
Comparison of Multiple Strategies for Precision Transgene Knock-In in Gallus gallus Genome via Microhomology-Mediated End Joining


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
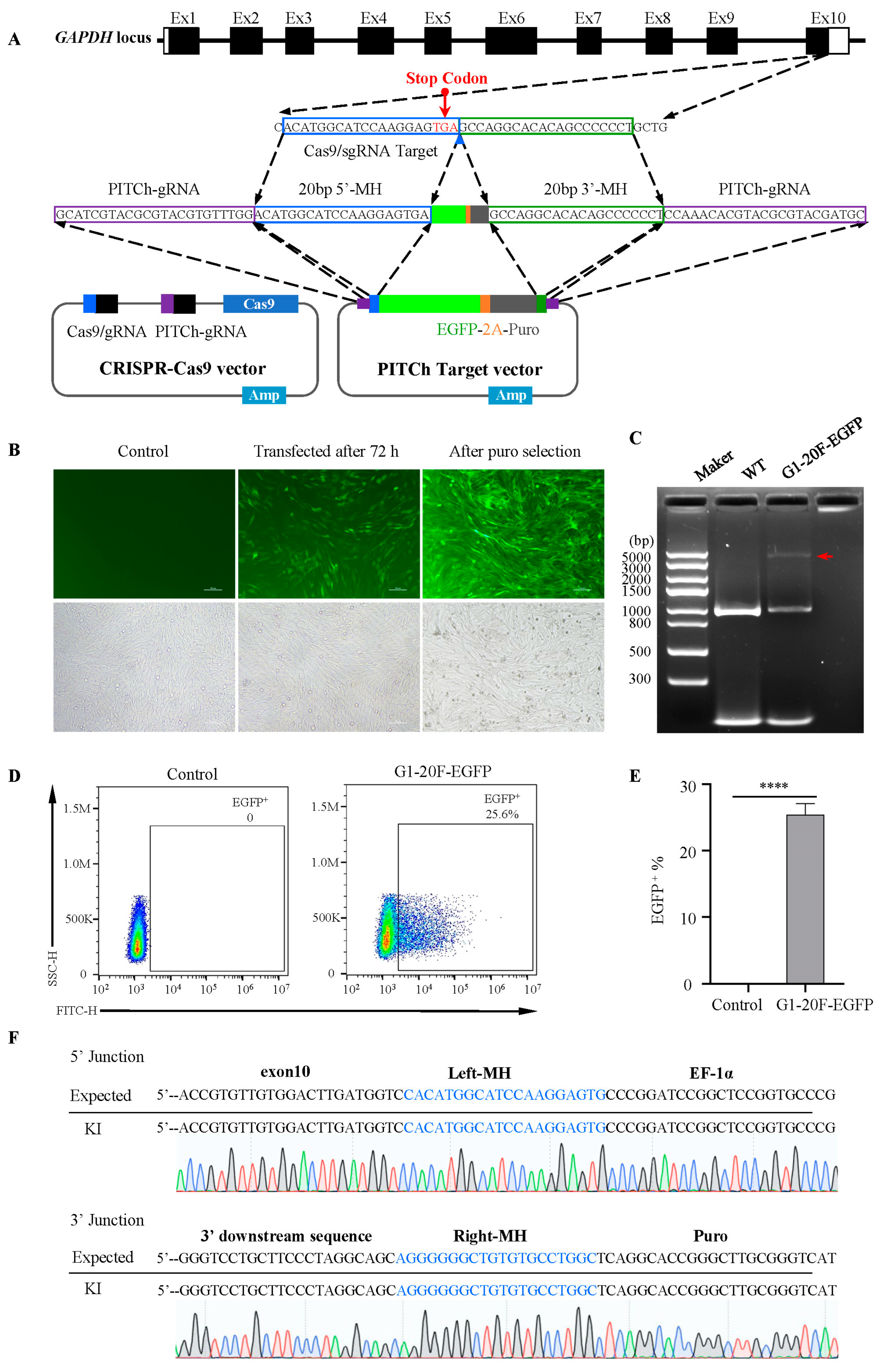
2.1. MMEJ-Mediated Efficient Foreign Gene Integration into DF-1 Cell
2.2. Reverse Knock-In More Productively Performs Than Forward Insertion
2.3. Short Homologous Arms Are More Efficient in Large-Fragment Integration
2.4. Different Transgene Insertion Sites Caused Diverse Effects on Integration Efficiency, Gene Expression, and Cell Viability
2.5. Broad Applicability of MMEJ-Assisted Targeting Vector for Gene Knock-In in the Chicken Genome
3. Discussion
4. Materials and Methods
4.1. Construction of PITCh Plasmids
4.2. Cell Culture and Transfection
4.3. T7E1 Assay
4.4. Genotyping and Sequencing of Knock-In Junctions
4.5. FACS Analysis
4.6. Western Blot Analysis
4.7. Off-Target Analysis of sgRNA
4.8. Growth Curve
4.9. Determination of Cell Proliferation
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sid, H.; Schusser, B. Applications of Gene Editing in Chickens: A New Era Is on the Horizon. Front. Genet. 2018, 9, 456. [Google Scholar] [CrossRef]
- Whitworth, K.M.; Green, J.A.; Redel, B.K.; Geisert, R.D.; Lee, K.; Telugu, B.P.; Wells, K.D.; Prather, R.S. Improvements in pig agriculture through gene editing. CABI Agric. Biosci. 2022, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.R.; Yang, H.; Byun, S.J.; Park, T.S. Research Note: Development of a chicken experimental model platform for induced pluripotent stem cells by using CRISPR/Cas9-mediated NANOG knock-in reporter DF1 cells. Poult. Sci. 2023, 102, 102425. [Google Scholar] [CrossRef] [PubMed]
- Yousefi Taemeh, S.; Dehdilani, N.; Goshayeshi, L.; Rival-Gervier, S.; Mehrzad, J.; Pain, B.; Dehghani, H. Study of the regulatory elements of the Ovalbumin gene promoter using CRISPR technology in chicken cells. J. Biol. Eng. 2023, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Koslová, A.; Trefil, P.; Mucksová, J.; Krchlíková, V.; Plachý, J.; Krijt, J.; Reinišová, M.; Kučerová, D.; Geryk, J.; Kalina, J.; et al. Knock-Out of Retrovirus Receptor Gene Tva in the Chicken Confers Resistance to Avian Leukosis Virus Subgroups A and K and Affects Cobalamin (Vitamin B12)-Dependent Level of Methylmalonic Acid. Viruses 2021, 13, 2504. [Google Scholar] [CrossRef]
- Yuan, M.; Zhang, J.; Gao, Y.; Yuan, Z.; Zhu, Z.; Wei, Y.; Wu, T.; Han, J.; Zhang, Y. HMEJ-based safe-harbor genome editing enables efficient generation of cattle with increased resistance to tuberculosis. J. Biol. Chem. 2021, 296, 100497. [Google Scholar] [CrossRef]
- Collarini, E.; Leighton, P.; Pedersen, D.; Harriman, B.; Jacob, R.; Mettler-Izquierdo, S.; Yi, H.; van de Lavoir, M.-C.; Etches, R.J. Inserting random and site-specific changes into the genome of chickens. Poult. Sci. 2015, 94, 799–803. [Google Scholar] [CrossRef]
- Macdonald, J.; Taylor, L.; Sherman, A.; Kawakami, K.; Takahashi, Y.; Sang, H.M.; McGrew, M.J. Efficient genetic modification and germ-line transmission of primordial germ cells using piggyBac and Tol2 transposons. Proc. Natl. Acad. Sci. USA 2012, 109, E1466–E1472. [Google Scholar] [CrossRef]
- Leighton, P.A.; van de Lavoir, M.C.; Diamond, J.H.; Xia, C.; Etches, R.J. Genetic modification of primordial germ cells by gene trapping, gene targeting, and phiC31 integrase. Mol. Reprod. Dev. 2008, 75, 1163–1175. [Google Scholar] [CrossRef]
- Tyack, S.G.; Jenkins, K.A.; O’Neil, T.E.; Wise, T.G.; Morris, K.R.; Bruce, M.P.; McLeod, S.; Wade, A.J.; McKay, J.; Moore, R.J.; et al. A new method for producing transgenic birds via direct in vivo transfection of primordial germ cells. Transgenic Res. 2013, 22, 1257–1264. [Google Scholar] [CrossRef]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2011, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; van Ooyen, A.; Cox, D.; Fung, Y.K.T.; Varmus, H. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature 1984, 307, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Nakade, S.; Sakane, Y.; Suzuki, K.T.; Yamamoto, T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2016, 11, 118–133. [Google Scholar] [CrossRef]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Sun, J.; Mo, L.; Xu, T.; Shahzad, Q.; Chen, D.; Yang, W.; Liao, Y.; Lu, Y. HMEJ-mediated efficient site-specific gene integration in chicken cells. J. Biol. Eng. 2019, 13, 90. [Google Scholar] [CrossRef]
- Schusser, B.; Collarini, E.J.; Yi, H.; Izquierdo, S.M.; Fesler, J.; Pedersen, D.; Klasing, K.C.; Kaspers, B.; Harriman, W.D.; van de Lavoir, M.C.; et al. Immunoglobulin knockout chickens via efficient homologous recombination in primordial germ cells. Proc. Natl. Acad. Sci. USA 2013, 110, 20170–20175. [Google Scholar] [CrossRef]
- Schusser, B.; Collarini, E.J.; Pedersen, D.; Yi, H.; Ching, K.; Izquierdo, S.; Thoma, T.; Lettmann, S.; Kaspers, B.; Etches, R.J.; et al. Expression of heavy chain-only antibodies can support B-cell development in light chain knockout chickens. Eur. J. Immunol. 2016, 46, 2137–2148. [Google Scholar] [CrossRef]
- Taleei, R.; Nikjoo, H. Biochemical DSB-repair model for mammalian cells in G1 and early S phases of the cell cycle. Mutat. Res. 2013, 756, 206–212. [Google Scholar] [CrossRef]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. 2018, 809, 81–87. [Google Scholar] [CrossRef]
- Brambati, A.; Sacco, O.; Porcella, S.; Heyza, J.; Kareh, M.; Schmidt, J.C.; Sfeir, A. RHINO directs MMEJ to repair DNA breaks in mitosis. Science 2023, 381, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Nakamae, K.; Nishimura, Y.; Takenaga, M.; Nakade, S.; Sakamoto, N.; Ide, H.; Sakuma, T.; Yamamoto, T. Establishment of expanded and streamlined pipeline of PITCh knock-in—A web-based design tool for MMEJ-mediated gene knock-in, PITCh designer, and the variations of PITCh, PITCh-TG and PITCh-KIKO. Bioengineered 2017, 8, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Ezaki, R.; Ichikawa, K.; Matsuzaki, M.; Horiuchi, H. Targeted Knock-in of a Fluorescent Protein Gene into the Chicken Vasa Homolog Locus of Chicken Primordial Germ Cells using CRIS-PITCh Method. J. Poult. Sci. 2022, 59, 182–190. [Google Scholar] [CrossRef]
- Kotin, R.M.; Linden, R.M.; Berns, K.I. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 1992, 11, 5071–5078. [Google Scholar] [CrossRef] [PubMed]
- Irion, S.; Luche, H.; Gadue, P.; Fehling, H.J.; Kennedy, M.; Keller, G. Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat. Biotechnol. 2007, 25, 1477–1482. [Google Scholar] [CrossRef]
- Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999, 21, 70–71. [Google Scholar] [CrossRef]
- Friedrich, G.; Soriano, P. Promoter traps in embryonic stem cells: A genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991, 5, 1513–1523. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kato-Itoh, M.; Yamaguchi, T.; Tamura, C.; Sanbo, M.; Hirabayashi, M.; Nakauchi, H. Identification of rat Rosa26 locus enables generation of knock-in rat lines ubiquitously expressing tdTomato. Stem Cells Dev. 2012, 21, 2981–2986. [Google Scholar] [CrossRef]
- Yang, D.; Song, J.; Zhang, J.; Xu, J.; Zhu, T.; Wang, Z.; Lai, L.; Chen, Y.E. Identification and characterization of rabbit ROSA26 for gene knock-in and stable reporter gene expression. Sci. Rep. 2016, 6, 25161. [Google Scholar] [CrossRef]
- Wu, M.; Wei, C.; Lian, Z.; Liu, R.; Zhu, C.; Wang, H.; Cao, J.; Shen, Y.; Zhao, F.; Zhang, L.; et al. Rosa26-targeted sheep gene knock-in via CRISPR-Cas9 system. Sci. Rep. 2016, 6, 24360. [Google Scholar] [CrossRef]
- Fischer, K.; Kind, A.; Schnieke, A. Assembling multiple xenoprotective transgenes in pigs. Xenotransplantation 2018, 25, e12431. [Google Scholar] [CrossRef] [PubMed]
- Park, T.S.; Lee, H.G.; Moon, J.K.; Lee, H.J.; Yoon, J.W.; Yun, B.N.; Kang, S.C.; Kim, J.; Kim, H.; Han, J.Y.; et al. Deposition of bioactive human epidermal growth factor in the egg white of transgenic hens using an oviduct-specific minisynthetic promoter. FASEB J. 2015, 29, 2386–2396. [Google Scholar] [CrossRef]
- Cao, D.; Wu, H.; Li, Q.; Sun, Y.; Liu, T.; Fei, J.; Zhao, Y.; Wu, S.; Hu, X.; Li, N. Expression of recombinant human lysozyme in egg whites of transgenic hens. PLoS ONE 2015, 10, e0118626. [Google Scholar] [CrossRef]
- Shi, M.; Kawabe, Y.; Ito, A.; Kamihira, M. Targeted knock-in into the OVA locus of chicken cells using CRISPR/Cas9 system with homology-independent targeted integration. J. Biosci. Bioeng. 2020, 129, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Oishi, I.; Yoshii, K.; Miyahara, D.; Tagami, T. Efficient production of human interferon beta in the white of eggs from ovalbumin gene-targeted hens. Sci. Rep. 2018, 8, 10203. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Xiao, N.; Xu, Y.; Yang, F.; Wang, X.; Hu, H.; Liu, Q.; Cui, K.; Tang, X. Efficient knock-in at the chicken ovalbumin locus using adenovirus as a CRISPR/Cas9 delivery system. 3 Biotech 2019, 9, 454. [Google Scholar] [CrossRef]
- Kim, Y.M.; Shim, J.H.; Park, J.S.; Choi, H.J.; Jung, K.M.; Lee, K.Y.; Park, K.J.; Han, J.Y. Sequential verification of exogenous protein production in OVA gene-targeted chicken bioreactors. Poult. Sci. 2023, 102, 102247. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Guo, Y.; Du, W.; Yin, Y.; Zhang, T.; Lu, H. Enhancing Targeted Genomic DNA Editing in Chicken Cells Using the CRISPR/Cas9 System. PLoS ONE 2017, 12, e0169768. [Google Scholar] [CrossRef]
- Wragg, D.; Mason, A.S.; Yu, L.; Kuo, R.; Lawal, R.A.; Desta, T.T.; Mwacharo, J.M.; Cho, C.Y.; Kemp, S.; Burt, D.W.; et al. Genome-wide analysis reveals the extent of EAV-HP integration in domestic chicken. BMC Genom. 2015, 16, 784. [Google Scholar] [CrossRef]
- Antonova, E.; Glazova, O.; Gaponova, A.; Eremyan, A.; Zvereva, S.; Grebenkina, N.; Volkova, N.; Volchkov, P. Successful CRISPR/Cas9 mediated homologous recombination in a chicken cell line. F1000Research 2018, 7, 238. [Google Scholar] [CrossRef]
- Dehdilani, N.; Goshayeshi, L.; Yousefi Taemeh, S.; Bahrami, A.R.; Rival Gervier, S.; Pain, B.; Dehghani, H. Integrating Omics and CRISPR Technology for Identification and Verification of Genomic Safe Harbor Loci in the Chicken Genome. Biol. Proced. Online 2023, 25, 18. [Google Scholar] [CrossRef]
- Abu-Bonsrah, K.D.; Zhang, D.; Newgreen, D.F. CRISPR/Cas9 Targets Chicken Embryonic Somatic Cells In Vitro and In Vivo and generates Phenotypic Abnormalities. Sci. Rep. 2016, 6, 34524. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Johnson, W.E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Mo, G.; Wei, P.; Hu, B.; Nie, Q.; Zhang, X. Advances on genetic and genomic studies of ALV resistance. J. Anim. Sci. Biotechnol. 2022, 13, 123. [Google Scholar] [CrossRef]
- Barr, S.D.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. Integration targeting by avian sarcoma-leukosis virus and human immunodeficiency virus in the chicken genome. J. Virol. 2005, 79, 12035–12044. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Sun, J.; Liu, Z.; Zheng, Q.; Wang, G. Comparison of Multiple Strategies for Precision Transgene Knock-In in Gallus gallus Genome via Microhomology-Mediated End Joining. Int. J. Mol. Sci. 2023, 24, 15731. https://doi.org/10.3390/ijms242115731
Wang L, Sun J, Liu Z, Zheng Q, Wang G. Comparison of Multiple Strategies for Precision Transgene Knock-In in Gallus gallus Genome via Microhomology-Mediated End Joining. International Journal of Molecular Sciences. 2023; 24(21):15731. https://doi.org/10.3390/ijms242115731
Chicago/Turabian StyleWang, Lijuan, Jiaxin Sun, Zhipeng Liu, Qiang Zheng, and Guojun Wang. 2023. "Comparison of Multiple Strategies for Precision Transgene Knock-In in Gallus gallus Genome via Microhomology-Mediated End Joining" International Journal of Molecular Sciences 24, no. 21: 15731. https://doi.org/10.3390/ijms242115731