Mitochondrial Impairment: A Link for Inflammatory Responses Activation in the Cardiorenal Syndrome Type 4

 ,
,  , , ,
, , ,  , ,
, ,  and
and
Abstract
:1. Cardiorenal Syndrome Overview
The Kidney-Heart Crosstalk
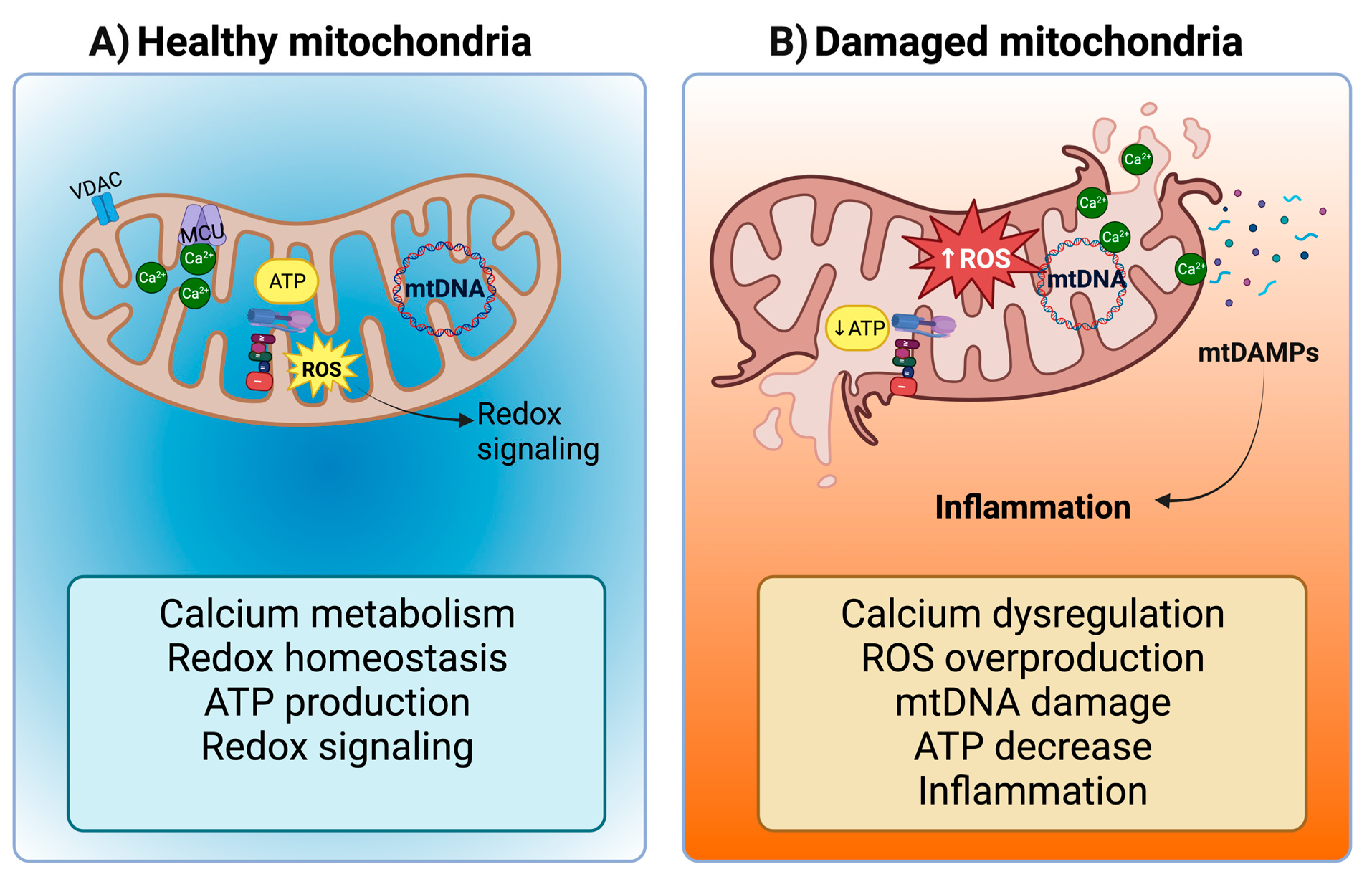
2. Mitochondrial Dysfunction and Inflammatory Alterations in CRS Type 4
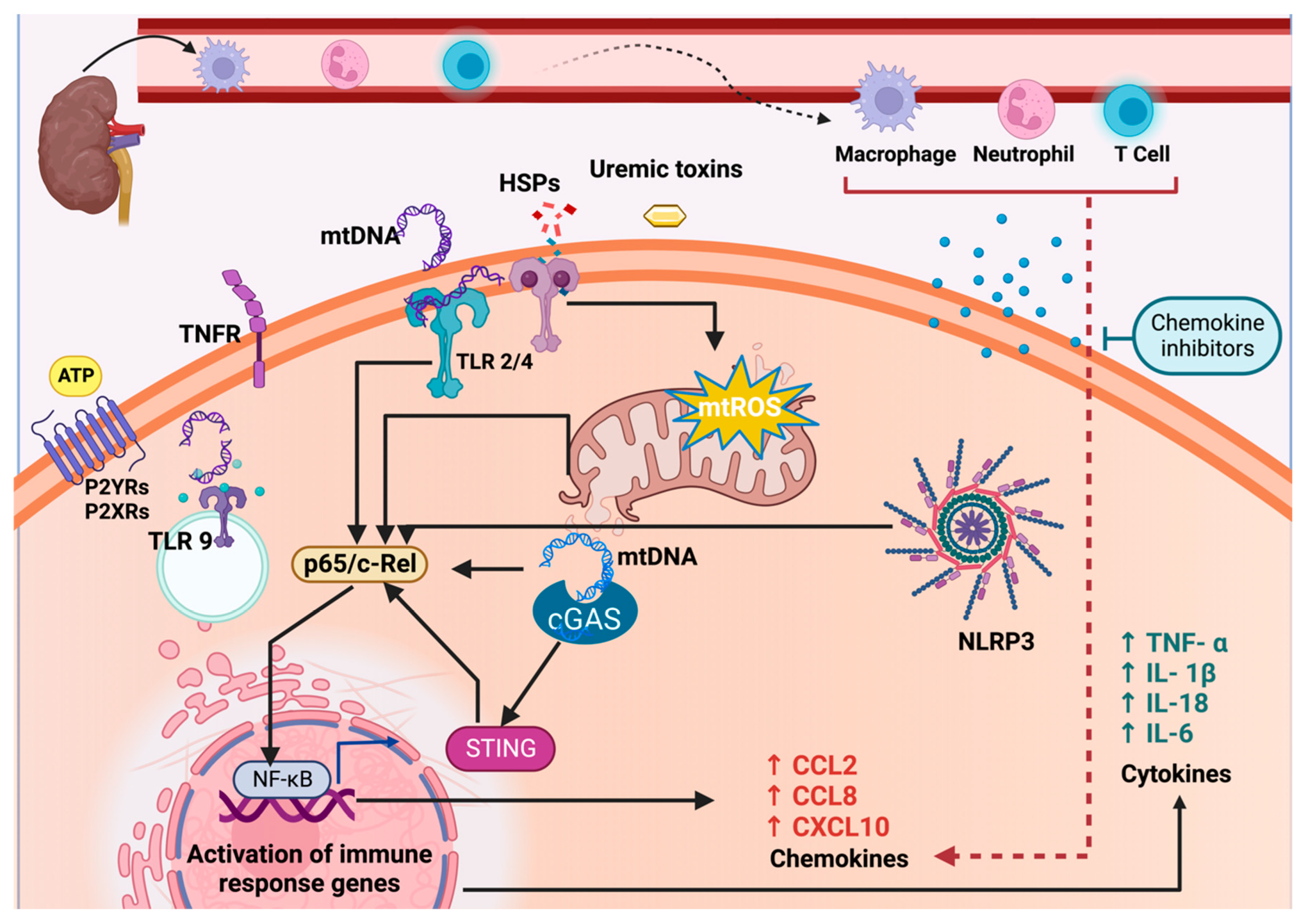
2.1. Mitochondrial Dysfunction in CKD Activates the NLRP3-NF-κB Pathway
2.2. The Release of mtDAMPs during CKD and the Establishment of CRS Type 4
2.3. NLRP3-NF-κB Pathway Activation in the Heart by CKD-Derived mtDAMPs and ROS
2.4. Involvement of NLRP3 Inflammasome and Toll-like Receptors 2 and 4 in CRS Type 4
2.5. Role of TLR9 in Inflammation and Its Implication in CRS Type 4
2.6. Extracellular Vesicles (EV) and Their Role in Inflammation
2.7. The Role of Autophagy and Mitophagy in NLRP3 Signaling Pathway in CRS Type 4
2.8. MAVS and NLRP3-NF-kB Signaling in CRS Type 4
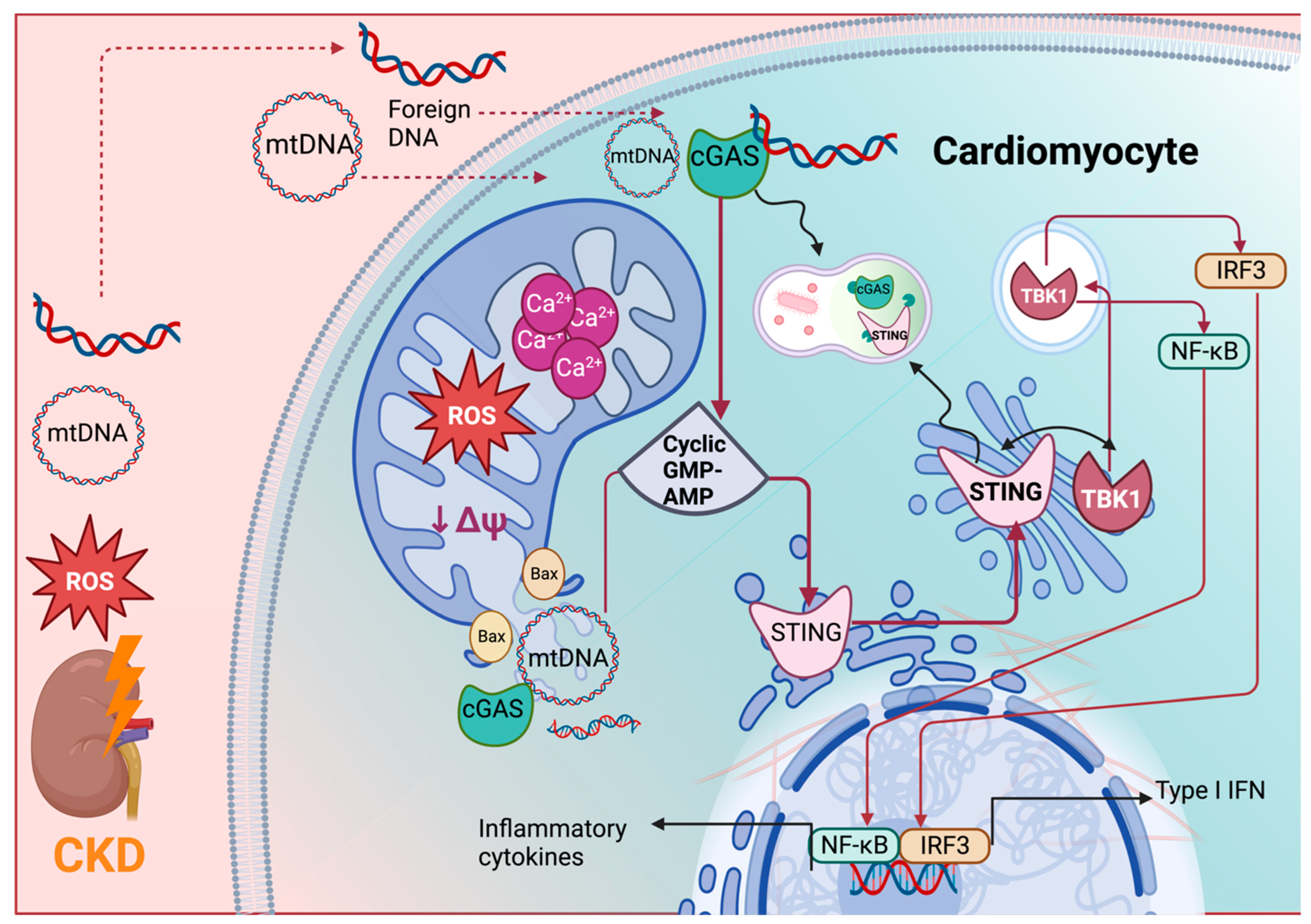
3. The Role of the cGAS-STING Pathway in CRS Type 4
3.1. The cGAS-STING Pathway
3.2. The Activation of the cGAS-STING-NF-κB Axis by mtDNA Release in CKD
3.3. The Activation of the cGAS-STING-NF-κB Axis by mtDNA Release in CRS Type 4
4. Chemokines Activation and the Pathophysiology of CRS Type 4
4.1. Chemokines Overview
4.2. The Role of Chemokines and Receptors in the Pathophysiology of CKD
4.2.1. Monocyte Chemoattractant Protein-1 (MCP-1)/CCL2 and CCR2 Receptor in CKD
4.2.2. C-C Motif Chemokine 8 (CCL8/MCP-2) in CKD
4.2.3. Chemokine Interferon-γ-Inducible Protein 10 (IP-10)/Chemokine (C-X-C Motif) Ligand (CXCL)10 in CKD
4.3. Chemokines and Receptors in the Pathophysiology of CRS Type 4
4.3.1. Monocyte Chemoattractant Protein-1 (MCP-1)/CCL2 and CCR2 in CRS Type 4
4.3.2. The Role of C-C Motif Chemokine 8 (CCL8/MCP-2) in CRS Type 4
4.3.3. IP-10/CXCL10 in CRS Type 4
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bright, R. Cases and Observations, Illustrative of Renal Disease, Accompanied with the Secretion of Albuminous Urine. Guy’s Hosp. Rep. 1836, 1, 338–379. [Google Scholar]
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-Renal Syndromes: Report from the Consensus Conference of the Acute Dialysis Quality Initiative. Eur. Heart J. 2010, 31, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.; Anker, S.D.; Mebazaa, A.; Palazzuoli, A.; Vescovo, G.; Bellomo, R.; Ponikowski, P.; Anand, I.; Aspromonte, N.; Bagshaw, S.; et al. ADQI 7: The Clinical Management of the Cardio-Renal Syndromes: Work Group Statements from the 7th ADQI Consensus Conference. Nephrol. Dial. Transplant. 2010, 25, 2077–2089. [Google Scholar] [CrossRef] [PubMed]
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement from the American Heart Association. Circulation 2019, 139, E840–E878. [Google Scholar] [CrossRef]
- Yogasundaram, H.; Chappell, M.C.; Braam, B.; Oudit, G.Y. Cardiorenal Syndrome and Heart Failure—Challenges and Opportunities. Can. J. Cardiol. 2019, 35, 1208–1219. [Google Scholar] [CrossRef]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A Single Number for Advocacy and Communication—Worldwide More than 850 Million Individuals Have Kidney Diseases. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2019, 34, 1803–1805. [Google Scholar] [CrossRef]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, Regional, and National Burden of Chronic Kidney Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Lopez-Diaz, A.M.; Guerrero-Mauvecin, J.; Miguel, V.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Tubular Mitochondrial Dysfunction, Oxidative Stress, and Progression of Chronic Kidney Disease. Antioxidants 2022, 11, 1356. [Google Scholar] [CrossRef]
- Andrassy, K.M. Comments on ‘KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease’. Kidney Int. 2013, 84, 622–623. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group. KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2020, 98, S1–S115. [Google Scholar] [CrossRef]
- McCullough, P.A. Cardiorenal Syndromes: Pathophysiology to Prevention. Int. J. Nephrol. 2011, 2011, 762590. [Google Scholar] [CrossRef] [PubMed]
- Peesapati, V.S.R.; Sadik, M.; Verma, S.; Attallah, M.A.; Khan, S. Panoramic Dominance of the Immune System in Cardiorenal Syndrome Type I. Cureus 2020, 12, e9869. [Google Scholar] [CrossRef] [PubMed]
- De Vecchis, R.; Baldi, C. Cardiorenal Syndrome Type 2: From Diagnosis to Optimal Management. Ther. Clin. Risk Manag. 2014, 10, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Jois, P.; Mebazaa, A. Cardio-Renal Syndrome Type 2: Epidemiology, Pathophysiology, and Treatment. Semin. Nephrol. 2012, 32, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, S.M.; Hoste, E.A.; Braam, B.; Briguori, C.; Kellum, J.A.; McCullough, P.A.; Ronco, C. Cardiorenal Syndrome Type 3: Pathophysiologic and Epidemiologic Considerations. In Contributions to Nephrology; McCullough, P.A., Kellum, J.A., Mehta, R.L., Murray, P.T., Ronco, C., Eds.; S. Karger AG: Basel, Switzerland, 2013; Volume 182, pp. 137–157. ISBN 978-3-318-02406-7. [Google Scholar]
- Chuasuwan, A.; Kellum, J.A. Cardio-Renal Syndrome Type 3: Epidemiology, Pathophysiology, and Treatment. Semin. Nephrol. 2012, 32, 31–39. [Google Scholar] [CrossRef]
- Da Silva, A.L.P.; da Silva, M.J.V. Type 4 Cardiorenal Syndrome. Rev. Port. Cardiol. 2016, 35, 601–616. [Google Scholar] [CrossRef]
- Pateinakis, P.; Papagianni, A. Cardiorenal Syndrome Type 4—Cardiovascular Disease in Patients with Chronic Kidney Disease: Epidemiology, Pathogenesis, and Management. Int. J. Nephrol. 2011, 2011, 938651. [Google Scholar] [CrossRef]
- Tumlin, J.A.; Costanzo, M.R.; Chawla, L.S.; Herzog, C.A.; Kellum, J.A.; McCullough, P.A.; Ronco, C. Cardiorenal Syndrome Type 4: Insights on Clinical Presentation and Pathophysiology from the Eleventh Consensus Conference of the Acute Dialysis Quality Initiative (ADQI). In Contributions to Nephrology; McCullough, P.A., Kellum, J.A., Mehta, R.L., Murray, P.T., Ronco, C., Eds.; S. Karger AG: Basel, Switzerland, 2013; Volume 182, pp. 158–173. ISBN 978-3-318-02406-7. [Google Scholar]
- Di Lullo, L.; Gorini, A.; Russo, D.; Santoboni, A.; Ronco, C. Left Ventricular Hypertrophy in Chronic Kidney Disease Patients: From Pathophysiology to Treatment. Cardiorenal Med. 2015, 5, 254–266. [Google Scholar] [CrossRef]
- Kumar, S.; Bogle, R.; Banerjee, D. Why Do Young People with Chronic Kidney Disease Die Early? World J. Nephrol. 2014, 3, 143–155. [Google Scholar] [CrossRef]
- Mittalhenkle, A.; Stehman-Breen, C.O.; Shlipak, M.G.; Fried, L.F.; Katz, R.; Young, B.A.; Seliger, S.; Gillen, D.; Newman, A.B.; Psaty, B.M.; et al. Cardiovascular Risk Factors and Incident Acute Renal Failure in Older Adults: The Cardiovascular Health Study. Clin. J. Am. Soc. Nephrol. CJASN 2008, 3, 450–456. [Google Scholar] [CrossRef]
- Hager, M.R.; Narla, A.D.; Tannock, L.R. Dyslipidemia in Patients with Chronic Kidney Disease. Rev. Endocr. Metab. Disord. 2017, 18, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Katz, R.; Robinson-Cohen, C.; Odden, M.C.; Dalrymple, L.; Shlipak, M.G.; Sarnak, M.J.; Siscovick, D.S.; Zelnick, L.; Psaty, B.M.; et al. Absolute Rates of Heart Failure, Coronary Heart Disease, and Stroke in Chronic Kidney Disease: An Analysis of 3 Community-Based Cohort Studies. JAMA Cardiol. 2017, 2, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.C.; Kuo, K.-L. Oxidative Stress in Chronic Kidney Disease. Ren. Replace. Ther. 2018, 4, 53. [Google Scholar] [CrossRef]
- Cozzolino, M.; Marin, D.; Sisti, G. New Frontiers in IVF: mtDNA and Autologous Germline Mitochondrial Energy Transfer. Reprod. Biol. Endocrinol. 2019, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Rysz, J.; Franczyk, B.; Ławiński, J.; Gluba-Brzózka, A. Oxidative Stress in ESRD Patients on Dialysis and the Risk of Cardiovascular Diseases. Antioxidants 2020, 9, 1079. [Google Scholar] [CrossRef] [PubMed]
- Moradi, H.; Sica, D.A.; Kalantar-Zadeh, K. Cardiovascular Burden Associated with Uremic Toxins in Patients with Chronic Kidney Disease. Am. J. Nephrol. 2013, 38, 136–148. [Google Scholar] [CrossRef]
- Schophuizen, C.M.S.; Wilmer, M.J.; Jansen, J.; Gustavsson, L.; Hilgendorf, C.; Hoenderop, J.G.J.; van den Heuvel, L.P.; Masereeuw, R. Cationic Uremic Toxins Affect Human Renal Proximal Tubule Cell Functioning through Interaction with the Organic Cation Transporter. Pflügers Arch.-Eur. J. Physiol. 2013, 465, 1701–1714. [Google Scholar] [CrossRef]
- Kopel, T.; Kaufman, J.S.; Hamburg, N.; Sampalis, J.S.; Vita, J.A.; Dember, L.M. Endothelium-Dependent and -Independent Vascular Function in Advanced Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1588–1594. [Google Scholar] [CrossRef]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef]
- Claridge, B.; Rai, A.; Fang, H.; Matsumoto, A.; Luo, J.; McMullen, J.R.; Greening, D.W. Proteome Characterisation of Extracellular Vesicles Isolated from Heart. Proteomics 2021, 21, 2100026. [Google Scholar] [CrossRef]
- Ranches, G.; Zeidler, M.; Kessler, R.; Hoelzl, M.; Hess, M.W.; Vosper, J.; Perco, P.; Schramek, H.; Kummer, K.K.; Kress, M.; et al. Exosomal Mitochondrial tRNAs and miRNAs as Potential Predictors of Inflammation in Renal Proximal Tubular Epithelial Cells. Mol. Ther.-Nucleic Acids 2022, 28, 794–813. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular Disease in Dialysis Patients. Nephrol. Dial. Transplant. 2018, 33, iii28–iii34. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Ma, G.; Wang, Z.; Liang, W.; Gao, W. Mechanisms of Kidney Cell Pyroptosis in Chronic Kidney Disease and the Effects of Traditional Chinese Medicine. Evid. Based Complement. Altern. Med. 2021, 2021, 1173324. [Google Scholar] [CrossRef] [PubMed]
- Cachofeiro, V.; Goicochea, M.; de Vinuesa, S.G.; Oubiña, P.; Lahera, V.; Luño, J. Oxidative Stress and Inflammation, a Link between Chronic Kidney Disease and Cardiovascular Disease. Kidney Int. 2008, 74, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, K.F.; Pothineni, N.V.K.; Rutland, J.; Ding, Z.; Mehta, J.L. Immunity, Inflammation, and Oxidative Stress in Heart Failure: Emerging Molecular Targets. Cardiovasc. Drugs Ther. 2017, 31, 593–608. [Google Scholar] [CrossRef] [PubMed]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial Dysfunction and the Inflammatory Response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- Schefold, J.C.; Filippatos, G.; Hasenfuss, G.; Anker, S.D.; von Haehling, S. Heart Failure and Kidney Dysfunction: Epidemiology, Mechanisms and Management. Nat. Rev. Nephrol. 2016, 12, 610–623. [Google Scholar] [CrossRef]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Sandhir, R.; Halder, A.; Sunkaria, A. Mitochondria as a Centrally Positioned Hub in the Innate Immune Response. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1090–1097. [Google Scholar] [CrossRef]
- Zhao, W.; Zhou, L.; Novák, P.; Shi, X.; Lin, C.B.; Zhu, X.; Yin, K. Metabolic Dysfunction in the Regulation of the NLRP3 Inflammasome Activation: A Potential Target for Diabetic Nephropathy. J. Diabetes Res. 2022, 2022, 2193768. [Google Scholar] [CrossRef]
- Chin, L.-H.; Hsu, Y.-J.; Hsu, S.-C.; Chen, Y.-H.; Chang, Y.-L.; Huang, S.-M.; Tsai, C.-S.; Lin, C.-Y. The Regulation of NLRP3 Inflammasome Expression during the Development of Cardiac Contractile Dysfunction in Chronic Kidney Disease. Oncotarget 2017, 8, 113303–113317. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Du, C.; Zhu, Y.; Ran, L.; Wang, Y.; Xiong, J.; Wu, Y.; Lan, Q.; Wang, Y.; Wang, L.; et al. Targeting Myocardial Mitochondria-STING-Polyamine Axis Prevents Cardiac Hypertrophy in Chronic Kidney Disease. JACC Basic Transl. Sci. 2022, 7, 820–840. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Zhang, A.; Cheng, Y.; Chen, Y. The cGAS-STING Pathway: A Promising Immunotherapy Target. Front. Immunol. 2021, 12, 795048. [Google Scholar] [CrossRef] [PubMed]
- Skopelja-Gardner, S.; An, J.; Elkon, K.B. Role of the cGAS–STING Pathway in Systemic and Organ-Specific Diseases. Nat. Rev. Nephrol. 2022, 18, 558–572. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR Signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 Inflammasome: From a Danger Signal Sensor to a Regulatory Node of Oxidative Stress and Inflammatory Diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Hemmers, C.; Schulte, C.; Wollenhaupt, J.; Wong, D.W.L.; Harlacher, E.; Orth-Alampour, S.; Klinkhammer, B.M.; Schirmer, S.H.; Böhm, M.; Marx, N.; et al. Chemokine CCL9 Is Upregulated Early in Chronic Kidney Disease and Counteracts Kidney Inflammation and Fibrosis. Biomedicines 2022, 10, 420. [Google Scholar] [CrossRef]
- Lee, J.; Lee, Y.; Kim, K.-H.; Kim, D.-K.; Joo, K.-W.; Shin, S.-J.; Kim, Y.-S.; Yang, S.-H. Chemokine (C-C Motif) Ligand 8 and Tubulo-Interstitial Injury in Chronic Kidney Disease. Cells 2022, 11, 658. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef]
- Amador-Martínez, I.; García-Ballhaus, J.; Buelna-Chontal, M.; Cortés-González, C.; Massó, F.; Jaisser, F.; Barrera-Chimal, J. Early Inflammatory Changes and CC Chemokine Ligand-8 Upregulation in the Heart Contribute to Uremic Cardiomyopathy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21761. [Google Scholar] [CrossRef]
- Mawhin, M.A.; Bright, R.G.; Fourre, J.D.; Vloumidi, E.I.; Tomlinson, J.; Sardini, A.; Pusey, C.D.; Woollard, K.J. Chronic Kidney Disease Mediates Cardiac Dysfunction Associated with Increased Resident Cardiac Macrophages. BMC Nephrol. 2022, 23, 47. [Google Scholar] [CrossRef] [PubMed]
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as Therapeutic Targets in Cardiovascular Disease: The Road Behind, The Road Ahead. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Frangogiannis, N.G. Chemokines in Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 14, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Mejia, E.M.; Hatch, G.M. Mitochondrial Phospholipids: Role in Mitochondrial Function. J. Bioenerg. Biomembr. 2016, 48, 99–112. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial Metabolism of Reactive Oxygen Species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- Doi, K.; Noiri, E. Mitochondrial Dysfunction in Cardiorenal Syndrome. Antioxid. Redox Signal. 2016, 25, 200–207. [Google Scholar] [CrossRef]
- Shi, S.; Zhang, B.; Li, Y.; Xu, X.; Lv, J.; Jia, Q.; Chai, R.; Xue, W.; Li, Y.; Wang, Y.; et al. Mitochondrial Dysfunction: An Emerging Link in the Pathophysiology of Cardiorenal Syndrome. Front. Cardiovasc. Med. 2022, 9, 837270. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of Mitochondrial Dynamics in Acute Kidney Injury in Cell Culture and Rodent Models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Mitochondrial Bioenergetics, Redox State, Dynamics and Turnover Alterations in Renal Mass Reduction Models of Chronic Kidney Diseases and Their Possible Implications in the Progression of This Illness. Pharmacol. Res. 2018, 135, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W. Mitochondrial Dysfunction in Cardiovascular Disease. Free Radic. Biol. Med. 2005, 38, 1278–1295. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, Z.; Liang, W.; Wei, Z.; Ding, G. Roles of Mitochondrial DNA Damage in Kidney Diseases: A New Biomarker. Int. J. Mol. Sci. 2022, 23, 15166. [Google Scholar] [CrossRef]
- Nakahira, K.; Hisata, S.; Choi, A.M.K. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef]
- Feehan, K.T.; Gilroy, D.W. Is Resolution the End of Inflammation? Trends Mol. Med. 2019, 25, 198–214. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Junho, C.V.C.; Trentin-Sonoda, M.; Panico, K.; dos Santos, R.S.N.; Abrahão, M.V.; Vernier, I.C.S.; Fürstenau, C.R.; Carneiro-Ramos, M.S. Cardiorenal Syndrome: Long Road between Kidney and Heart. Heart Fail. Rev. 2022, 27, 2137–2153. [Google Scholar] [CrossRef]
- Schett, G.; Neurath, M.F. Resolution of Chronic Inflammatory Disease: Universal and Tissue-Specific Concepts. Nat. Commun. 2018, 9, 3261. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.-M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhang, Y.; Zhang, Y.; Ma, Y. Chronic Kidney Disease and NLRP3 Inflammasome: Pathogenesis, Development and Targeted Therapeutic Strategies. Biochem. Biophys. Rep. 2023, 33, 101417. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Xu, K.; Li, C.; Qi, C.; Fang, Y.; Zhu, N.; Bao, J.; Zhao, Z.; Yu, Q.; Wu, H.; et al. NLRP3 Associated with Chronic Kidney Disease Progression after Ischemia/Reperfusion-Induced Acute Kidney Injury. Cell Death Discov. 2021, 7, 324. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Zaza, G.; Simone, S.; Villani, G.; Latorre, D.; Pontrelli, P.; Carella, M.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Mitochondrial Dysregulation and Oxidative Stress in Patients with Chronic Kidney Disease. BMC Genom. 2009, 10, 388. [Google Scholar] [CrossRef]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular Mechanisms Regulating NLRP3 Inflammasome Activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in Health and Disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Kinoshita, T.; Imamura, R.; Kushiyama, H.; Suda, T. NLRP3 Mediates NF-κB Activation and Cytokine Induction in Microbially Induced and Sterile Inflammation. PLoS ONE 2015, 10, e0119179. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Vringer, E.; Tait, S.W.G. Mitochondria and Cell Death-Associated Inflammation. Cell Death Differ. 2023, 30, 304–312. [Google Scholar] [CrossRef]
- Zhong, F.; Liang, S.; Zhong, Z. Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol. 2019, 40, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z. Crosstalk of Reactive Oxygen Species and NF-κB Signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Tapia, E.; Molina-Jijón, E.; Medina-Campos, O.N.; Macías-Ruvalcaba, N.A.; León-Contreras, J.C.; Hernández-Pando, R.; García-Arroyo, F.E.; Cristóbal, M.; Sánchez-Lozada, L.G.; et al. Curcumin Prevents Mitochondrial Dynamics Disturbances in Early 5/6 Nephrectomy: Relation to Oxidative Stress and Mitochondrial Bioenergetics: Curcumin Prevents Mitochondrial Dynamics Disturbances. BioFactors 2017, 43, 293–310. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial Dysfunction in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Nguyen, T.-V.; Penfold, S.A.; Higgins, G.C.; Thallas-Bonke, V.; Tan, S.M.; Van Bergen, N.J.; Sourris, K.C.; Harcourt, B.E.; Thorburn, D.R.; et al. Mapping Time-Course Mitochondrial Adaptations in the Kidney in Experimental Diabetes. Clin. Sci. 2016, 130, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Hallan, S.; Afkarian, M.; Zelnick, L.R.; Kestenbaum, B.; Sharma, S.; Saito, R.; Darshi, M.; Barding, G.; Raftery, D.; Ju, W.; et al. Metabolomics and Gene Expression Analysis Reveal Down-Regulation of the Citric Acid (TCA) Cycle in Non-Diabetic CKD Patients. EBioMedicine 2017, 26, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Uribe, A.P.; Bellido, B.; Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Hernández-Santos, J.A.; Fernández-Valverde, F.; Hernández-Cruz, E.Y.; Orozco-Ibarra, M.; Pedraza-Chaverri, J. Temporal Characterization of Mitochondrial Impairment in the Unilateral Ureteral Obstruction Model in Rats. Free Radic. Biol. Med. 2021, 172, 358–371. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective Effects of N-Acetyl-Cysteine in Mitochondria Bioenergetics, Oxidative Stress, Dynamics and S-Glutathionylation Alterations in Acute Kidney Damage Induced by Folic Acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef]
- Hui, Y.; Lu, M.; Han, Y.; Zhou, H.; Liu, W.; Li, L.; Jin, R. Resveratrol Improves Mitochondrial Function in the Remnant Kidney from 5/6 Nephrectomized Rats. Acta Histochem. 2017, 119, 392–399. [Google Scholar] [CrossRef]
- Correa, F.; Buelna-Chontal, M.; Hernández-Reséndiz, S.; García-Niño, W.R.; Roldán, F.J.; Soto, V.; Silva-Palacios, A.; Amador, A.; Pedraza-Chaverrí, J.; Tapia, E.; et al. Curcumin Maintains Cardiac and Mitochondrial Function in Chronic Kidney Disease. Free Radic. Biol. Med. 2013, 61, 119–129. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Rojas-Morales, P.; Avila-Rojas, S.H.; León-Contreras, J.C.; Hernández-Pando, R.; Jiménez-Uribe, A.P.; Prieto-Carrasco, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Temporal Alterations in Mitochondrial β-Oxidation and Oxidative Stress Aggravate Chronic Kidney Disease Development in 5/6 Nephrectomy Induced Renal Damage. Int. J. Mol. Sci. 2020, 21, 6512. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Groß, C.J.; Mishra, R.; Schneider, K.S.; Médard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.-A.; Fromm, S.; et al. K + Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective Fatty Acid Oxidation in Renal Tubular Epithelial Cells Has a Key Role in Kidney Fibrosis Development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, W.; He, Q.; Xue, J.; Wang, J.; Xiong, C.; Pu, X.; Nie, Z. Mass Spectrometry Imaging of Kidney Tissue Sections of Rat Subjected to Unilateral Ureteral Obstruction. Sci. Rep. 2017, 7, 41954. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, X.; Yao, Q.; Liu, Y.; Zhang, H.; Dong, Z. HIF-1-Mediated Production of Exosomes during Hypoxia Is Protective in Renal Tubular Cells. Am. J. Physiol. Renal Physiol. 2017, 313, F906–F913. [Google Scholar] [CrossRef]
- Olona, A.; Leishman, S.; Anand, P.K. The NLRP3 Inflammasome: Regulation by Metabolic Signals. Trends Immunol. 2022, 43, 978–989. [Google Scholar] [CrossRef]
- Corcoran, S.E.; O’Neill, L.A.J. HIF1α and Metabolic Reprogramming in Inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Yao, H.; Chen, X.; Shang, L.; Li, P.; Cui, X.; Zeng, J. Peroxisome-Generated Succinate Induces Lipid Accumulation and Oxidative Stress in the Kidneys of Diabetic Mice. J. Biol. Chem. 2022, 298, 101660. [Google Scholar] [CrossRef]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of Mitochondria Prevents High-Fat Diet–Induced Glomerulopathy and Proximal Tubular Injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.C.P.; Bocharov, A.V.; Baranova, I.N.; Vishnyakova, T.G.; Huang, Y.G.; Wilkins, K.J.; Hu, X.; Street, J.M.; Alvarez-Prats, A.; Mullick, A.E.; et al. Antagonism of Scavenger Receptor CD36 by 5A Peptide Prevents Chronic Kidney Disease Progression in Mice Independent of Blood Pressure Regulation. Kidney Int. 2016, 89, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Okamura, D.M.; Pennathur, S.; Pasichnyk, K.; López-Guisa, J.M.; Collins, S.; Febbraio, M.; Heinecke, J.; Eddy, A.A. CD36 Regulates Oxidative Stress and Inflammation in Hypercholesterolemic CKD. J. Am. Soc. Nephrol. 2009, 20, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Ceja-Galicia, Z.A.; García-Arroyo, F.E.; Aparicio-Trejo, O.E.; El-Hafidi, M.; Gonzaga-Sánchez, G.; León-Contreras, J.C.; Hernández-Pando, R.; Guevara-Cruz, M.; Tovar, A.R.; Rojas-Morales, P.; et al. Therapeutic Effect of Curcumin on 5/6Nx Hypertriglyceridemia: Association with the Improvement of Renal Mitochondrial β-Oxidation and Lipid Metabolism in Kidney and Liver. Antioxidants 2022, 11, 2195. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.-K.; Park, K.-S. Oxidative Stress and Calcium Dysregulation by Palmitate in Type 2 Diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty Acid–Induced NLRP3-ASC Inflammasome Activation Interferes with Insulin Signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Tapia, E.; Rojas-Morales, P.; León-Contreras, J.C.; Martínez-Klimova, E.; Hernández-Pando, R.; Sánchez- Lozada, L.G.; Pedraza-Chaverri, J. Chronic Impairment of Mitochondrial Bioenergetics and β-Oxidation Promotes Experimental AKI-to-CKD Transition Induced by Folic Acid. Free Radic. Biol. Med. 2020, 154, 18–32. [Google Scholar] [CrossRef]
- Feng, B.; Meng, R.; Huang, B.; Bi, Y.; Shen, S.; Zhu, D. Silymarin Protects against Renal Injury through Normalization of Lipid Metabolism and Mitochondrial Biogenesis in High Fat-Fed Mice. Free Radic. Biol. Med. 2017, 110, 240–249. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered Renal Lipid Metabolism and Renal Lipid Accumulation in Human Diabetic Nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in Chronic Kidney Disease: Novel Insights and Therapeutic Opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- Helal, I.; Fick-Brosnahan, G.M.; Reed-Gitomer, B.; Schrier, R.W. Glomerular Hyperfiltration: Definitions, Mechanisms and Clinical Implications. Nat. Rev. Nephrol. 2012, 8, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Ertuglu, L.A.; Afsar, B.; Ozdogan, E.; Kucuksumer, Z.S.; Ortiz, A.; Covic, A.; Kuwabara, M.; Cherney, D.Z.I.; van Raalte, D.H.; et al. Renal Hyperfiltration Defined by High Estimated Glomerular Filtration Rate: A Risk Factor for Cardiovascular Disease and Mortality. Diabetes Obes. Metab. 2019, 21, 2368–2383. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria and Calcium: From Cell Signalling to Cell Death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Prieto-Carrasco, R.; García-Arroyo, F.E.; Aparicio-Trejo, O.E.; Rojas-Morales, P.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Tapia, E.; Pedraza-Chaverri, J. Progressive Reduction in Mitochondrial Mass Is Triggered by Alterations in Mitochondrial Biogenesis and Dynamics in Chronic Kidney Disease Induced by 5/6 Nephrectomy. Biology 2021, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between Albuminuria, Kidney Function, and Inflammatory Biomarker Profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. CJASN 2012, 7, 1938–1946. [Google Scholar] [CrossRef]
- Jassim, A.H.; Inman, D.M.; Mitchell, C.H. Crosstalk Between Dysfunctional Mitochondria and Inflammation in Glaucomatous Neurodegeneration. Front. Pharmacol. 2021, 12, 699623. [Google Scholar] [CrossRef]
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The Uremic Toxin Hippurate Promotes Endothelial Dysfunction via the Activation of Drp1-Mediated Mitochondrial Fission. Redox Biol. 2018, 16, 303–313. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Jeong, S.-Y.; Seol, D.-W. The Role of Mitochondria in Apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef]
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Ortiz, A. Regulated Cell Death Pathways in Kidney Disease. Nat. Rev. Nephrol. 2023, 19, 281–299. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release Mechanisms of Major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef]
- Junho, C.V.C.; Azevedo, C.A.B.; Da Cunha, R.S.; De Yurre, A.R.; Medei, E.; Stinghen, A.E.M.; Carneiro-Ramos, M.S. Heat Shock Proteins: Connectors between Heart and Kidney. Cells 2021, 10, 1939. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Cao, X.; Zou, J.; Shen, B.; Zhang, X.; Liu, Z.; Lv, W.; Teng, J.; Ding, X. Indoxyl Sulfate, a Valuable Biomarker in Chronic Kidney Disease and Dialysis. Hemodial. Int. Int. Symp. Home Hemodial. 2017, 21, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. BioMed Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Minciunescu, A.; Genovese, L.; deFilippi, C. Cardiovascular Alterations and Structural Changes in the Setting of Chronic Kidney Disease: A Review of Cardiorenal Syndrome Type 4. SN Compr. Clin. Med. 2022, 5, 15. [Google Scholar] [CrossRef]
- Delgado-Valero, B.; Cachofeiro, V.; Martínez-Martínez, E. Fibrosis, the Bad Actor in Cardiorenal Syndromes: Mechanisms Involved. Cells 2021, 10, 1824. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yisireyili, M.; Goto, S.; Cheng, X.W.; Nakayama, T.; Matsushita, T.; Niwa, T.; Murohara, T.; Takeshita, K. Indoxyl Sulfate Activates NLRP3 Inflammasome to Induce Cardiac Contractile Dysfunction Accompanied by Myocardial Fibrosis and Hypertrophy. Cardiovasc. Toxicol. 2022, 22, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.; Dos Santos, E.P.; Barbosa, M.R.; Arioni, S.; Santos, T.C.D.O.; Silva, D.T.D.R.E.; Agudelo, J.H.; Franco, M.P.; Fernandez, R.; Pereira, R.; et al. Thioredoxin-Interacting Protein: The Redoxissome Complex in Glomerular Lesion. Eur. J. Biol. 2022, 81, 274–280. [Google Scholar] [CrossRef]
- Nishida, K.; Watanabe, H.; Murata, R.; Tokumaru, K.; Fujimura, R.; Oshiro, S.; Nagasaki, T.; Miyahisa, M.; Hiramoto, Y.; Nosaki, H.; et al. Recombinant Long-Acting Thioredoxin Ameliorates AKI to CKD Transition via Modulating Renal Oxidative Stress and Inflammation. Int. J. Mol. Sci. 2021, 22, 5600. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Whayne, T.F.; Parinandi, N.; Maulik, N. Thioredoxins in Cardiovascular Disease. Can. J. Physiol. Pharmacol. 2015, 93, 903–911. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Itani, H.A.; Nazarewicz, R.R.; McMaster, W.G.; Flynn, C.R.; Uzhachenko, R.; Fessel, J.P.; Gamboa, J.L.; Harrison, D.G.; Dikalov, S.I. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ. Res. 2017, 121, 564–574. [Google Scholar] [CrossRef]
- Ho, H.-J.; Shirakawa, H. Oxidative Stress and Mitochondrial Dysfunction in Chronic Kidney Disease. Cells 2022, 12, 88. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, H.; Qiao, Z.; Wang, Y.; Zhang, P.; Yang, D. Loss of Thioredoxin 2 Alters Mitochondrial Respiratory Function and Induces Cardiomyocyte Hypertrophy. Exp. Cell Res. 2018, 372, 61–72. [Google Scholar] [CrossRef]
- Darwesh, A.M.; Jamieson, K.L.; Wang, C.; Samokhvalov, V.; Seubert, J.M. Cardioprotective Effects of CYP-Derived Epoxy Metabolites of Docosahexaenoic Acid Involve Limiting NLRP3 Inflammasome Activation. Can. J. Physiol. Pharmacol. 2019, 97, 544–556. [Google Scholar] [CrossRef]
- Shateri, H.; Manafi, B.; Tayebinia, H.; Karimi, J.; Khodadadi, I. Imbalance in Thioredoxin System Activates NLRP3 Inflammasome Pathway in Epicardial Adipose Tissue of Patients with Coronary Artery Disease. Mol. Biol. Rep. 2021, 48, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.M.; Shehatou, G.S.G.; Nader, M.A.; Abdelaziz, R.R. Piperine Mitigates Aortic Vasculopathy in Streptozotocin-Diabetic Rats via Targeting TXNIP-NLRP3 Signaling. Life Sci. 2023, 314, 121275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Peng, T.; Zhu, H.; Zheng, X.; Zhang, X.; Jiang, N.; Cheng, X.; Lai, X.; Shunnar, A.; Singh, M.; et al. Prevention of Hyperglycemia-Induced Myocardial Apoptosis by Gene Silencing of Toll-like Receptor-4. J. Transl. Med. 2010, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Bugyei-Twum, A.; Abadeh, A.; Thai, K.; Zhang, Y.; Mitchell, M.; Kabir, G.; Connelly, K.A. Suppression of NLRP3 Inflammasome Activation Ameliorates Chronic Kidney Disease-Induced Cardiac Fibrosis and Diastolic Dysfunction. Sci. Rep. 2016, 6, 39551. [Google Scholar] [CrossRef]
- Anders, H.-J.; Muruve, D.A. The Inflammasomes in Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 1007–1018. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Aranda-Rivera, A.K.; Osorio-Alonso, H.; Martínez-Klimova, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Extracellular Vesicles in Redox Signaling and Metabolic Regulation in Chronic Kidney Disease. Antioxidants 2022, 11, 356. [Google Scholar] [CrossRef]
- Liu, R.-M.; Desai, L.P. Reciprocal Regulation of TGF-β and Reactive Oxygen Species: A Perverse Cycle for Fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef]
- Song, B.; Estrada, K.D.; Lyons, K.M. Smad Signaling in Skeletal Development and Regeneration. Cytokine Growth Factor Rev. 2009, 20, 379–388. [Google Scholar] [CrossRef]
- Patel, B.; Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Prabhu, S.D. Mononuclear Phagocytes Are Dispensable for Cardiac Remodeling in Established Pressure-Overload Heart Failure. PLoS ONE 2017, 12, e0170781. [Google Scholar] [CrossRef]
- Gullestad, L.; Ueland, T.; Vinge, L.E.; Finsen, A.; Yndestad, A.; Aukrust, P. Inflammatory Cytokines in Heart Failure: Mediators and Markers. Cardiology 2012, 122, 23–35. [Google Scholar] [CrossRef]
- Korn, S.H.; Wouters, E.F.M.; Vos, N.; Janssen-Heininger, Y.M.W. Cytokine-Induced Activation of Nuclear Factor-κB Is Inhibited by Hydrogen Peroxide through Oxidative Inactivation of IκB Kinase. J. Biol. Chem. 2001, 276, 35693–35700. [Google Scholar] [CrossRef]
- An, N.; Gao, Y.; Si, Z.; Zhang, H.; Wang, L.; Tian, C.; Yuan, M.; Yang, X.; Li, X.; Shang, H.; et al. Regulatory Mechanisms of the NLRP3 Inflammasome, a Novel Immune-Inflammatory Marker in Cardiovascular Diseases. Front. Immunol. 2019, 10, 1592. [Google Scholar] [CrossRef] [PubMed]
- Mavrogonatou, E.; Konstantinou, A.; Kletsas, D. Long-Term Exposure to TNF-α Leads Human Skin Fibroblasts to a P38 MAPK- and ROS-Mediated Premature Senescence. Biogerontology 2018, 19, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.-Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial Reactive Oxygen Species Promote Production of Proinflammatory Cytokines and Are Elevated in TNFR1-Associated Periodic Syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Kouri, V.-P.; Olkkonen, J.; Nurmi, K.; Peled, N.; Ainola, M.; Mandelin, J.; Nordström, D.C.; Eklund, K.K. IL-17A and TNF Synergistically Drive Expression of Proinflammatory Mediators in Synovial Fibroblasts via IκBζ-Dependent Induction of ELF3. Rheumatology 2022, 62, 872–885. [Google Scholar] [CrossRef]
- Fan, Z.; Dong, J.; Mu, Y.; Liu, X. Nesfatin-1 Protects against Diabetic Cardiomyopathy in the Streptozotocin-Induced Diabetic Mouse Model via the P38-MAPK Pathway. Bioengineered 2022, 13, 14670–14681. [Google Scholar] [CrossRef] [PubMed]
- Tripepi, G.; Mallamaci, F.; Zoccali, C. Inflammation Markers, Adhesion Molecules, and All-Cause and Cardiovascular Mortality in Patients with ESRD: Searching for the Best Risk Marker by Multivariate Modeling. J. Am. Soc. Nephrol. JASN 2005, 16 (Suppl. 1), S83–S88. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Qureshi, A.R.; Heimbürger, O.; Barany, P.; Wang, K.; Pecoits-Filho, R.; Stenvinkel, P.; Lindholm, B. Serum Albumin, C-Reactive Protein, Interleukin 6, and Fetuin a as Predictors of Malnutrition, Cardiovascular Disease, and Mortality in Patients with ESRD. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2006, 47, 139–148. [Google Scholar] [CrossRef]
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 Inflammasome in the Pathogenesis of Cardiovascular Diseases. Basic Res. Cardiol. 2018, 113, 5. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef]
- Piccinini, A.M.; Midwood, K.S. DAMPening Inflammation by Modulating TLR Signalling. Mediat. Inflamm. 2010, 2010, 672395. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Xia, H.; Liang, Y.; Ye, Y.; Lu, Y.; Xu, X.; Duan, A.; He, J.; Chen, Z.; Wu, Y.; et al. Toll-like Receptor 9 Can Be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci. Rep. 2016, 6, 22579. [Google Scholar] [CrossRef] [PubMed]
- Garibotto, G.; Carta, A.; Picciotto, D.; Viazzi, F.; Verzola, D. Toll-like Receptor-4 Signaling Mediates Inflammation and Tissue Injury in Diabetic Nephropathy. J. Nephrol. 2017, 30, 719–727. [Google Scholar] [CrossRef]
- Verzola, D.; Bonanni, A.; Sofia, A.; Montecucco, F.; D’Amato, E.; Cademartori, V.; Parodi, E.L.; Viazzi, F.; Venturelli, C.; Brunori, G.; et al. Toll-like Receptor 4 Signalling Mediates Inflammation in Skeletal Muscle of Patients with Chronic Kidney Disease: Toll-like Receptor 4 in Muscle of Chronic Kidney Disease Patients. J. Cachexia Sarcopenia Muscle 2017, 8, 131–144. [Google Scholar] [CrossRef]
- Anders, H.-J.; Suarez-Alvarez, B.; Grigorescu, M.; Foresto-Neto, O.; Steiger, S.; Desai, J.; Marschner, J.A.; Honarpisheh, M.; Shi, C.; Jordan, J.; et al. The Macrophage Phenotype and Inflammasome Component NLRP3 Contributes to Nephrocalcinosis-Related Chronic Kidney Disease Independent from IL-1–Mediated Tissue Injury. Kidney Int. 2018, 93, 656–669. [Google Scholar] [CrossRef]
- Trentin-Sonoda, M.; Da Silva, R.C.; Kmit, F.V.; Abrahão, M.V.; Cahli, G.M.; Brasil, G.V.; Muzi-Filho, H.; Silva, P.A.; Tovar-Moll, F.F.; Vieyra, A.; et al. Knockout of Toll-Like Receptors 2 and 4 Prevents Renal Ischemia-Reperfusion-Induced Cardiac Hypertrophy in Mice. PLoS ONE 2015, 10, e0139350. [Google Scholar] [CrossRef]
- Wang, A.Y.-M.; Wang, M.; Woo, J.; Lam, C.W.-K.; Lui, S.-F.; Li, P.K.-T.; Sanderson, J.E. Inflammation, Residual Kidney Function, and Cardiac Hypertrophy Are Interrelated and Combine Adversely to Enhance Mortality and Cardiovascular Death Risk of Peritoneal Dialysis Patients. J. Am. Soc. Nephrol. JASN 2004, 15, 2186–2194. [Google Scholar] [CrossRef]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-Shock Protein 70 Inhibits Apoptosis by Preventing Recruitment of Procaspase-9 to the Apaf-1 Apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar] [CrossRef]
- Vega, V.L.; Rodríguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 Translocates into the Plasma Membrane after Stress and Is Released into the Extracellular Environment in a Membrane-Associated Form That Activates Macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, M.; Triantafilou, K. Heat-Shock Protein 70 and Heat-Shock Protein 90 Associate with Toll-like Receptor 4 in Response to Bacterial Lipopolysaccharide. Biochem. Soc. Trans. 2004, 32, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Kornej, J.; Reinhardt, C.; Kosiuk, J.; Arya, A.; Hindricks, G.; Adams, V.; Husser, D.; Bollmann, A. Response of Circulating Heat Shock Protein 70 and Anti-Heat Shock Protein 70 Antibodies to Catheter Ablation of Atrial Fibrillation. J. Transl. Med. 2013, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, Z.; Zhou, L.; Chen, Y.; He, M.; Cheng, L.; Hu, F.B.; Tanguay, R.M.; Wu, T. Plasma Levels of Hsp70 and Anti-Hsp70 Antibody Predict Risk of Acute Coronary Syndrome. Cell Stress Chaperones 2010, 15, 675–686. [Google Scholar] [CrossRef]
- Ramírez, V.; Mejía-Vilet, J.M.; Hernández, D.; Gamba, G.; Bobadilla, N.A. Radicicol, a Heat Shock Protein 90 Inhibitor, Reduces Glomerular Filtration Rate. Am. J. Physiol.-Ren. Physiol. 2008, 295, F1044–F1051. [Google Scholar] [CrossRef]
- Amador-Martínez, I.; Pérez-Villalva, R.; Uribe, N.; Cortés-González, C.; Bobadilla, N.A.; Barrera-Chimal, J. Reduced Endothelial Nitric Oxide Synthase Activation Contributes to Cardiovascular Injury during Chronic Kidney Disease Progression. Am. J. Physiol.-Ren. Physiol. 2019, 317, F275–F285. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and P38 Protein Kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Ries, M.; Schuster, P.; Thomann, S.; Donhauser, N.; Vollmer, J.; Schmidt, B. Identification of Novel Oligonucleotides from Mitochondrial DNA That Spontaneously Induce Plasmacytoid Dendritic Cell Activation. J. Leukoc. Biol. 2013, 94, 123–135. [Google Scholar] [CrossRef]
- Brinkmann, M.M.; Spooner, E.; Hoebe, K.; Beutler, B.; Ploegh, H.L.; Kim, Y.-M. The Interaction between the ER Membrane Protein UNC93B and TLR3, 7, and 9 Is Crucial for TLR Signaling. J. Cell Biol. 2007, 177, 265–275. [Google Scholar] [CrossRef]
- Fukui, R.; Saitoh, S.-I.; Kanno, A.; Onji, M.; Shibata, T.; Ito, A.; Onji, M.; Matsumoto, M.; Akira, S.; Yoshida, N.; et al. Unc93B1 Restricts Systemic Lethal Inflammation by Orchestrating Toll-like Receptor 7 and 9 Trafficking. Immunity 2011, 35, 69–81. [Google Scholar] [CrossRef]
- Sasai, M.; Linehan, M.M.; Iwasaki, A. Bifurcation of Toll-Like Receptor 9 Signaling by Adaptor Protein 3. Science 2010, 329, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Ewald, S.E.; Lee, B.L.; Lau, L.; Wickliffe, K.E.; Shi, G.-P.; Chapman, H.A.; Barton, G.M. The Ectodomain of Toll-like Receptor 9 Is Cleaved to Generate a Functional Receptor. Nature 2008, 456, 658–662. [Google Scholar] [CrossRef]
- Feng, W.; Zhang, K.; Liu, Y.; Chen, J.; Cai, Q.; He, W.; Zhang, Y.; Wang, M.-H.; Wang, J.; Huang, H. Advanced Oxidation Protein Products Aggravate Cardiac Remodeling via Cardiomyocyte Apoptosis in Chronic Kidney Disease. Am. J. Physiol.-Heart Circ. Physiol. 2018, 314, H475–H483. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Li, H.; Kim, M.; Shlomchik, M.J.; Lee, H.T. Kidney Proximal Tubular TLR9 Exacerbates Ischemic Acute Kidney Injury. J. Immunol. 2018, 201, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Bliksøen, M.; Mariero, L.H.; Torp, M.K.; Baysa, A.; Ytrehus, K.; Haugen, F.; Seljeflot, I.; Vaage, J.; Valen, G.; Stensløkken, K.-O. Extracellular mtDNA Activates NF-κB via Toll-like Receptor 9 and Induces Cell Death in Cardiomyocytes. Basic Res. Cardiol. 2016, 111, 42. [Google Scholar] [CrossRef]
- Baysa, A.; Maghazachi, A.A.; Sand, K.L.; Campesan, M.; Zaglia, T.; Mongillo, M.; Giorgio, M.; Di Lisa, F.; Gullestad, L.; Mariero, L.H.; et al. Toll-like Receptor 9 Signaling after Myocardial Infarction: Role of p66ShcA Adaptor Protein. Biochem. Biophys. Res. Commun. 2023, 644, 70–78. [Google Scholar] [CrossRef]
- McCarthy, C.G.; Wenceslau, C.F.; Goulopoulou, S.; Ogbi, S.; Baban, B.; Sullivan, J.C.; Matsumoto, T.; Webb, R.C. Circulating Mitochondrial DNA and Toll-like Receptor 9 Are Associated with Vascular Dysfunction in Spontaneously Hypertensive Rats. Cardiovasc. Res. 2015, 107, 119–130. [Google Scholar] [CrossRef]
- Lv, L.-L.; Feng, Y.; Tang, T.-T.; Liu, B.-C. New Insight into the Role of Extracellular Vesicles in Kidney Disease. J. Cell. Mol. Med. 2019, 23, 731–739. [Google Scholar] [CrossRef]
- Karpman, D.; Ståhl, A.; Arvidsson, I. Extracellular Vesicles in Renal Disease. Nat. Rev. Nephrol. 2017, 13, 545–562. [Google Scholar] [CrossRef]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide Signalling during Inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Dou, G.; Tian, R.; Liu, X.; Yuan, P.; Ye, Q.; Liu, J.; Liu, S.; Zhou, J.; Deng, Z.; Chen, X.; et al. Chimeric Apoptotic Bodies Functionalized with Natural Membrane and Modular Delivery System for Inflammation Modulation. Sci. Adv. 2020, 6, eaba2987. [Google Scholar] [CrossRef] [PubMed]
- Vénéreau, E.; Ceriotti, C.; Bianchi, M.E. DAMPs from Cell Death to New Life. Front. Immunol. 2015, 6, 422. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Zuo, Z.; Jing, K.; Wu, H.; Wang, S.; Ye, L.; Li, Z.; Yang, C.; Pan, Q.; Liu, W.J.; Liu, H.F. Mechanisms and Functions of Mitophagy and Potential Roles in Renal Disease. Front. Physiol. 2020, 11, 935. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Mohammad, N.S.; Nazli, R.; Zafar, H.; Fatima, S. Effects of lipid based Multiple Micronutrients Supplement on the birth outcome of underweight pre-eclamptic women: A randomized clinical trial. Pak. J. Med. Sci. 2022, 38, 219–226. [Google Scholar] [CrossRef]
- Gao, Y.; Dai, X.; Li, Y.; Li, G.; Lin, X.; Ai, C.; Cao, Y.; Li, T.; Lin, B. Role of Parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J. Transl. Med. 2020, 18, 114. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef]
- Zhuang, Y.; Ding, G.; Zhao, M.; Bai, M.; Yang, L.; Ni, J.; Wang, R.; Jia, Z.; Huang, S.; Zhang, A. NLRP3 inflammasome mediates albumin-induced renal tubular injury through impaired mitochondrial function. J. Biol. Chem. 2014, 289, 25101–25111. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Liu, T.; Bi, X.; Zhang, Z. Mitochondria-Targeted Antioxidant Mito-Tempo Protects Against Aldosterone-Induced Renal Injury In Vivo. Cell. Physiol. Biochem. 2017, 44, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Song, Y.; Chen, L.; Chen, P.; Yuan, M.; Meng, Y.; Wang, Q.; Zheng, G.; Qiu, Z. Urolithin A Attenuates Hyperuricemic Nephropathy in Fructose-Fed Mice by Impairing STING-NLRP3 Axis-Mediated Inflammatory Response via Restoration of Parkin-Dependent Mitophagy. Front. Pharmacol. 2022, 13, 907209. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, X.; Du, W.; Xue, Z.; Huang, W.; Guan, Z.; Wang, H. BI-1 ameliorates myocardial injury by activating the mitochondrial unfolded protein response and FUNDC1-related mitophagy in cardiorenal syndrome type 3. Cell. Signal. 2022, 91, 110218. [Google Scholar] [CrossRef]
- Khan, S.; Basu, S.; Raj, D.; Lahiri, A. Role of Mitochondria in Regulating Immune Response during Bacterial Infection. Int. Rev. Cell Mol. Biol. 2023, 374, 159–200. [Google Scholar] [CrossRef]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Srivastava, A.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Mulay, S.R.; Scholze, A. Involvement of Inflammasome Components in Kidney Disease. Antioxidants 2022, 11, 246. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein That Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The Mitochondrial Antiviral Protein MAVS Associates with NLRP3 and Regulates Its Inflammasome Activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, W.; Gong, F.; Chen, Y.; Chen, E. The Role and Mechanism of Pyroptosis and Potential Therapeutic Targets in Sepsis: A Review. Front. Immunol. 2021, 12, 711939. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, W.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis Is Driven by Non-Selective Gasdermin-D Pore and Its Morphology Is Different from MLKL Channel-Mediated Necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.-D. Mitochondria: Diversity in the Regulation of the NLRP3 Inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, Z.-M.; Ji, J.-L.; Gan, W.; Zhang, A.; Shi, H.-J.; Wang, H.; Lv, L.; Li, Z.; Tang, T.; et al. Macrophage-Derived Exosomal Mir-155 Regulating Cardiomyocyte Pyroptosis and Hypertrophy in Uremic Cardiomyopathy. JACC Basic Transl. Sci. 2020, 5, 148–166. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, C.; Rudemiller, N.P.; Abais, J.M.; Mattson, D.L. Inflammation and Hypertension: New Understandings and Potential Therapeutic Targets. Curr. Hypertens. Rep. 2015, 17, 507. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Ma, X.M.; Geng, K.; Law, B.Y.-K.; Wang, P.; Pu, Y.L.; Chen, Q.; Xu, H.W.; Tan, X.Z.; Jiang, Z.Z.; Xu, Y. Lipotoxicity-Induced mtDNA Release Promotes Diabetic Cardiomyopathy by Activating the cGAS-STING Pathway in Obesity-Related Diabetes. Cell Biol. Toxicol. 2022, 39, 277–299. [Google Scholar] [CrossRef]
- Zang, N.; Cui, C.; Guo, X.; Song, J.; Hu, H.; Yang, M.; Xu, M.; Wang, L.; Hou, X.; He, Q.; et al. cGAS-STING Activation Contributes to Podocyte Injury in Diabetic Kidney Disease. iScience 2022, 25, 105145. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 2015, 18, 157–168. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.-M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273.e6. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Dhillon, P.; Huang, S.; Sheng, X.; Shrestha, R.; Qiu, C.; Kaufman, B.A.; Park, J.; Pei, L.; Baur, J.; et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019, 30, 784–799.e5. [Google Scholar] [CrossRef] [PubMed]
- Mitrofanova, A.; Fontanella, A.; Tolerico, M.; Mallela, S.; David, J.M.; Zuo, Y.; Boulina, M.; Kim, J.-J.; Santos, J.; Ge, M.; et al. Activation of Stimulator of IFN Genes (STING) Causes Proteinuria and Contributes to Glomerular Diseases. J. Am. Soc. Nephrol. 2022, 33, 2153–2173. [Google Scholar] [CrossRef]
- Nishimoto, S.; Sata, M.; Fukuda, D. Expanding Role of Deoxyribonucleic Acid-Sensing Mechanism in the Development of Lifestyle-Related Diseases. Front. Cardiovasc. Med. 2022, 9, 881181. [Google Scholar] [CrossRef]
- Gong, W.; Lu, L.; Zhou, Y.; Liu, J.; Ma, H.; Fu, L.; Huang, S.; Zhang, Y.; Zhang, A.; Jia, Z. The Novel STING Antagonist H151 Ameliorates Cisplatin-Induced Acute Kidney Injury and Mitochondrial Dysfunction. Am. J. Physiol.-Ren. Physiol. 2021, 320, F608–F616. [Google Scholar] [CrossRef]
- Hernández-Reséndiz, S.; Correa, F.; García-Niño, W.R.; Buelna-Chontal, M.; Roldán, F.J.; Ramírez-Camacho, I.; Delgado-Toral, C.; Carbó, R.; Pedraza-Chaverrí, J.; Tapia, E.; et al. Cardioprotection by Curcumin Post-Treatment in Rats with Established Chronic Kidney Disease. Cardiovasc. Drugs Ther. 2015, 29, 111–120. [Google Scholar] [CrossRef]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond–Mitochondrial Performance in Apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Renault, T.T.; Chipuk, J.E. Death upon a Kiss: Mitochondrial Outer Membrane Composition and Organelle Communication Govern Sensitivity to BAK/BAX-Dependent Apoptosis. Chem. Biol. 2014, 21, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, K.; Hertlein, V.; Jenner, A.; Dellmann, T.; Gojkovic, M.; Peña-Blanco, A.; Dadsena, S.; Wajngarten, N.; Danial, J.S.H.; Thevathasan, J.V.; et al. The Interplay between BAX and BAK Tunes Apoptotic Pore Growth to Control Mitochondrial-DNA-Mediated Inflammation. Mol. Cell 2022, 82, 933–949.e9. [Google Scholar] [CrossRef] [PubMed]
- Khedr, S.; Dissanayake, L.V.; Palygin, O.; Staruschenko, A. Potential Role of cGAS-STING Pathway in the Induction of Diabetic Kidney Disease. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Gulen, M.F.; Koch, U.; Haag, S.M.; Schuler, F.; Apetoh, L.; Villunger, A.; Radtke, F.; Ablasser, A. Signalling Strength Determines Proapoptotic Functions of STING. Nat. Commun. 2017, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 Mitigate STING-Induced Inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Chen, K.; Dai, H.; Yuan, J.; Chen, J.; Lin, L.; Zhang, W.; Wang, L.; Zhang, J.; Li, K.; He, Y. Optineurin-Mediated Mitophagy Protects Renal Tubular Epithelial Cells against Accelerated Senescence in Diabetic Nephropathy. Cell Death Dis. 2018, 9, 105. [Google Scholar] [CrossRef]
- Tang, C.; He, L.; Liu, J.; Dong, Z. Mitophagy: Basic Mechanism and Potential Role in Kidney Diseases. Kidney Dis. 2015, 1, 71–79. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Kwak, M.-J.; Ge, J.; Rodgers, M.A.; Shi, M.; Leslie, B.J.; Hopfner, K.-P.; Ha, T.; et al. Crosstalk between the cGAS DNA Sensor and Beclin-1 Autophagy Protein Shapes Innate Antimicrobial Immune Responses. Cell Host Microbe 2014, 15, 228–238. [Google Scholar] [CrossRef]
- Liu, C.; Wang, X.; Wang, X.; Zhang, Y.; Min, W.; Yu, P.; Miao, J.; Shen, W.; Chen, S.; Zhou, S.; et al. A New LKB1 Activator, Piericidin Analogue S14, Retards Renal Fibrosis through Promoting Autophagy and Mitochondrial Homeostasis in Renal Tubular Epithelial Cells. Theranostics 2022, 12, 7158–7179. [Google Scholar] [CrossRef]
- Escames, G.; López, L.C.; García, J.A.; García-Corzo, L.; Ortiz, F.; Acuña-Castroviejo, D. Mitochondrial DNA and Inflammatory Diseases. Hum. Genet. 2012, 131, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Nakashima, R.; Matsuda, K.; Okamaoto, K.; Hirano, R.; Kawasaki, H.; Miuma, S.; Miyaaki, H.; Malhi, H.; Abiru, S.; et al. Detection of DNA Damage Response in Nonalcoholic Fatty Liver Disease via P53-Binding Protein 1 Nuclear Expression. Mod. Pathol. 2019, 32, 997–1007. [Google Scholar] [CrossRef]
- Wang, X.; Rao, H.; Zhao, J.; Wee, A.; Li, X.; Fei, R.; Huang, R.; Wu, C.; Liu, F.; Wei, L. STING Expression in Monocyte-Derived Macrophages Is Associated with the Progression of Liver Inflammation and Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Lab. Investig. 2020, 100, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C. Inflammatory Markers and Type 2 Diabetes. Diabetes Technol. Ther. 2006, 8, 1–6. [Google Scholar] [CrossRef]
- Bhandari, S. Risk Factors and Metabolic Mechanisms in the Pathogenesis of Uraemic Cardiac Disease. Front. Biosci. 2011, 16, 1364–1387. [Google Scholar] [CrossRef]
- Charo, I.F.; Ransohoff, R.M. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Bandow, K.; Kusuyama, J.; Shamoto, M.; Kakimoto, K.; Ohnishi, T.; Matsuguchi, T. LPS-Induced Chemokine Expression in Both MyD88-Dependent and -Independent Manners Is Regulated by Cot/Tpl2-ERK Axis in Macrophages. FEBS Lett. 2012, 586, 1540–1546. [Google Scholar] [CrossRef]
- Nomiyama, H.; Osada, N.; Yoshie, O. Systematic Classification of Vertebrate Chemokines Based on Conserved Synteny and Evolutionary History. Genes Cells 2013, 18, 1–16. [Google Scholar] [CrossRef]
- Chang, T.-T.; Chen, C.; Chen, J.-W. CCL7 as a Novel Inflammatory Mediator in Cardiovascular Disease, Diabetes Mellitus, and Kidney Disease. Cardiovasc. Diabetol. 2022, 21, 185. [Google Scholar] [CrossRef]
- Miao, M.; De Clercq, E.; Li, G. Clinical Significance of Chemokine Receptor Antagonists. Expert Opin. Drug Metab. Toxicol. 2020, 16, 11–30. [Google Scholar] [CrossRef] [PubMed]
- DeVries, M.E.; Kelvin, A.A.; Xu, L.; Ran, L.; Robinson, J.; Kelvin, D.J. Defining the Origins and Evolution of the Chemokine/Chemokine Receptor System. J. Immunol. 2006, 176, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-T.; Chen, J.-W. The Role of Chemokines and Chemokine Receptors in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 3172. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A. NF-κB, Chemokine Gene Transcription and Tumour Growth. Nat. Rev. Immunol. 2002, 2, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Moser, B.; Loetscher, P. Lymphocyte Traffic Control by Chemokines. Nat. Immunol. 2001, 2, 123–128. [Google Scholar] [CrossRef]
- Dimeloe, S.; Burgener, A.; Grählert, J.; Hess, C. T-cell Metabolism Governing Activation, Proliferation and Differentiation; A Modular View. Immunology 2017, 150, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.L.; Hogaboam, C.M.; Kunkel, S.L. Chemokines Provide the Sustained Inflammatory Bridge between Innate and Acquired Immunity. Cytokine Growth Factor Rev. 2005, 16, 553–560. [Google Scholar] [CrossRef]
- Segerer, S.; Nelson, P.J.; Schlöndorff, D. Chemokines, Chemokine Receptors, and Renal Disease: From Basic Science to Pathophysiologic and Therapeutic Studies. J. Am. Soc. Nephrol. 2000, 11, 152–176. [Google Scholar] [CrossRef]
- Chung, A.C.K.; Lan, H.Y. Chemokines in Renal Injury. J. Am. Soc. Nephrol. 2011, 22, 802–809. [Google Scholar] [CrossRef]
- Galliera, E.; Corsi, M.; Bonecchi, R.; Locati, M.; Mantovani, A. Chemokines as Pharmacological Targets. Mini-Rev. Med. Chem. 2008, 8, 638–646. [Google Scholar] [CrossRef]
- Vielhauer, V.; Anders, H.-J. Chemokines and Chemokine Receptors as Therapeutic Targets in Chronic Kidney Disease. Front. Biosci. 2009, S1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.W.; Lai, K.N. The Pathogenic Role of the Renal Proximal Tubular Cell in Diabetic Nephropathy. Nephrol. Dial. Transplant. 2012, 27, 3049–3056. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J.; Ryu, M. Renal Microenvironments and Macrophage Phenotypes Determine Progression or Resolution of Renal Inflammation and Fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Weidenbusch, M.; Anders, H.-J. Tissue Microenvironments Define and Get Reinforced by Macrophage Phenotypes in Homeostasis or during Inflammation, Repair and Fibrosis. J. Innate Immun. 2012, 4, 463–477. [Google Scholar] [CrossRef]
- Wang, Y.; Rangan, G.K.; Tay, Y.-C.; Wang, Y.; Harris, D.C.H. Induction of Monocyte Chemoattractant Protein-1 by Albumin Is Mediated by Nuclear Factor κB in Proximal Tubule Cells. J. Am. Soc. Nephrol. 1999, 10, 1204–1213. [Google Scholar] [CrossRef]
- Zoja, C.; Donadelli, R.; Colleoni, S.; Figliuzzi, M.; Bonazzola, S.; Morigi, M.; Remuzzi, G. Protein Overload Stimulates RANTES Production by Proximal Tubular Cells Depending on NF-kB Activation. Kidney Int. 1998, 53, 1608–1615. [Google Scholar] [CrossRef]
- Huang, X.R.; Chung, A.C.K.; Zhou, L.; Wang, X.J.; Lan, H.Y. Latent TGF-β1 Protects against Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. JASN 2008, 19, 233–242. [Google Scholar] [CrossRef]
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral Obstruction as a Model of Renal Interstitial Fibrosis and Obstructive Nephropathy. Kidney Int. 2009, 75, 1145–1152. [Google Scholar] [CrossRef]
- Chung, A.C.K.; Huang, X.R.; Zhou, L.; Heuchel, R.; Lai, K.N.; Lan, H.Y. Disruption of the Smad7 Gene Promotes Renal Fibrosis and Inflammation in Unilateral Ureteral Obstruction (UUO) in Mice. Nephrol. Dial. Transplant. 2009, 24, 1443–1454. [Google Scholar] [CrossRef]
- Okada, H.; Moriwaki, K.; Kalluri, R.; Imai, H.; Ban, S.; Takahama, M.; Suzuki, H. Inhibition of Monocyte Chemoattractant Protein-1 Expression in Tubular Epithelium Attenuates Tubulointerstitial Alteration in Rat Goodpasture Syndrome. Kidney Int. 2000, 57, 927–936. [Google Scholar] [CrossRef]
- Panzer, U.; Steinmetz, O.M.; Reinking, R.R.; Meyer, T.N.; Fehr, S.; Schneider, A.; Zahner, G.; Wolf, G.; Helmchen, U.; Schaerli, P.; et al. Compartment-Specific Expression and Function of the Chemokine IP-10/CXCL10 in a Model of Renal Endothelial Microvascular Injury. J. Am. Soc. Nephrol. 2006, 17, 454–464. [Google Scholar] [CrossRef] [PubMed]
- DeVrieze, B.W.; Hurley, J.A. Goodpasture Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Papayianni, A.; Alexopoulos, E.; Giamalis, P.; Gionanlis, L.; Belechri, A.; Koukoudis, P.; Memmos, D. Circulating Levels of ICAM-1, VCAM-1, and MCP-1 Are Increased in Haemodialysis Patients: Association with Inflammation, Dyslipidaemia, and Vascular Events. Nephrol. Dial. Transplant. 2002, 17, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. T Helper Cell Differentiation, Heterogeneity, and Plasticity. Cold Spring Harb. Perspect. Biol. 2018, 10, a030338. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Takahashi, N.; Ushiki, M.; Monya, M.; Aihara, F.; Kuboki, E.; Moriyama, S.; Iida, M.; Kitamura, H.; Qiu, C.-H.; et al. Intestinal CD169+ Macrophages Initiate Mucosal Inflammation by Secreting CCL8 That Recruits Inflammatory Monocytes. Nat. Commun. 2015, 6, 7802. [Google Scholar] [CrossRef]
- Islam, S.A.; Chang, D.S.; Colvin, R.A.; Byrne, M.H.; McCully, M.L.; Moser, B.; Lira, S.A.; Charo, I.F.; Luster, A.D. Mouse CCL8, a CCR8 Agonist, Promotes Atopic Dermatitis by Recruiting IL-5+ TH2 Cells. Nat. Immunol. 2011, 12, 167–177. [Google Scholar] [CrossRef]
- Akchurin, O.M.; Kaskel, F. Update on Inflammation in Chronic Kidney Disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.; Park, M.; Seo, A.; Kim, K.H.; Kim, S.; Kang, M.; Kang, E.; Yoo, K.D.; Lee, S.; et al. Inflammatory Chemokine (C-C Motif) Ligand 8 Inhibition Ameliorates Peritoneal Fibrosis. FASEB J. 2023, 37, e22632. [Google Scholar] [CrossRef]
- Dangi, A.; Husain, I.; Jordan, C.Z.; Yu, S.; Natesh, N.; Shen, X.; Kwun, J.; Luo, X. Blocking CCL8-CCR8–Mediated Early Allograft Inflammation Improves Kidney Transplant Function. J. Am. Soc. Nephrol. 2022, 33, 1876–1890. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Stiles, J.K. The Emerging Role of CXCL10 in Cancer (Review). Oncol. Lett. 2011, 2, 583–589. [Google Scholar] [CrossRef]
- Luster, A.D.; Ravetch, J.V. Biochemical Characterization of a Gamma Interferon-Inducible Cytokine (IP-10). J. Exp. Med. 1987, 166, 1084–1097. [Google Scholar] [CrossRef]
- Dyer, K.D.; Percopo, C.M.; Fischer, E.R.; Gabryszewski, S.J.; Rosenberg, H.F. Pneumoviruses Infect Eosinophils and Elicit MyD88-Dependent Release of Chemoattractant Cytokines and Interleukin-6. Blood 2009, 114, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, G. Mitochondrial DNA in Uremia and New Targets to Treat Myocardial Hypertrophy in the Cardiorenal Syndrome. JACC Basic Transl. Sci. 2022, 7, 841–843. [Google Scholar] [CrossRef]
- Niwa, T. Role of Indoxyl Sulfate in the Progression of Chronic Kidney Disease and Cardiovascular Disease: Experimental and Clinical Effects of Oral Sorbent AST-120. Ther. Apher. Dial. 2011, 15, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl Sulfate Upregulates Expression of ICAM-1 and MCP-1 by Oxidative Stress-Induced NF-κB Activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Behr, T.M.; Wang, X.; Aiyar, N.; Coatney, R.W.; Li, X.; Koster, P.; Angermann, C.E.; Ohlstein, E.; Feuerstein, G.Z.; Winaver, J. Monocyte Chemoattractant Protein-1 Is Upregulated in Rats With Volume-Overload Congestive Heart Failure. Circulation 2000, 102, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, K.; Chen, F.; Liu, Q.; Ni, J.; Cao, W.; Hua, Y.; He, F.; Liu, Z.; Li, L.; et al. Role of the CCL2-CCR2 Axis in Cardiovascular Disease: Pathogenesis and Clinical Implications. Front. Immunol. 2022, 13, 975367. [Google Scholar] [CrossRef]
- Hayashidani, S.; Tsutsui, H.; Shiomi, T.; Ikeuchi, M.; Matsusaka, H.; Suematsu, N.; Wen, J.; Egashira, K.; Takeshita, A. Anti-Monocyte Chemoattractant Protein-1 Gene Therapy Attenuates Left Ventricular Remodeling and Failure After Experimental Myocardial Infarction. Circulation 2003, 108, 2134–2140. [Google Scholar] [CrossRef]
- Xia, Y.; Frangogiannis, N.G. MCP-1/CCL2 as a Therapeutic Target in Myocardial Infarction and Ischemic Cardiomyopathy. Inflamm. Allergy-Drug Targets 2007, 6, 101–107. [Google Scholar] [CrossRef]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Takeya, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Hypertensive Myocardial Fibrosis and Diastolic Dysfunction: Another Model of Inflammation? Hypertension 2004, 43, 739–745. [Google Scholar] [CrossRef]
- Majmudar, M.D.; Keliher, E.J.; Heidt, T.; Leuschner, F.; Truelove, J.; Sena, B.F.; Gorbatov, R.; Iwamoto, Y.; Dutta, P.; Wojtkiewicz, G.; et al. Monocyte-Directed RNAi Targeting CCR2 Improves Infarct Healing in Atherosclerosis-Prone Mice. Circulation 2013, 127, 2038–2046. [Google Scholar] [CrossRef]
- Dimas, G.G.; Didangelos, T.P.; Grekas, D.M. Matrix Gelatinases in Atherosclerosis and Diabetic Nephropathy: Progress and Challenges. Curr. Vasc. Pharmacol. 2017, 15, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.H.; Xie, D.; Kelly, K.J. Cardiac effects of renal ischemia. Am. J. Physiol.-Ren. Physiol. 2023, 324, F64–F74. [Google Scholar] [CrossRef] [PubMed]
- Hobson, S.; Arefin, S.; Witasp, A.; Hernandez, L.; Kublickiene, K.; Shiels, P.G.; Stenvinkel, P. Accelerated Vascular Aging in Chronic Kidney Disease: The Potential for Novel Therapies. Circ. Res. 2023, 132, 950–969. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Guan, W.; Zhang, Y.; Deng, Q.; Li, J.; Ye, H.; Deng, S.; Han, W.; Yu, Y. Large-Scale Gene Analysis of Rabbit Atherosclerosis to Discover New Biomarkers for Coronary Artery Disease. Open Biol. 2019, 9, 180238. [Google Scholar] [CrossRef]
- Ardigo, D.; Assimes, T.L.; Fortmann, S.P.; Go, A.S.; Hlatky, M.; Hytopoulos, E.; Iribarren, C.; Tsao, P.S.; Tabibiazar, R.; Quertermous, T. Circulating Chemokines Accurately Identify Individuals with Clinically Significant Atherosclerotic Heart Disease. Physiol. Genom. 2007, 31, 402–409. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Mach, F.; Steffens, S. The Specific Role of Chemokines in Atherosclerosis. Thromb. Haemost. 2007, 97, 714–721. [Google Scholar] [CrossRef]
- Dai, S.; Zhang, J.; Xu, Z. Silencing CCL8 Inhibited the Proliferation and Migration of PDGF-BB-Stimulated Human Aortic Smooth Muscle Cells. Biosci. Biotechnol. Biochem. 2020, 84, 1585–1593. [Google Scholar] [CrossRef]
- Van den Borne, P.; Quax, P.H.A.; Hoefer, I.E.; Pasterkamp, G. The Multifaceted Functions of CXCL10 in Cardiovascular Disease. BioMed Res. Int. 2014, 2014, 893106. [Google Scholar] [CrossRef]
- Altara, R.; Mallat, Z.; Booz, G.W.; Zouein, F.A. The CXCL10/CXCR3 Axis and Cardiac Inflammation: Implications for Immunotherapy to Treat Infectious and Noninfectious Diseases of the Heart. J. Immunol. Res. 2016, 2016, 4396368. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRS Subtype | Description | References |
|---|---|---|
| Type 1 | It develops when there is an acute deterioration of cardiac function due to conditions such as cardiogenic shock, ADHF, cardiac surgery, and acute coronary syndrome leading to AKI (defined by an increase in serum creatinine ≥ 0.3 mg/dL) or renal dysfunction. | [11,12] |
| Type 2 | It characterizes chronic CVD, such as chronic HF, that leads to CKD. CKD increases the frequency of hospitalizations and deaths from pump failure and arrhythmia. | [2,13,14] |
| Type 3 | Describes a sudden worsening of renal function, such as AKI or glomerulonephritis, causing acute cardiac dysfunction (e.g., HF, arrhythmia, or pulmonary edema). | [15,16] |
| Type 4 | It defines CKD as leading to the progression of CVD. CVD may include decreased cardiac function, diastolic dysfunction, ventricular hypertrophy, or increased risk of adverse cardiovascular events due to pressure and fluid overload, representing a risk factor for death. | [17,18] |
| Type 5 | This syndrome appears when an acute or chronic systemic disease such as diabetes mellitus, sepsis, systemic lupus erythematosus, vasculitis, and sarcoidosis, leads to simultaneous cardiac and renal dysfunction. | [11] |
| Chemokine/ Chemokine Receptor | Inhibitory Strategy | Species (Mice or Rats) | CKD Type | Beneficial Effects | Reference |
|---|---|---|---|---|---|
| MCP-1/ CCL2 | Blocking of MCP-1 by injecting antisense oligodeoxynucleotide | Rats | Two months model of Goodpasteur syndrome. | ↓ MCP-1 mRNA. ↓ Mononuclear cell infiltration. ↓ Monocity/macrophages in the interstitium. ↓ Tubulointerstitial damage. | [272] |
| CCL8 | Anti-CCL8 mAb | Mice | UUO mouse model (14 days). End-stage CKD in the obstructed kidney. | ↓ Fibrosis and apoptosis. ↓ E-cadherin and BCL-2. ↓ Fibronectin. ↓ CCR2. | [50] |
| CXCL10/ IP-10 | Inhibition by anti-IP-10/CXCL10 antibody | Rats | Rat model of renal endothelial microvascular injury in CKD. | ↓ Tubulointerstitial T cell recruitment. Improved renal function. ↓ Serum creatinine. ↓ BUN. | [273] |
| Chemokine/ Chemokine Receptor | Inhibitory Strategy | Species (Mice or Rats) | Model | Beneficial Effects | References |
|---|---|---|---|---|---|
| CCL8 | Anti-CCL8 in early CKD | Mice | Uremic cardiomyopathy induced by 5/6 nephrectomy. | ↓ Attenuated infiltration of TCD4+, lymphocytes and macrophages. ↓ Cardiac remodeling. ↓ Inflammation. ↓ Cardiac dysfunction. | [52] |
| CXCL10 | CCR2-/- mice or Anti-CXCL10 antibody | Mice | Uremic cardiomyopathy induced by 5/6 nephrectomy or intraperitoneal folate (240 mg/kg body weight). | ↓ Monocyte infiltration in the heart. ↓ Cardiac alterations. ↓ Macrophage local proliferation. ↓ Cardiac hypertrophy. ↓ Cardiac dysfunction. | [53] |
| PRR Type | DAMPs or tDAMPs That Activate PRRs | Associated Cytokines/ Chemokines | Therapeutic Effect of the PPR Inhibition in the CRS Type 4 | References |
|---|---|---|---|---|
| NLRP3 | ↑ ROS Extracellular ATP Cardiolipin EVs Apoptotic bodies | IL-1β, IL-18, TGF-β, TNF-α CCL8, CXCL10 | ↓ Cardiac dysfunction ↓ Cardiac fibrosis ↓ Cardiac hypertrophy | [121,135,136,142,144,145,146,149,158,192]. |
| TLR2/ TLR4 | Cell debris Nucleic acid fragments ↑ Oxidative products ↑ Uremic toxins HSPs | IL-1β, IL-18, IFN-γ, IL-6, TNF-α CCL2, CCL8 | ↓ Cardiac hypertrophy ↓ BNP levels ↓ α-actin levels | [45,71,131,145,167,168,169,170,171,172,173,174,175,176]. |
| TLR9 | mtDNA EVs | IL-1β, IL-18, IL-6 Chemokines? | ? | [180,181,182,187,189,193]. |
| cGAS-STING | dsDNA Neutrophil DNA-protein complexes mtDNA | IL-18, IL-1β CCL8, CXCL10 | ↓ Cardiac hypertrophy | [44,217,218,226,233]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amador-Martínez, I.; Aparicio-Trejo, O.E.; Bernabe-Yepes, B.; Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Mitochondrial Impairment: A Link for Inflammatory Responses Activation in the Cardiorenal Syndrome Type 4. Int. J. Mol. Sci. 2023, 24, 15875. https://doi.org/10.3390/ijms242115875
Amador-Martínez I, Aparicio-Trejo OE, Bernabe-Yepes B, Aranda-Rivera AK, Cruz-Gregorio A, Sánchez-Lozada LG, Pedraza-Chaverri J, Tapia E. Mitochondrial Impairment: A Link for Inflammatory Responses Activation in the Cardiorenal Syndrome Type 4. International Journal of Molecular Sciences. 2023; 24(21):15875. https://doi.org/10.3390/ijms242115875
Chicago/Turabian StyleAmador-Martínez, Isabel, Omar Emiliano Aparicio-Trejo, Bismarck Bernabe-Yepes, Ana Karina Aranda-Rivera, Alfredo Cruz-Gregorio, Laura Gabriela Sánchez-Lozada, José Pedraza-Chaverri, and Edilia Tapia. 2023. "Mitochondrial Impairment: A Link for Inflammatory Responses Activation in the Cardiorenal Syndrome Type 4" International Journal of Molecular Sciences 24, no. 21: 15875. https://doi.org/10.3390/ijms242115875
APA StyleAmador-Martínez, I., Aparicio-Trejo, O. E., Bernabe-Yepes, B., Aranda-Rivera, A. K., Cruz-Gregorio, A., Sánchez-Lozada, L. G., Pedraza-Chaverri, J., & Tapia, E. (2023). Mitochondrial Impairment: A Link for Inflammatory Responses Activation in the Cardiorenal Syndrome Type 4. International Journal of Molecular Sciences, 24(21), 15875. https://doi.org/10.3390/ijms242115875