
Involvement of Lipophagy and Chaperone-Mediated Autophagy in the Pathogenesis of Non-Alcoholic Fatty Liver Disease by Regulation of Lipid Droplets

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
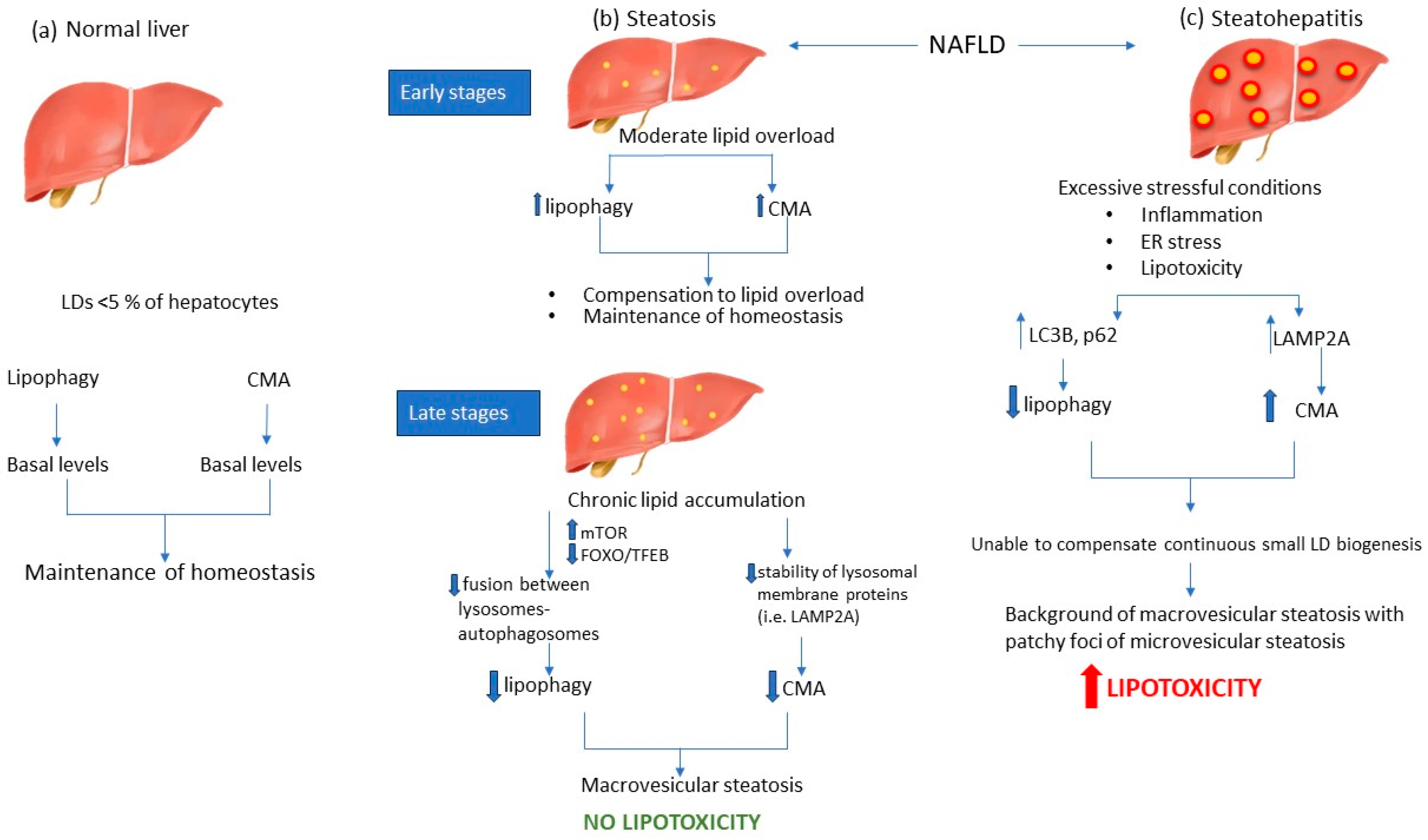
2. Pathogenesis of NAFLD
3. Role of LDs in NAFLD Progression
3.1. Biogenesis of LDs
3.2. Structural Changes in LDs Affecting NAFLD Progression
3.3. Perilipins: The Gatekeeper of LDs Surface and Their Role in NAFLD
3.4. Breakdown of Lipid Droplets
3.4.1. Degradation of PLINs via CMA
3.4.2. Degradation of the Lipid Core via Lipolysis and Lipophagy
4. Autophagy Modulation during NAFLD
4.1. Lipophagy Modulation during NAFLD
4.2. CMA Is Involved in LD Degradation during NAFLD
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Grefhorst, A.; van de Peppel, I.P.; Larsen, L.E.; Jonker, J.W.; Holleboom, A.G. The Role of Lipophagy in the Development and Treatment of Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2020, 11, 601627. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 451–496. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y. Review article: Is non-alcoholic fatty liver disease a spectrum, or are steatosis and non-alcoholic steatohepatitis distinct conditions? Aliment. Pharmacol. Ther. 2012, 36, 815–823. [Google Scholar] [CrossRef]
- Loomba, R.; Adams, L.A. The 20% Rule of NASH Progression: The Natural History of Advanced Fibrosis and Cirrhosis Caused by NASH. Hepatology 2019, 70, 1885–1888. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Ding, H.; Ge, G.; Tseng, Y.; Ma, Y.; Zhang, J.; Liu, J. Hepatic autophagy fluctuates during the development of non-alcoholic fatty liver disease. Ann. Hepatol. 2020, 19, 516–522. [Google Scholar] [CrossRef]
- Niture, S.; Lin, M.; Rios-Colon, L.; Qi, Q.; Moore, J.T.; Kumar, D. Emerging Roles of Impaired Autophagy in Fatty Liver Disease and Hepatocellular Carcinoma. Int. J. Hepatol. 2021, 2021, 6675762. [Google Scholar] [CrossRef]
- Tasset, I.; Cuervo, A.M. Role of chaperone-mediated autophagy in metabolism. FEBS J. 2016, 283, 2403–2413. [Google Scholar] [CrossRef]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Autophagy in health and disease. 1. Regulation and significance of autophagy: An overview. Am. J. Physiol. Physiol. 2010, 298, C776–C785. [Google Scholar] [CrossRef] [PubMed]
- Carotti, S.; Aquilano, K.; Zalfa, F.; Ruggiero, S.; Valentini, F.; Zingariello, M.; Francesconi, M.; Perrone, G.; Alletto, F.; Antonelli-Incalzi, R.; et al. Lipophagy Impairment Is Associated With Disease Progression in NAFLD. Front. Physiol. 2020, 11, 850. [Google Scholar] [CrossRef] [PubMed]
- Hubert, V.; Weiss, S.; Rees, A.J.; Kain, R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells 2022, 11, 2562. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Czaja, M.J. Autophagy in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Matos, A.F.; Júnior, W.S.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Walther, T.C.; Chung, J.; Farese, R.V., Jr. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef]
- Barrows, B.R.; Parks, E.J. Contributions of Different Fatty Acid Sources to Very Low-Density Lipoprotein-Triacylglycerol in the Fasted and Fed States. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease—Revisited after a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K. Role of Insulin Resistance and Lipotoxicity in Non-Alcoholic Steatohepatitis. Clin. Liver Dis. 2009, 13, 545–563. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut–liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef]
- Feldstein, A.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; Sauver, J.S.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The Natural History of Nonalcoholic Fatty Liver Disease: A Population-Based Cohort Study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Filozof, C.; Goldstein, B.J.; Williams, R.N.; Sanyal, A. Non-Alcoholic Steatohepatitis: Limited Available Treatment Options but Promising Drugs in Development and Recent Progress Towards a Regulatory Approval Pathway. Drugs 2015, 75, 1373–1392. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.A.; Silver, D.L. Cytosolic lipid droplets: From mechanisms of fat storage to disease. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 304–326. [Google Scholar] [CrossRef] [PubMed]
- Thiam, A.R.; Forêt, L. The physics of lipid droplet nucleation, growth and budding. Biochim. Biophys. Acta 2016, 1861 Pt A, 715–722. [Google Scholar] [CrossRef]
- Kounakis, K.; Chaniotakis, M.; Markaki, M.; Tavernarakis, N. Emerging Roles of Lipophagy in Health and Disease. Front. Cell Dev. Biol. 2019, 7, 185. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Landis, C.S.; Jin, G.-Y.; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated With Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef]
- Rasineni, K.; McVicker, B.L.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Rab GTPases Associate with Isolated Lipid Droplets (LDs) and Show Altered Content After Ethanol Administration: Potential Role in Alcohol-Impaired LD Metabolism. Alcohol. Clin. Exp. Res. 2014, 38, 327–335. [Google Scholar] [CrossRef]
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Ünalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659. [Google Scholar] [CrossRef]
- Celebi, G.; Cicek, A.F.; Gurel, H.; Genc, H.; Kirik, A.; Ercin, C.N.; Dogru, T. Microvesicular steatosis: A missed item in the management of nonalcoholic fatty liver disease? Acta Gastroenterol. Belg. 2020, 83, 565–570. [Google Scholar]
- Germano, C.W.; Mega, P.F.; Mattosinho, T.J.A.P.; Dias, L.L.C.; Gestic, M.A.; Utrini, M.P.; Chaim, F.D.M.; Callejas-Neto, F.; Chaim, E.A.; Cazzo, E. Microvesicular Steatosis in Individuals with Obesity: A Histological Marker of Non-alcoholic Fatty Liver Disease Severity. Obes. Surg. 2023, 33, 813–820. [Google Scholar] [CrossRef]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Brasaemle, D.L.; Wolins, N.E. Isolation of Lipid Droplets from Cells by Density Gradient Centrifugation. Curr. Protoc. Cell Biol. 2006, 29, 3–15. [Google Scholar] [CrossRef]
- Sun, Z.; Gong, J.; Wu, H.; Xu, W.; Wu, L.; Xu, D.; Gao, J.; Wu, J.-W.; Yang, H.; Yang, M.; et al. Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat. Commun. 2013, 4, 1594. [Google Scholar] [CrossRef] [PubMed]
- Straub, B.K.; Stoeffel, P.; Heid, H.; Zimbelmann, R.; Schirmacher, P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 2008, 47, 1936–1946. [Google Scholar] [CrossRef] [PubMed]
- Pawella, L.M.; Hashani, M.; Eiteneuer, E.; Renner, M.; Bartenschlager, R.; Schirmacher, P.; Straub, B.K. Perilipin discerns chronic from acute hepatocellular steatosis. J. Hepatol. 2013, 60, 633–642. [Google Scholar] [CrossRef]
- McManaman, J.L.; Bales, E.S.; Orlicky, D.J.; Jackman, M.; MacLean, P.S.; Cain, S.; Crunk, A.E.; Mansur, A.; Graham, C.E.; Bowman, T.; et al. Perilipin-2-null mice are protected against diet-induced obesity, adipose inflammation, and fatty liver disease. J. Lipid Res. 2013, 54, 1346–1359. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef]
- Straub, B.K.; Gyoengyoesi, B.; Koenig, M.; Hashani, M.; Pawella, L.M.; Herpel, E.; Mueller, W.; Macher-Goeppinger, S.; Heid, H.; Schirmacher, P. Adipophilin/perilipin-2 as a lipid droplet-specific marker for metabolically active cells and diseases associated with metabolic dysregulation. Histopathology 2013, 62, 617–631. [Google Scholar] [CrossRef]
- Najt, C.P.; Devarajan, M.; Mashek, D.G. Perilipins at a glance. J. Cell Sci. 2022, 135, jcs259501. [Google Scholar] [CrossRef]
- Sanchez, P.B.M.; Krizanac, M.; Weiskirchen, R.; Asimakopoulos, A. Understanding the Role of Perilipin 5 in Non-Alcoholic Fatty Liver Disease and Its Role in Hepatocellular Carcinoma: A Review of Novel Insights. Int. J. Mol. Sci. 2021, 22, 5284. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Laurens, C.; Bourlier, V.; Mairal, A.; Louche, K.; Badin, P.-M.; Mouisel, E.; Montagner, A.; Marette, A.; Tremblay, A.; Weisnagel, J.S.; et al. Perilipin 5 fine-tunes lipid oxidation to metabolic demand and protects against lipotoxicity in skeletal muscle. Sci. Rep. 2016, 6, 38310. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.Y.; Sun, K.; Zhang, M.; Zhou, X.; Zheng, X.; Tian, S.; Liu, Y.; Chen, L.; Gao, X.; Ye, J.; et al. Disruption of Plin5 degradation by CMA causes lipid homeostasis imbalance in NAFLD. Liver Int. 2020, 40, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Ikura, Y.; Arimoto, J.; Sugioka, K.; Iezzoni, J.C.; Park, S.H.; Naruko, T.; Itabe, H.; Kawada, N.; Caldwell, S.H.; et al. Expression of Perilipin and Adipophilin in Nonalcoholic Fatty Liver Disease; Relevance to Oxidative Injury and Hepatocyte Ballooning. J. Atheroscler. Thromb. 2009, 16, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol. Metab. 2021, 50, 101115. [Google Scholar] [CrossRef]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta 2016, 1861, 269–284. [Google Scholar] [CrossRef]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. et Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862 Pt B, 1221–1232. [Google Scholar] [CrossRef]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. 2018, 9, 778. [Google Scholar] [CrossRef]
- Granneman, J.G.; Moore, H.-P.H.; Mottillo, E.P.; Zhu, Z.; Zhou, L. Interactions of Perilipin-5 (Plin5) with Adipose Triglyceride Lipase. J. Biol. Chem. 2011, 286, 5126–5135. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Rout, A.K.; Strub, M.-P.; Piszczek, G.; Tjandra, N. Structure of Transmembrane Domain of Lysosome-associated Membrane Protein Type 2a (LAMP-2A) Reveals Key Features for Substrate Specificity in Chaperone-mediated Autophagy. J. Biol. Chem. 2014, 289, 35111–35123. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient Chaperone-Mediated Autophagy in Liver Leads to Metabolic Dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall’Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, E705–E714. [Google Scholar] [CrossRef]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS—Lipases and Lipolysis in Lipid Metabolism and Signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef]
- Zhang, S.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Liu, J.; He, G.; Zheng, H.; Yang, L.; et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. 2022, 13, 132. [Google Scholar] [CrossRef]
- Shin, D.W. Lipophagy: Molecular Mechanisms and Implications in Metabolic Disorders. Mol. Cells 2020, 43, 686–693. [Google Scholar] [CrossRef]
- Sathyanarayan, A.; Mashek, M.T.; Mashek, D.G. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Tsai, T.-H.; Chen, E.; Li, L.; Saha, P.; Lee, H.-J.; Huang, L.-S.; Shelness, G.S.; Chan, L.; Chang, B.H.-J. The constitutive lipid droplet protein PLIN2 regulates autophagy in liver. Autophagy 2017, 13, 1130–1144. [Google Scholar] [CrossRef]
- Schroeder, B.; Schulze, R.J.; Weller, S.G.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015, 61, 1896–1907. [Google Scholar] [CrossRef]
- Khambu, B.; Yan, S.; Huda, N.; Liu, G.; Yin, X.-M. Autophagy in non-alcoholic fatty liver disease and alcoholic liver disease. Liver Res. 2018, 2, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Kashima, J.; Shintani-Ishida, K.; Nakajima, M.; Maeda, H.; Unuma, K.; Uchiyama, Y.; Yoshida, K.-I. Immunohistochemical study of the autophagy marker microtubule-associated protein 1 light chain 3 in normal and steatotic human livers. Hepatol. Res. 2014, 44, 779–787. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, Á.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wang, T.; Yang, L.; Wang, H.-Y. Ginsenoside Rb2 Alleviates Hepatic Lipid Accumulation by Restoring Autophagy via Induction of Sirt1 and Activation of AMPK. Int. J. Mol. Sci. 2017, 18, 1063. [Google Scholar] [CrossRef]
- Stayrook, K.R.; Bramlett, K.S.; Savkur, R.S.; Ficorilli, J.; Cook, T.; Christe, M.E.; Michael, L.F.; Burris, T.P. Regulation of Carbohydrate Metabolism by the Farnesoid X Receptor. Endocrinology 2005, 146, 984–991. [Google Scholar] [CrossRef]
- Oberkofler, H.; Pfeifenberger, A.; Soyal, S.; Felder, T.; Hahne, P.; Miller, K.; Krempler, F.; Patsch, W. Aberrant hepatic TRIB3 gene expression in insulin-resistant obese humans. Diabetologia 2010, 53, 1971–1975. [Google Scholar] [CrossRef]
- Wu, W.K.K.; Zhang, L.; Chan, M.T.V. Autophagy, NAFLD and NAFLD-Related HCC. Adv. Exp. Med. Biol. 2018, 1061, 127–138. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Dash, S.; Aydin, Y.; Moroz, K. Chaperone-Mediated Autophagy in the Liver: Good or Bad? Cells 2019, 8, 1308. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Seth, R.K.; Kumar, A.; Kadiiska, M.B.; Michelotti, G.; Diehl, A.M.; Chatterjee, S. Purinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitis. Am. J. Physiol. Liver Physiol. 2013, 305, G950–G963. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Function of Autophagy in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1304–1313. [Google Scholar] [CrossRef]
- Le, S.; Fu, X.; Pang, M.; Zhou, Y.; Yin, G.; Zhang, J.; Fan, D. The Antioxidative Role of Chaperone-Mediated Autophagy as a Downstream Regulator of Oxidative Stress in Human Diseases. Technol. Cancer Res. Treat. 2022, 21, 15330338221114178. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Vucur, M.; Luedde, T.; Schneiders, S.; Kalampoka, S.; Weiss, T.S.; Weiskirchen, R. Perilipin 5 and Lipocalin 2 Expression in Hepatocellular Carcinoma. Cancers 2019, 11, 385. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic Autophagy Is Suppressed in the Presence of Insulin Resistance and Hyperinsulinemia: Inhibition of FoxO1-Dependent Expression of Key Autophagy Genes by Insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef]
- Álvarez-Mercado, A.I.; Rojano-Alfonso, C.; Micó-Carnero, M.; Caballeria-Casals, A.; Peralta, C.; Casillas-Ramírez, A. New Insights Into the Role of Autophagy in Liver Surgery in the Setting of Metabolic Syndrome and Related Diseases. Front. Cell Dev. Biol. 2021, 9, 670273. [Google Scholar] [CrossRef]
- Kuo, N.-C.; Huang, S.-Y.; Yang, C.-Y.; Shen, H.-H.; Lee, Y.-M. Involvement of HO-1 and Autophagy in the Protective Effect of Magnolol in Hepatic Steatosis-Induced NLRP3 Inflammasome Activation In Vivo and In Vitro. Antioxidants 2020, 9, 924. [Google Scholar] [CrossRef]
- Ramos, V.d.M.; Kowaltowski, A.J.; Kakimoto, P.A. Autophagy in Hepatic Steatosis: A Structured Review. Front. Cell Dev. Biol. 2021, 9, 657389. [Google Scholar] [CrossRef]
- He, Q.; Sha, S.; Sun, L.; Zhang, J.; Dong, M. GLP-1 analogue improves hepatic lipid accumulation by inducing autophagy via AMPK/mTOR pathway. Biochem. Biophys. Res. Commun. 2016, 476, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Schon, H.-T.; Bartneck, M.; Borkham-Kamphorst, E.; Nattermann, J.; Lammers, T.; Tacke, F.; Weiskirchen, R. Pharmacological Intervention in Hepatic Stellate Cell Activation and Hepatic Fibrosis. Front. Pharmacol. 2016, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Ueno, M.; Suzuki, J.; Hirose, M.; Sato, S.; Imagawa, M.; Zenimaru, Y.; Takahashi, S.; Ikuyama, S.; Koizumi, T.; Konoshita, T.; et al. Cardiac overexpression of perilipin 2 induces dynamic steatosis: Prevention by hormone-sensitive lipase. Am. J. Physiol. Metab. 2017, 313, E699–E709. [Google Scholar] [CrossRef] [PubMed]
- Mardani, I.; Dalen, K.T.; Drevinge, C.; Miljanovic, A.; Ståhlman, M.; Klevstig, M.; Täng, M.S.; Fogelstrand, P.; Levin, M.; Ekstrand, M.; et al. Plin2-deficiency reduces lipophagy and results in increased lipid accumulation in the heart. Sci. Rep. 2019, 9, 6909. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mastoridou, E.M.; Goussia, A.C.; Kanavaros, P.; Charchanti, A.V. Involvement of Lipophagy and Chaperone-Mediated Autophagy in the Pathogenesis of Non-Alcoholic Fatty Liver Disease by Regulation of Lipid Droplets. Int. J. Mol. Sci. 2023, 24, 15891. https://doi.org/10.3390/ijms242115891
Mastoridou EM, Goussia AC, Kanavaros P, Charchanti AV. Involvement of Lipophagy and Chaperone-Mediated Autophagy in the Pathogenesis of Non-Alcoholic Fatty Liver Disease by Regulation of Lipid Droplets. International Journal of Molecular Sciences. 2023; 24(21):15891. https://doi.org/10.3390/ijms242115891
Chicago/Turabian StyleMastoridou, Eleftheria M., Anna C. Goussia, Panagiotis Kanavaros, and Antonia V. Charchanti. 2023. "Involvement of Lipophagy and Chaperone-Mediated Autophagy in the Pathogenesis of Non-Alcoholic Fatty Liver Disease by Regulation of Lipid Droplets" International Journal of Molecular Sciences 24, no. 21: 15891. https://doi.org/10.3390/ijms242115891
APA StyleMastoridou, E. M., Goussia, A. C., Kanavaros, P., & Charchanti, A. V. (2023). Involvement of Lipophagy and Chaperone-Mediated Autophagy in the Pathogenesis of Non-Alcoholic Fatty Liver Disease by Regulation of Lipid Droplets. International Journal of Molecular Sciences, 24(21), 15891. https://doi.org/10.3390/ijms242115891