding, angiogenesis, response to hypoxia, positive regulation of endothelial cell proliferation, inflammatory response, negative regulation of angiogenesis, extracellular region, extracellular space, extracellular exosome | |
| NM_003803.3:c.4580G>T:p.(Gly1527Val) | VUS | missense_variant | 0.000008027 | MYOM1 | Structural constituent of muscle, extraocular skeletal muscle development, positive regulation of gene expression, positive regulation of protein secretion, striated muscle myosin thick filament, M band | |
| NM_003803.3:c.3032T>C:p.(Val1011Ala) | VUS | missense_variant | 0 | MYOM1 | Structural constituent of muscle, extraocular skeletal muscle development, positive regulation of gene expression, positive regulation of protein secretion, striated muscle myosin thick filament, M band | |
| NM_033282.3:c.1411_1412insT:p.(Ser473fs) | VUS | frameshift_variant | 0.0009697 | OPN4 | 11-cis retinal binding, G-protein-coupled photoreceptor activity, visual perception, phototransduction, optokinetic behavior, detection of visible light, regulation of circadian rhythm, retina development in camera-type eye, photoreceptor disc membrane, phototransduction, cellular response to light stimulus | |
| NM_003247.3:c.799G>A:p.(Glu267Lys) | VUS | missense_variant | 0.0002277 | THBS2 | Extracellular matrix structural constituent, calcium ion binding, protein binding, negative regulation of angiogenesis, positive regulation of synapse assembly, extracellular region, collagen-containing extracellular matrix | |
| OFT-00268 | NM_033225.5:c.9089C>G:p.(Pro3030Arg) | VUS | missense_variant | 0 | CSMD1 | Male gonad development, female gonad development, oviduct epithelium development |
| NM_033225.5:c.7050T>A:p.(Ser2350Arg) | VUS | missense_variant | 0.00001019 | CSMD1 | Male gonad development, female gonad development, oviduct epithelium development | |
| OFT-00332 | NM_173728.3:c.380C>G:p.(Pro127Arg) | VUS | missense_variant | 0.00001197 | ARHGEF15 | Guanyl-nucleotide exchange factor activity, GTPase activator activity, retina vasculature morphogenesis in camera-type eye, regulation of postsynapse assembly, negative regulation of synapse maturation, dendrite, postsynapse, glutamatergic synapse |
| NM_000186.3:c.481G>T:p.(Ala161Ser) | VUS | missense_variant | 0.00009160 | CFH | Heparan sulfate proteoglycan binding, complement activation, extracellular region, extracellular space, extracellular exosome | |
| NM_013291.2:c.2383G>A:p.(Glu795Lys) | VUS | missense_variant&splice_region_variant | 0 | CPSF1 | mRNA 3’-UTR AU-rich region binding, mRNA polyadenylation, co-transcriptional RNA 3’-end processing, cleavage and polyadenylation pathway, mRNA cleavage and polyadenylation specificity factor complex, nucleus, mRNA polyadenylation | |
| NM_005529.6:c.4078A>G:p.(Asn1360Asp) | VUS | missense_variant | 0.0001388 | HSPG2 | Collagen-containing extracellular matrix, amyloid-beta binding, calcium ion binding, extracellular matrix structural constituent conferring compression resistance, collagen V binding, angiogenesis, response to hypoxia, positive regulation of endothelial cell proliferation, inflammatory response, negative regulation of angiogenesis, extracellular region, extracellular space, extracellular exosome | |
| NM_002332.2:c.11930C>T:p.(Ser3977Leu) | VUS | missense_variant | 0.0003749 | LRP1 | Negative regulation of metallopeptidase activity, RNA binding, heparan sulfate proteoglycan binding, negative regulation of gene expression, regulation of extracellular matrix disassembly, negative regulation of Wnt signaling pathway | |
| NM_201269.2:c.1366A>T:p.(Thr456Ser) | VUS | missense_variant | 0 | ZNF644 | RNA polymerase II cis-regulatory region sequence-specific DNA binding, DNA-binding transcription factor activity, RNA polymerase II-specific, transcription corepressor binding, regulation of transcription by RNA polymerase II, nucleus, DNA-binding transcription factor activity | |
| OFT-00403 | NM_006292.3:c.942C>T:p.(Ile314Ile) | VUS | synonymous_variant | 0.0001450 | TSG101 | DNA binding, transcription corepressor activity, ubiquitin protein ligase binding, negative regulation of transcription by RNA polymerase II, regulation of cell growth, extracellular transport, negative regulation of epidermal-growth-factor-activated receptor activity, negative regulation of cell population proliferation |
| OFT-00429 | NM_013281.3:c.325T>G:p.(Leu109Val) | VUS | missense_variant | 0.0001205 | FLRT3 | Fibroblast growth factor receptor binding, axon guidance, synapse assembly, fibroblast growth factor receptor signaling pathway, neuron projection development, extracellular matrix, extracellular space |
| NM_005529.6:c.742_744delCTT:p.(Leu248del) | VUS | conservative_inframe_deletion | 0 | HSPG2 | Collagen-containing extracellular matrix, amyloid-beta binding, calcium ion binding, extracellular matrix structural constituent conferring compression resistance, collagen V binding, angiogenesis, response to hypoxia, positive regulation of endothelial cell proliferation, inflammatory response, negative regulation of angiogenesis, extracellular region, extracellular space, extracellular exosome | |
| NM_005529.6:c.738delT:p.(Leu247fs) | LP | frameshift_variant | 0 | HSPG2 | Collagen-containing extracellular matrix, amyloid-beta binding, calcium ion binding, extracellular matrix structural constituent conferring compression resistance, collagen V binding, angiogenesis, response to hypoxia, positive regulation of endothelial cell proliferation, inflammatory response, negative regulation of angiogenesis, extracellular region, extracellular space, extracellular exosome | |
| NM_002337.3:c.298G>A:p.(Gly100Ser) | VUS | missense_variant | 0.0003393 | LRPAP1 | Amyloid-beta binding, signaling receptor binding, endoplasmic reticulum-Golgi intermediate compartment, endosome | |
| NM_004994.2:c.1270C>A:p.(Arg424Ser) | VUS | missense_variant | 0 | MMP9 | Metalloendopeptidase activity, serine-type endopeptidase activity, extracellular matrix disassembly, collagen catabolic process, cellular response to reactive oxygen species, negative regulation of intrinsic apoptotic signaling pathway, negative regulation of cation channel activity, collagen-containing extracellular matrix, extracellular exosome, extracellular matrix organization, collagen catabolic process | |
| OFT-00474 | NM_014907.2:c.925G>A:p.(Ala309Thr) | VUS | missense_variant | 0.0007006 | FRMPD1 | Regulation of G-protein-coupled receptor signaling pathway |
| NM_005559.3:c.781A>G:p.(Ile261Val) | VUS | missense_variant | 0.00009679 | LAMA1 | Collagen-containing extracellular matrix, extracellular matrix structural constituent, glycosphingolipid binding, morphogenesis of an epithelial sheet, neuron projection development, establishment of epithelial cell apical/basal polarity, retinal blood vessel morphogenesis, positive regulation of integrin-mediated signaling pathway, extracellular region, extracellular space, extracellular matrix, collagen-containing extracellular matrix, protein complex involved in cell–matrix adhesion, laminin-1 complex, laminin-3 complex | |
| NM_152683.3:c.1380T>C:p.(Cys460Cys) | VUS | splice_region_variant&synonymous_variant | 0.0008670 | PRIMPOL | Chromatin binding, DNA-directed DNA polymerase activity, DNA primase activity, mitochondrial DNA replication, synthesis of RNA primer, replication fork processing | |
| OFT-00477 | NM_006292.3:c.942C>T:p.(Ile314Ile) | VUS | synonymous_variant | 0.0001450 | TSG101 | DNA binding, transcription corepressor activity, ubiquitin protein ligase binding, negative regulation of transcription by RNA polymerase II, regulation of cell growth, extracellular transport, negative regulation of epidermal-growth-factor-activated receptor activity, negative regulation of cell population proliferation |
| OFT-00506 | NM_001853.3:c.1511C>T:p.(Pro504Leu) | VUS | missense_variant | 0.00006417 | COL9A3 | Extracellular matrix structural constituent conferring tensile strength, protein homodimerization activity, extracellular region, collagen-containing extracellular matrix, collagen type IX trimer, extracellular matrix organization, extracellular space |
| OFT-00546 | NM_000350.2:c.6148G>C:p.(Val2050Leu) | VUS | missense_variant&splice_region_variant | 0.002857 | ABCA4 | Retinoid binding, 11-cis retinal binding, all-trans retinal binding, retinol transmembrane transporter activity, ABC-type transporter activity, visual perception, phototransduction, visible light, retinol transport, retinal metabolic process, photoreceptor cell maintenance, photoreceptor outer segment, photoreceptor disc membrane, rod photoreceptor disc membrane, ATPase-coupled intramembrane lipid transporter activity |
| NM_198576.3:c.2737G>A:p.(Val913Met) | VUS | missense_variant | 0.0001489 | AGRN | Collagen-containing extracellular matrix, extracellular matrix structural constituent, laminin binding, heparan sulfate proteoglycan binding, clustering of voltage-gated sodium channels, positive regulation of transcription by RNA polymerase II, extracellular region, collagen-containing extracellular matrix, extracellular exosome, neuromuscular junction development | |
| NM_013281.3:c.1135G>A:p.(Gly379Arg) | VUS | missense_variant | 0.00001594 | FLRT3 | Fibroblast growth factor receptor binding, axon guidance, synapse assembly, fibroblast growth factor receptor signaling pathway, neuron projection development, extracellular matrix, extracellular space | |
| NM_000426.3:c.6880G>T:p.(Val2294Leu) | VUS | missense_variant | 0 | LAMA2 | Maintenance of blood–brain barrier, structural molecule activity, extracellular matrix structural constituent, axon guidance, positive regulation of integrin-mediated signaling pathway, extracellular region, collagen-containing extracellular matrix, protein complex involved in cell-matrix adhesion | |
| NM_005560.4:c.11063G>A:p.(Gly3688Glu) | VUS | missense_variant | 0.000004051 | LAMA5 | Collagen-containing extracellular matrix, integrin binding, extracellular matrix structural constituent, morphogenesis of a polarized epithelium, integrin-mediated signaling pathway, morphogenesis of embryonic epithelium, regulation of epithelial cell proliferation, postsynapse organization, extracellular region, synaptic cleft, collagen-containing extracellular matrix, extracellular exosome, extracellular matrix of synaptic cleft, laminin-5 complex, laminin-10 complex, laminin-11 complex | |
| NM_000428.2:c.3998G>C:p.(Gly1333Ala) | VUS | missense_variant | 0 | LTBP2 | Supramolecular fiber organization, collagen-containing extracellular matrix, extracellular matrix structural constituent, calcium ion binding, growth factor binding, transforming growth factor beta receptor signaling pathway, protein secretion, extracellular region, extracellular space, extracellular matrix, collagen-containing extracellular matrix, extracellular exosome, supramolecular fiber organization | |
| OFT-00586 | NM_001080512.2:c.1425G>C:p.(Leu475Phe) | VUS | missense_variant | 0.00003186 | BICC1 | RNA binding, determination of left/right symmetry, negative regulation of canonical Wnt signaling pathway |
| NM_001206955.1:c.2128G>A:p.(Gly710Arg) | VUS | missense_variant | 0.00004244 | CNTN4 | Neuron cell–cell adhesion, nervous system development, axonogenesis, axon guidance, axonal fasciculation, neuron projection development, negative regulation of neuron differentiation, regulation of synaptic plasticity, extracellular region, plasma membrane, axon | |
| NM_000428.2:c.1487G>A:p.(Gly496Asp) | VUS | missense_variant | 0.0003103 | LTBP2 | Supramolecular fiber organization, collagen-containing extracellular matrix, extracellular matrix structural constituent, calcium ion binding, growth factor binding, transforming growth factor beta receptor signaling pathway, protein secretion, extracellular region, extracellular space, extracellular matrix, collagen-containing extracellular matrix, extracellular exosome, supramolecular fiber organization | |
| NM_006292.3:c.307G>A:p.(Val103Ile) | VUS | missense_variant | 0.00001916 | TSG101 | DNA binding, transcription corepressor activity, ubiquitin protein ligase binding, negative regulation of transcription by RNA polymerase II, regulation of cell growth, extracellular transport, negative regulation of epidermal-growth-factor-activated receptor activity, negative regulation of cell population proliferation | |
| OFT-00601 | NM_033225.5:c.5600C>G:p.(Pro1867Arg) | VUS | missense_variant | 0 | CSMD1 | Male gonad development, female gonad development, oviduct epithelium development |
| OFT-00710 | NM_000138.4:c.8195G>C:p.(Ser2732Thr) | VUS | missense_variant | 0 | FBN1 | Collagen-containing extracellular matrix, integrin binding, extracellular matrix structural constituent, extracellular matrix constituent conferring elasticity, sequestering of BMP in extracellular matrix, sequestering of TGFbeta in extracellular matrix, camera-type eye development, embryonic eye morphogenesis, post-embryonic eye morphogenesis, cellular response to transforming growth factor beta stimulus, anatomical structure morphogenesis |
| NM_000428.2:c.2512G>A:p.(Val838Met) | VUS | missense_variant | 0.00002578 | LTBP2 | Supramolecular fiber organization, collagen-containing extracellular matrix, extracellular matrix structural constituent, calcium ion binding, growth factor binding, transforming growth factor beta receptor signaling pathway, protein secretion, extracellular region, extracellular space, extracellular matrix, collagen-containing extracellular matrix, extracellular exosome, supramolecular fiber organization | |
| NM_206933.2:c.9776T>C:p.(Val3259Ala) | VUS | missense_variant | 0 | USH2A | Photoreceptor inner segment, photoreceptor connecting cilium, neuronal cell body, collagen binding, visual perception, photoreceptor cell maintenance, sensory perception of light stimulus, extracellular region, USH2 complex |
Variants found in each family with its classification, functional annotation, frequencies in gnomAD global population and main pathways related to each gene affected.
References
- Kloss, B.A.; Tompson, S.W.; Whisenhunt, K.N.; Quow, K.L.; Huang, S.J.; Pavelec, D.M.; Rosenberg, T.; Young, T.L. Exome Sequence Analysis of 14 Families with High Myopia. Investig. Opthalmol. Vis. Sci. 2017, 58, 1982–1990. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Summers, J.A.; Schaeffel, F.; Marcos, S.; Wu, H.; Tkatchenko, A.V. Functional integration of eye tissues and refractive eye development: Mechanisms and pathways. Exp. Eye Res. 2021, 209, 108693. [Google Scholar] [CrossRef] [PubMed]
- González-Iglesias, E.; López-Vázquez, A.; Noval, S.; Nieves-Moreno, M.; Granados-Fernández, M.; Arruti, N.; Rosa-Pérez, I.; Pacio-Míguez, M.; Montaño, V.E.F.; Rodríguez-Solana, P.; et al. Next-Generation Sequencing Screening of 43 Families with Non-Syndromic Early-Onset High Myopia: A Clinical and Genetic Study. Int. J. Mol. Sci. 2022, 23, 4233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-M.; Lu, S.-Y.; Zhang, X.-J.; Chen, L.-J.; Pang, C.-P.; Yam, J.C. Myopia Genetics and Heredity. Children 2022, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Morgan, I.G.; Ohno-Matsui, K.; Saw, S.-M. Myopia. Lancet 2012, 379, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Holden, B.A.; Fricke, T.R.; Wilson, D.A.; Jong, M.; Naidoo, K.S.; Sankaridurg, P.; Wong, T.Y.; Naduvilath, T.; Resnikoff, S. Global Prevalence of Myopia and High Myopia and Temporal Trends from 2000 through 2050. Ophthalmology 2016, 123, 1036–1042. [Google Scholar] [CrossRef]
- Tedja, M.S.; Haarman, A.E.G.; Meester-Smoor, M.A.; Kaprio, J.; Mackey, D.A.; Guggenheim, J.A.; Hammond, C.J.; Verhoeven, V.J.M.; Klaver, C.C.W.; for the CREAM Consortium. IMI–Myopia Genetics Report. Investig. Opthalmol. Vis. Sci. 2019, 60, M89–M105. [Google Scholar] [CrossRef]
- Mojarrad, N.G.; Plotnikov, D.; Williams, C.; Guggenheim, J.A.; for the UK Biobank Eye and Vision Consortium. Association Between Polygenic Risk Score and Risk of Myopia. JAMA Ophthalmol. 2020, 138, 7–13. [Google Scholar] [CrossRef]
- OMIM—Online Mendelian Inheritance in Man. Available online: https://www.omim.org/ (accessed on 31 January 2022).
- Utz, V.M.; Pfeifer, W.; Longmuir, S.Q.; Olson, R.J.; Wang, K.; Drack, A.V. Presentation of TRPM1-Associated Congenital Stationary Night Blindness in Children. JAMA Ophthalmol. 2018, 136, 389–398. [Google Scholar] [CrossRef]
- Zhou, L.; Li, T.; Xing, Y.-Q.; Li, Y.; Wu, Q.-S.; Zhang, M.-J. Novel TRPM1 mutations in two Chinese families with early-onset high myopia, with or without complete congenital stationary night blindness. Int. J. Ophthalmol. 2016, 9, 1396–1402. [Google Scholar] [CrossRef]
- Al-Hujaili, H.; Taskintuna, I.; Neuhaus, C.; Bergmann, C.; Schatz, P. Long-term follow-up of retinal function and structure in TRPM1-associated complete congenital stationary night blindness. Mol. Vis. 2019, 25, 851–858. [Google Scholar] [PubMed]
- Al Oreany, A.A.; Al Hadlaq, A.; Schatz, P. Congenital stationary night blindness with hypoplastic discs, negative electroretinogram and thinning of the inner nuclear layer. Graefe’s Arch. Clin. Exp. Ophthalmol. 2016, 254, 1951–1956. [Google Scholar] [CrossRef]
- The Gene Ontology Home Page. Available online: http://geneontology.org/ (accessed on 1 August 2012).
- GeneCards Human Gene Database. GeneCards—Human Genes. Available online: https://www.genecards.org/ (accessed on 2 February 2023).
- Loosli, F. ArhGEF18 regulated Rho signaling in vertebrate retina development. Small GTPases 2013, 4, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Iwagawa, T.; Honda, H.; Watanabe, S. Jmjd3 Plays Pivotal Roles in the Proper Development of Early-Born Retinal Lineages: Amacrine, Horizontal, and Retinal Ganglion Cells. Investig. Opthalmol. Vis. Sci. 2020, 61, 43. [Google Scholar] [CrossRef] [PubMed]
- Basit, S.; Albalawi, A.M.; Alharby, E.; Khoshhal, K.I. Exome sequencing identified rare variants in genes HSPG2 and ATP2B4 in a family segregating developmental dysplasia of the hip. BMC Med. Genet. 2017, 18, 34. [Google Scholar] [CrossRef]
- Wan, L.; Deng, B.; Wu, Z.; Chen, X. Exome sequencing study of 20 patients with high myopia. PeerJ 2018, 6, e5552. [Google Scholar] [CrossRef] [PubMed]
- Kjellström, U.; Martell, S.; Brobeck, C.; Andréasson, S. Autosomal recessive Stickler syndrome associated with homozygous mutations in the COL9A2 gene. Ophthalmic Genet. 2020, 42, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; McFadden, S.A.; Morgan, I.; Cui, D.; Hu, J.; Wan, W.; Zeng, J. All-trans retinoic acid regulates the expression of the extracellular matrix protein fibulin-1 in the guinea pig sclera and human scleral fibroblasts. Mol. Vis. 2010, 16, 689–697. [Google Scholar]
- Sun, W.; Huang, L.; Xu, Y.; Xiao, X.; Li, S.; Jia, X.; Gao, B.; Wang, P.; Guo, X.; Zhang, Q. Exome Sequencing on 298 Probands With Early-Onset High Myopia: Approximately One-Fourth Show Potential Pathogenic Mutations in RetNet Genes. Investig. Opthalmol. Vis. Sci. 2015, 56, 8365–8372. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.-J.; Piao, S.-Y.; Shen, R.-J.; Ma, Y.; Xue, Z.-Q.; Zhang, W.; Liu, J.; Jin, Z.-B.; Zhuang, W.-J. Whole-Exome Sequencing in a Cohort of High Myopia Patients in Northwest China. Front. Cell Dev. Biol. 2021, 9, 645501. [Google Scholar] [CrossRef]
- Yang, E.; Yu, J.; Liu, X.; Chu, H.; Li, L. Familial Whole Exome Sequencing Study of 30 Families with Early-Onset High Myopia. Investig. Opthalmol. Vis. Sci. 2023, 64, 10. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Zhang, F.J.; Zhu, S.Q.; Duan, H.; Li, Y.; Zhou, Z.J.; Ma, W.X.; Wang, N.L. The association of a single nucleotide polymorphism in the promoter region of the LAMA1 gene with susceptibility to Chinese high myopia. Mol. Vis. 2011, 17, 1003–1010. [Google Scholar] [PubMed]
- Sasaki, S.; Ota, M.; Meguro, A.; Nishizaki, R.; Okada, E.; Mok, J.; Kimura, T.; Oka, A.; Katsuyama, Y.; Ohno, S.; et al. A single nucleotide polymorphism analysis of the LAMA1 gene in Japanese patients with high myopia. Clin. Ophthalmol. 2007, 1, 289–295. [Google Scholar] [PubMed]
- Liang, Y.; Song, Y.; Zhang, F.; Sun, M.; Wang, N. Effect of a Single Nucleotide Polymorphism in the LAMA1 Promoter Region on Transcriptional Activity: Implication for Pathological Myopia. Curr. Eye Res. 2016, 41, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Frost, M.R.; Siegwart, J.T.; Norton, T.T. Patterns of mRNA and protein expression during minus-lens compensation and recovery in tree shrew sclera. Mol. Vis. 2011, 17, 903–919. [Google Scholar] [PubMed]
- Chen, C.-W.; Yao, J.-Y. RNA sequence analysis identified bone morphogenetic protein-2 (BMP2) as a biomarker underlying form deprivation myopia. Biochem. Biophys. Rep. 2022, 30, 101261. [Google Scholar] [CrossRef] [PubMed]
- Cases, O.; Joseph, A.; Obry, A.; Santin, M.D.; Ben-Yacoub, S.; Pâques, M.; Amsellem-Levera, S.; Bribian, A.; Simonutti, M.; Augustin, S.; et al. Foxg1-Cre Mediated Lrp2 Inactivation in the Developing Mouse Neural Retina, Ciliary and Retinal Pigment Epithelia Models Congenital High Myopia. PLoS ONE 2015, 10, e0129518. [Google Scholar] [CrossRef]
- Mongan, M.; Wang, J.; Liu, H.; Fan, Y.; Jin, C.; Kao, W.Y.-W.; Xia, Y. Loss of MAP3K1 enhances proliferation and apoptosis during retinal development. Development 2011, 138, 4001–4012. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kimura, E.; Mongan, M.; Xia, Y. Genetic Control of MAP3K1 in Eye Development and Sex Differentiation. Cells 2021, 11, 34. [Google Scholar] [CrossRef]
- Ren, Y.; Yang, X.; Luo, Z.; Wu, J.; Lin, H. HIF-1α aggravates pathologic myopia through the miR-150-5p/LAMA4/p38 MAPK signaling axis. Mol. Cell. Biochem. 2022, 477, 1065–1074. [Google Scholar] [CrossRef]
- Wen, K.; Shao, X.; Li, Y.; Li, Y.; Li, Y.; Wang, Q.; Su, R.; Zhang, L.; Cai, Y.; Sun, J.; et al. The plasminogen protein is associated with high myopia as revealed by the iTRAQ-based proteomic analysis of the aqueous humor. Sci. Rep. 2021, 11, 8789. [Google Scholar] [CrossRef] [PubMed]
- Onami, H.; Nagai, N.; Kaji, H.; Nishizawa, M.; Sato, Y.; Osumi, N.; Nakazawa, T.; Abe, T. Transscleral Sustained Vasohibin-1 Delivery by a Novel Device Suppressed Experimentally-Induced Choroidal Neovascularization. PLoS ONE 2013, 8, e58580. [Google Scholar] [CrossRef] [PubMed]
- Musolf, A.M.; Haarman, A.E.G.; Luben, R.N.; Ong, J.-S.; Patasova, K.; Trapero, R.H.; Marsh, J.; Jain, I.; Jain, R.; Wang, P.Z.; et al. Rare variant analyses across multiethnic cohorts identify novel genes for refractive error. Commun. Biol. 2023, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Haarman, A.E.G.; Thiadens, A.A.H.J.; van Tienhoven, M.; E Loudon, S.; Klein, J.E.M.M.A.d.; Brosens, E.; Polling, J.R.; van der Schoot, V.; Bouman, A.; A Kievit, A.J.; et al. Whole exome sequencing of known eye genes reveals genetic causes for high myopia. Hum. Mol. Genet. 2022, 31, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- Campla, C.K.; Bocchero, U.; Strickland, R.; Nellissery, J.; Advani, J.; Ignatova, I.; Srivastava, D.; Aponte, A.M.; Wang, Y.; Gumerson, J.; et al. Frmpd1 Facilitates Trafficking of G-Protein Transducin and Modulates Synaptic Function in Rod Photoreceptors of Mammalian Retina. eNeuro 2022, 9, ENEURO.0348-22.2022. [Google Scholar] [CrossRef] [PubMed]
- Skeie, J.M.; Hernandez, J.; Hinek, A.; Mullins, R.F. Molecular responses of choroidal endothelial cells to elastin derived peptides through the elastin-binding protein (GLB1). Matrix Biol. 2012, 31, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-H.; Cai, X.-B.; Xia, L.-Q.; Zhou, F.-Y.; Wen, X.-R.; Chen, D.-F.; Han, F.; Zhou, K.-J.; Jin, Z.-B.; Zhuang, W.-J.; et al. Mutational screening of AGRN, SLC39A5, SCO2, P4HA2, BSG, ZNF644, and CPSF1 in a Chinese cohort of 103 patients with nonsyndromic high myopia. Mol. Vis. 2021, 27, 706–717. [Google Scholar] [PubMed]
- Wang, H.; Su, S.; Yang, M.; Hu, N.; Yao, Y.; Zhu, R.; Zhou, J.; Liang, C.; Guan, H. Association of ZNF644, GRM6, and CTNND2 genes with high myopia in the Han Chinese population: Jiangsu Eye Study. Eye 2016, 30, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Meguro, A.; Yamane, T.; Takeuchi, M.; Miyake, M.; Fan, Q.; Zhao, W.; Wang, I.-J.; Mizuki, Y.; Yamada, N.; Nomura, N.; et al. Genome-Wide Association Study in Asians Identifies Novel Loci for High Myopia and Highlights a Nervous System Role in Its Pathogenesis. Ophthalmology 2020, 127, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Aldahmesh, M.A.; Khan, A.O.; Alkuraya, H.; Adly, N.; Anazi, S.; Al-Saleh, A.A.; Mohamed, J.Y.; Hijazi, H.; Prabakaran, S.; Tacke, M.; et al. Mutations in LRPAP1 Are Associated with Severe Myopia in Humans. Am. J. Hum. Genet. 2013, 93, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.; Zhang, Y.; Li, Y.; Wang, Q.; Sun, J. Comprehensive analysis of transcriptome-wide m6A methylome in the anterior capsule of the lens of high myopia patients. Epigenetics 2020, 16, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Ma, Y.; Qin, Y.-X.; Cai, B.; Ma, L.-M.; Ma, Z.; Liu, Y.; Jin, Z.-B.; Zhuang, W.-J. Mutational investigation of 17 causative genes in a cohort of 113 families with nonsyndromic early-onset high myopia in northwestern China. Mol. Genet. Genom. 2023, 298, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Sun, W.; Xiao, X.; Li, S.; Jia, X.; Zhou, L.; Wang, P.; Zhang, Q. CPSF1 mutations are associated with early-onset high myopia and involved in retinal ganglion cell axon projection. Hum. Mol. Genet. 2019, 28, 1959–1970. [Google Scholar] [CrossRef]
- Chakraborty, R.; Landis, E.G.; Mazade, R.; Yang, V.; Strickland, R.; Hattar, S.; Stone, R.A.; Iuvone, P.M.; Pardue, M.T. Melanopsin modulates refractive development and myopia. Exp. Eye Res. 2021, 214, 108866. [Google Scholar] [CrossRef]
- Rasool, S.; Dar, R.; Khan, M.S.; Ayoub, S.G.; Rashid, S.; Rehman, M.U.; Jan, T.; Qureshi, M.A.; Andrabi, K.I. MYP2 locus genes: Sequence variations, genetic association studies and haplotypic association in patients with High Myopia. Int. J. Biochem. Mol. Biol. 2021, 12, 35–48. [Google Scholar] [PubMed]
- Xiang, X.; Wang, T.; Tong, P.; Li, Y.; Guo, H.; Wan, A.; Xia, L.; Liu, Y.; Li, Y.; Tian, Q.; et al. New ZNF644 mutations identified in patients with high myopia. Mol. Vis. 2014, 20, 939–946. [Google Scholar]
- Shi, Y.; Li, Y.; Zhang, D.; Zhang, H.; Li, Y.; Lu, F.; Liu, X.; He, F.; Gong, B.; Cai, L.; et al. Exome Sequencing Identifies ZNF644 Mutations in High Myopia. PLOS Genet. 2011, 7, e1002084. [Google Scholar] [CrossRef] [PubMed]
- Kusuhara, S.; Fukushima, Y.; Fukuhara, S.; Jakt, L.M.; Okada, M.; Shimizu, Y.; Hata, M.; Nishida, K.; Negi, A.; Hirashima, M.; et al. Arhgef15 Promotes Retinal Angiogenesis by Mediating VEGF-Induced Cdc42 Activation and Potentiating RhoJ Inactivation in Endothelial Cells. PLoS ONE 2012, 7, e45858. [Google Scholar] [CrossRef]
- García-Gen, E.; Penadés, M.; Mérida, S.; Desco, C.; Araujo-Miranda, R.; Navea, A.; Bosch-Morell, F. High Myopia and the Complement System: Factor H in Myopic Maculopathy. J. Clin. Med. 2021, 10, 2600. [Google Scholar] [CrossRef] [PubMed]
- Le, D.; Lim, S.; Min, K.W.; Park, J.W.; Kim, Y.; Ha, T.; Moon, K.H.; Wagner, K.-U.; Kim, J.W. Tsg101 Is Necessary for the Establishment and Maintenance of Mouse Retinal Pigment Epithelial Cell Polarity. Mol. Cells 2021, 44, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Li, J.; Xiao, X.; Li, S.; Jia, X.; Sun, W.; Guo, X.; Zhang, Q. Detection of Mutations in LRPAP1, CTSH, LEPREL1, ZNF644, SLC39A5, and SCO2 in 298 Families With Early-Onset High Myopia by Exome Sequencing. Investig. Opthalmol. Vis. Sci. 2014, 56, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Zhang, P.; Gao, L.; Ma, Q.; Li, J.; Wang, S.; Liu, B.; Wang, X.; Meng, C. Genetic susceptibility to high myopia in Han Chinese population. Open Life Sci. 2022, 17, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Hall, N.F.; Gale, C.R.; Ye, S.; Martyn, C.N. Myopia and Polymorphisms in Genes for Matrix Metalloproteinases. Investig. Opthalmol. Vis. Sci. 2009, 50, 2632–2636. [Google Scholar] [CrossRef] [PubMed]
- Swierkowska, J.; Karolak, J.A.; Gambin, T.; Rydzanicz, M.; Frajdenberg, A.; Mrugacz, M.; Podfigurna-Musielak, M.; Stankiewicz, P.; Lupski, J.R.; Gajecka, M. Variants in FLRT3 and SLC35E2B identified using exome sequencing in seven high myopia families from Central Europe. Adv. Med. Sci. 2021, 66, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Keen, B.A.; Bailey, L.J.; Jozwiakowski, S.K.; Doherty, A.J. Human PrimPol mutation associated with high myopia has a DNA replication defect. Nucleic Acids Res. 2014, 42, 12102–12111. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Wang, Q.; Li, Y.; Cheng, S.; Liu, J.; Liu, Y. Concurrent pathogenic variants in SLC6A1/NOTCH1/PRIMPOL genes in a Chinese patient with myoclonic-atonic epilepsy, mild aortic valve stenosis and high myopia. BMC Med. Genet. 2020, 21, 93. [Google Scholar] [CrossRef]
- Zhao, F.; Wu, J.; Xue, A.; Su, Y.; Wang, X.; Lu, X.; Zhou, Z.; Qu, J.; Zhou, X. Exome sequencing reveals CCDC111 mutation associated with high myopia. Hum. Genet. 2013, 132, 913–921. [Google Scholar] [CrossRef]
- Lin, F.-Y.; Huang, Z.; Lu, N.; Chen, W.; Fang, H.; Han, W. Controversial opinion: Evaluation of EGR1 and LAMA2 loci for high myopia in Chinese populations. J. Zhejiang Univ. B 2016, 17, 225–235. [Google Scholar] [CrossRef]
- Clark, R.; Pozarickij, A.; Hysi, P.G.; Ohno-Matsui, K.; Williams, C.; Guggenheim, J.A. UK Biobank Eye and Vision Consortium Education interacts with genetic variants near GJD2, RBFOX1, LAMA2, KCNQ5 and LRRC4C to confer susceptibility to myopia. PLoS Genet. 2022, 18, e1010478. [Google Scholar] [CrossRef]
- Jin, W.; Hepei, L.; Mingkun, X.; Li, W. Assessment of BicC family RNA binding protein 1 and Ras protein specific guanine nucleotide releasing factor 1 as candidate genes for high myopia: A case–control study. Indian J. Ophthalmol. 2017, 65, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Li, F.F.; Lu, S.Y.; Tang, S.M.; Kam, K.W.; O S, T.P.; Yip, W.W.K.; Young, A.L.; Tham, C.C.; Pang, C.P.; Yam, J.C.; et al. Genetic associations of myopia severities and endophenotypes in children. Br. J. Ophthalmol. 2020, 105, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Hejtmancik, J.F.; Shiels, A. Overview of the Lens. Prog. Mol. Biol. Transl. Sci. 2015, 134, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.H.; Patel, B.C.; Tadi, P. Anatomy, Head and Neck: Eye Retina. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Habibi, M. Dopamine Receptors, Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780128093245. [Google Scholar] [CrossRef]
- Brown, D.M.; Kowalski, M.A.; Paulus, Q.M.; Yu, J.; Kumar, P.; Kane, M.A.; Patel, J.M.; Ethier, C.R.; Pardue, M.T. Altered Structure and Function of Murine Sclera in Form-Deprivation Myopia. Investig. Opthalmol. Vis. Sci. 2022, 63, 13. [Google Scholar] [CrossRef] [PubMed]





















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).