
Analysis of CD74 Occurrence in Oncogenic Fusion Proteins


Abstract
:1. Introduction
2. CD74 Fusion Proteins in Cancer
2.1. Analysis of CD74 Fusion Partners
2.2. CD74-ROS1 Expression
2.3. CD74-NTRK1 Expression
2.4. CD74-NRG1 Expression
2.5. CD74-PDGFRB Expression
2.6. CD74-NRG2a Expression
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ortiz de Mendíbil, I.; Vizmanos, J.L.; Novo, F.J. Signatures of Selection in Fusion Transcripts Resulting from Chromosomal Translocations in Human Cancer. PLoS ONE 2009, 4, e4805. [Google Scholar] [CrossRef]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G.W. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Zhang, W. Fusion genes in solid tumors: An emerging target for cancer diagnosis and treatment. Chin. J. Cancer 2013, 32, 594–603. [Google Scholar] [CrossRef]
- Farago, A.F.; Taylor, M.S.; Doebele, R.C.; Zhu, V.W.; Kummar, S.; Spira, A.I.; Boyle, T.A.; Haura, E.B.; Arcila, M.E.; Benayed, R.; et al. Clinicopathologic Features of Non-Small-Cell Lung Cancer Harboring an NTRK Gene Fusion. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Capitán, A.; Sánchez-Herrero, E.; Robado de Lope, L.; Aguilar-Hernández, A.; Sullivan, I.; Calvo, V.; Moya-Horno, I.; Viteri, S.; Cabrera, C.; Aguado, C.; et al. Detecting ALK, ROS1 and RET fusions and the METΔex14 splicing variant in liquid biopsies of non-small cell lung cancer patients using RNA-based techniques. Mol. Oncol. 2023, 17, 1884–1897. [Google Scholar] [CrossRef]
- Jonna, S.; Feldman, R.A.; Swensen, J.; Gatalica, Z.; Korn, W.M.; Borghaei, H.; Ma, P.C.; Nieva, J.J.; Spira, A.I.; Vanderwalde, A.M.; et al. Detection of NRG1 Gene Fusions in Solid Tumors. Clin. Cancer Res. 2019, 25, 4966–4972. [Google Scholar] [CrossRef]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF signal transduction initiated by binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Merk, M.; Zierow, S.; Leng, L.; Das, R.; Du, X.; Schulte, W.; Fan, J.; Lue, H.; Chen, Y.; Xiong, H.; et al. The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc. Natl. Acad. Sci. USA 2011, 108, E577–E585. [Google Scholar] [CrossRef]
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 2006, 25, 595–606. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]
- Schwartz, V.; Lue, H.; Kraemer, S.; Korbiel, J.; Krohn, R.; Ohl, K.; Bucala, R.; Weber, C.; Bernhagen, J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009, 583, 2749–2757. [Google Scholar] [CrossRef] [PubMed]
- Lue, H.; Kapurniotu, A.; Fingerle-Rowson, G.; Roger, T.; Leng, L.; Thiele, M.; Calandra, T.; Bucala, R.; Bernhagen, J. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell Signal. 2006, 18, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Gore, Y.; Starlets, D.; Maharshak, N.; Becker-Herman, S.; Kaneyuki, U.; Leng, L.; Bucala, R.; Shachar, I. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J. Biol. Chem. 2008, 283, 2784–2792. [Google Scholar] [CrossRef]
- Lue, H.; Thiele, M.; Franz, J.; Dahl, E.; Speckgens, S.; Leng, L.; Fingerle-Rowson, G.; Bucala, R.; Lüscher, B.; Bernhagen, J. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene 2007, 26, 5046–5059. [Google Scholar] [CrossRef] [PubMed]
- Lue, H.; Dewor, M.; Leng, L.; Bucala, R.; Bernhagen, J. Activation of the JNK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on CXCR4 and CD74. Cell Signal. 2011, 23, 135–144. [Google Scholar] [CrossRef]
- Starlets, D.; Gore, Y.; Binsky, I.; Haran, M.; Harpaz, N.; Shvidel, L.; Becker-Herman, S.; Berrebi, A.; Shachar, I. Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood 2006, 107, 4807–4816. [Google Scholar] [CrossRef]
- Cheng, S.P.; Liu, C.L.; Chen, M.J.; Chien, M.N.; Leung, C.H.; Lin, C.H.; Hsu, Y.C.; Lee, J.J. CD74 expression and its therapeutic potential in thyroid carcinoma. Endocr. Relat. Cancer 2015, 22, 179–190. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Rajendran, G.; Abbott, E.; Martin, A.; Amalraj, S.; Dennis, K.; Li, X.; Warrick, J.; Taylor, J.A., 3rd. Role of MIF1/MIF2/CD74 interactions in bladder cancer. J. Pathol. 2023, 259, 46–55. [Google Scholar] [CrossRef]
- Thavayogarajah, T.; Sinitski, D.; El Bounkari, O.; Torres-Garcia, L.; Lewinsky, H.; Harjung, A.; Chen, H.R.; Panse, J.; Vankann, L.; Shachar, I.; et al. CXCR4 and CD74 together enhance cell survival in response to macrophage migration-inhibitory factor in chronic lymphocytic leukemia. Exp. Hematol. 2022, 115, 30–43. [Google Scholar] [CrossRef]
- Burton, J.D.; Ely, S.; Reddy, P.K.; Stein, R.; Gold, D.V.; Cardillo, T.M.; Goldenberg, D.M. CD74 is expressed by multiple myeloma and is a promising target for therapy. Clin. Cancer Res. 2004, 10, 6606–6611. [Google Scholar] [CrossRef]
- Greenwood, C.; Metodieva, G.; Al-Janabi, K.; Lausen, B.; Alldridge, L.; Leng, L.; Bucala, R.; Fernandez, N.; Metodiev, M.V. Stat1 and CD74 overexpression is co-dependent and linked to increased invasion and lymph node metastasis in triple-negative breast cancer. J. Proteom. 2012, 75, 3031–3040. [Google Scholar] [CrossRef] [PubMed]
- Gold, D.V.; Stein, R.; Burton, J.; Goldenberg, D.M. Enhanced expression of CD74 in gastrointestinal cancers and benign tissues. Int. J. Clin. Exp. Pathol. 2010, 4, 1–12. [Google Scholar] [PubMed]
- McClelland, M.; Zhao, L.; Carskadon, S.; Arenberg, D. Expression of CD74, the receptor for macrophage migration inhibitory factor, in non-small cell lung cancer. Am. J. Pathol. 2009, 174, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Young, A.N.; Amin, M.B.; Moreno, C.S.; Lim, S.D.; Cohen, C.; Petros, J.A.; Marshall, F.F.; Neish, A.S. Expression profiling of renal epithelial neoplasms: A method for tumor classification and discovery of diagnostic molecular markers. Am. J. Pathol. 2001, 158, 1639–1651. [Google Scholar] [CrossRef]
- Meyer-Siegler, K.L.; Iczkowski, K.A.; Leng, L.; Bucala, R.; Vera, P.L. Inhibition of macrophage migration inhibitory factor or its receptor (CD74) attenuates growth and invasion of DU-145 prostate cancer cells. J. Immunol. 2006, 177, 8730–8739. [Google Scholar] [CrossRef]
- Koide, N.; Yamada, T.; Shibata, R.; Mori, T.; Fukuma, M.; Yamazaki, K.; Aiura, K.; Shimazu, M.; Hirohashi, S.; Nimura, Y.; et al. Establishment of perineural invasion models and analysis of gene expression revealed an invariant chain (CD74) as a possible molecule involved in perineural invasion in pancreatic cancer. Clin. Cancer Res. 2006, 12, 2419–2426. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Decker, P.A.; Morlan, B.W.; Wu, W.; Ballman, K.V.; Giannini, C.; Sarkaria, J.N. Expression of CD74 in high grade gliomas: A potential role in temozolomide resistance. J. Neurooncol. 2010, 100, 177–186. [Google Scholar] [CrossRef]
- Hong, W.C.; Lee, D.E.; Kang, H.W.; Kim, M.J.; Kim, M.; Kim, J.H.; Fang, S.; Kim, H.J.; Park, J.S. CD74 Promotes a Pro-Inflammatory Tumor Microenvironment by Inducing S100A8 and S100A9 Secretion in Pancreatic Cancer. Int. J. Mol. Sci. 2023, 24, 12993. [Google Scholar] [CrossRef]
- Xu, S.; Li, X.; Tang, L.; Liu, Z.; Yang, K.; Cheng, Q. CD74 Correlated with Malignancies and Immune Microenvironment in Gliomas. Front. Mol. Biosci. 2021, 8, 706949. [Google Scholar] [CrossRef]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef]
- Fernandez-Cuesta, L.; Plenker, D.; Osada, H.; Sun, R.; Menon, R.; Leenders, F.; Ortiz-Cuaran, S.; Peifer, M.; Bos, M.; Dassler, J.; et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov. 2014, 4, 415–422. [Google Scholar] [CrossRef]
- Liu, Y.F.; Wang, B.Y.; Zhang, W.N.; Huang, J.Y.; Li, B.S.; Zhang, M.; Jiang, L.; Li, J.F.; Wang, M.J.; Dai, Y.J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Claesson, L.; Larhammar, D.; Rask, L.; Peterson, P.A. cDNA clone for the human invariant gamma chain of class II histocompatibility antigens and its implications for the protein structure. Proc. Natl. Acad. Sci. USA 1983, 80, 7395–7399. [Google Scholar] [CrossRef] [PubMed]
- Strubin, M.; Mach, B.; Long, E.O. The complete sequence of the mRNA for the HLA-DR-associated invariant chain reveals a polypeptide with an unusual transmembrane polarity. EMBO J. 1984, 3, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Strubin, M.; Berte, C.; Mach, B. Alternative splicing and alternative initiation of translation explain the four forms of the Ia antigen-associated invariant chain. EMBO J. 1986, 5, 3483–3488. [Google Scholar] [CrossRef]
- O’Sullivan, D.M.; Larhammar, D.; Wilson, M.C.; Peterson, P.A.; Quaranta, V. Structure of the human Ia-associated invariant (gamma)-chain gene: Identification of 5′ sequences shared with major histocompatibility complex class II genes. Proc. Natl. Acad. Sci. USA 1986, 83, 4484–4488. [Google Scholar] [CrossRef]
- Koch, N.; Lauer, W.; Habicht, J.; Dobberstein, B. Primary structure of the gene for the murine Ia antigen-associated invariant chains (Ii). An alternatively spliced exon encodes a cysteine-rich domain highly homologous to a repetitive sequence of thyroglobulin. EMBO J. 1987, 6, 1677–1683. [Google Scholar] [CrossRef]
- Gedde-Dahl, M.; Freisewinkel, I.; Staschewski, M.; Schenck, K.; Koch, N.; Bakke, O. Exon 6 is essential for invariant chain trimerization and induction of large endosomal structures. J. Biol. Chem. 1997, 272, 8281–8287. [Google Scholar] [CrossRef]
- Kuwana, T.; Peterson, P.A.; Karlsson, L. Exit of major histocompatibility complex class II–invariant chain p35 complexes from the endoplasmic reticulum is modulated by phosphorylation. Proc. Natl. Acad. Sci. USA 1998, 95, 1056–1061. [Google Scholar] [CrossRef]
- Marks, M.S.; Blum, J.S.; Cresswell, P. Invariant chain trimers are sequestered in the rough endoplasmic reticulum in the absence of association with HLA class II antigens. J. Cell Biol. 1990, 111, 839–855. [Google Scholar] [CrossRef]
- Jasanoff, A.; Wagner, G.; Wiley, D.C. Structure of a trimeric domain of the MHC class II-associated chaperonin and targeting protein Ii. EMBO J. 1998, 17, 6812–6818. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Amaya, M.; Mellins, E.; Wiley, D.C. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature 1995, 378, 457–462. [Google Scholar] [CrossRef]
- Zhu, Y.; Rudensky, A.Y.; Corper, A.L.; Teyton, L.; Wilson, I.A. Crystal structure of MHC class II I-Ab in complex with a human CLIP peptide: Prediction of an I-Ab peptide-binding motif. J. Mol. Biol. 2003, 326, 1157–1174. [Google Scholar] [CrossRef]
- Günther, S.; Schlundt, A.; Sticht, J.; Roske, Y.; Heinemann, U.; Wiesmüller, K.H.; Jung, G.; Falk, K.; Rötzschke, O.; Freund, C. Bidirectional binding of invariant chain peptides to an MHC class II molecule. Proc. Natl. Acad. Sci. USA 2010, 107, 22219–22224. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Jayaraman, P.; Bergseng, E.; Madhusudhan, M.S.; Kim, C.Y.; Sollid, L.M. Unraveling the structural basis for the unusually rich association of human leukocyte antigen DQ2.5 with class-II-associated invariant chain peptides. J. Biol. Chem. 2017, 292, 9218–9228. [Google Scholar] [CrossRef]
- Guncar, G.; Pungercic, G.; Klemencic, I.; Turk, V.; Turk, D. Crystal structure of MHC class II-associated p41 Ii fragment bound to cathepsin L reveals the structural basis for differentiation between cathepsins L and S. EMBO J. 1999, 18, 793–803. [Google Scholar] [CrossRef]
- Kohsaka, S.; Hayashi, T.; Nagano, M.; Ueno, T.; Kojima, S.; Kawazu, M.; Shiraishi, Y.; Kishikawa, S.; Suehara, Y.; Takahashi, F.; et al. Identification of Novel CD74-NRG2alpha Fusion from Comprehensive Profiling of Lung Adenocarcinoma in Japanese Never or Light Smokers. J. Thorac. Oncol. 2020, 15, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Chiva, C.; Barthe, P.; Codina, A.; Gairí, M.; Molina, F.; Granier, C.; Pugnière, M.; Inui, T.; Nishio, H.; Nishiuchi, Y.; et al. Synthesis and NMR structure of p41icf, a potent inhibitor of human cathepsin L. J. Am. Chem. Soc. 2003, 125, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.L.; Niesvizky, R.; Stadtmauer, E.A.; Chanan-Khan, A.; Siegel, D.; Horne, H.; Wegener, W.A.; Goldenberg, D.M. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 163, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Furman, R.R.; Rutherford, S.; Ruan, J.; Ely, S.; Greenberg, J.; Coleman, M.; Goldsmith, S.J.; Leonard, J.P. Phase I study of the anti-CD74 monoclonal antibody milatuzumab (hLL1) in patients with previously treated B-cell lymphomas. Leuk. Lymphoma 2015, 56, 3065–3070. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, C.L.; Li, X.; Embry, M.; Yu, A.; Krimm, S.; Krueger, S.; Greenland, N.Y.; Wen, K.W.; Jones, C.; DeAlmeida, V.; et al. Targeting CD74 in multiple myeloma with the novel, site-specific antibody-drug conjugate STRO-001. Oncotarget 2018, 9, 37700–37714. [Google Scholar] [CrossRef]
- Hu, H.; Ding, N.; Zhou, H.; Wang, S.; Tang, L.; Xiao, Z. A novel CD74-ROS1 gene fusion in a patient with inflammatory breast cancer: A case report. J. Med. Case Rep. 2021, 15, 277. [Google Scholar] [CrossRef] [PubMed]
- Sadras, T.; Jalud, F.B.; Kosasih, H.J.; Horne, C.R.; Brown, L.M.; El-Kamand, S.; de Bock, C.E.; McAloney, L.; Ng, A.P.; Davidson, N.M.; et al. Unusual PDGFRB fusion reveals novel mechanism of kinase activation in Ph-like B-ALL. Leukemia 2023, 37, 905–909. [Google Scholar] [CrossRef]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef]
- Lan, S.; Li, H.; Liu, Y.; Xu, J.; Huang, Z.; Yan, S.; Zhang, Q.; Cheng, Y. A Novel ROS1-FBXL17 Fusion Co-Existing with CD74-ROS1 Fusion May Improve Sensitivity to Crizotinib and Prolong Progression-Free Survival of Patients with Lung Adenocarcinoma. Onco Targets Ther. 2020, 13, 11499–11504. [Google Scholar] [CrossRef]
- Nakaoku, T.; Tsuta, K.; Ichikawa, H.; Shiraishi, K.; Sakamoto, H.; Enari, M.; Furuta, K.; Shimada, Y.; Ogiwara, H.; Watanabe, S.; et al. Druggable oncogene fusions in invasive mucinous lung adenocarcinoma. Clin. Cancer Res. 2014, 20, 3087–3093. [Google Scholar] [CrossRef]
- Cai, W.; Li, W.; Ren, S.; Zheng, L.; Li, X.; Zhou, C. Coexistence of three variants involving two different fusion partners of ROS1 including a novel variant of ROS1 fusions in lung adenocarcinoma: A case report. J. Thorac. Oncol. 2014, 9, e43–e46. [Google Scholar] [CrossRef]
- Hashiguchi, M.H.; Sato, T.; Watanabe, R.; Kagyo, J.; Matsuzaki, T.; Domoto, H.; Kato, T.; Nakahara, Y.; Yokose, T.; Hiroshima, Y.; et al. A case of lung adenocarcinoma with a novel CD74-ROS1 fusion variant identified by comprehensive genomic profiling that responded to crizotinib and entrectinib. Thorac. Cancer 2021, 12, 2504–2507. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, K.; Piper-Vallillo, A.J.; Viray, H.; Khan, A.M.; Rangachari, D.; Costa, D.B. Cases of ROS1-rearranged lung cancer: When to use crizotinib, entrectinib, lorlatinib, and beyond? Precis. Cancer Med. 2020, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, B.; Wang, C.; Liao, C.; Wang, S.; Cao, R.; Ma, T.; Wang, K. Case report: A novel reciprocal ROS1-CD74 fusion in a NSCLC patient partially benefited from sequential tyrosine kinase inhibitors treatment. Front. Oncol. 2022, 12, 1021342. [Google Scholar] [CrossRef]
- Xia, H.; Xue, X.; Ding, H.; Ou, Q.; Wu, X.; Nagasaka, M.; Shao, Y.W.; Hu, X.; Ou, S.I. Evidence of NTRK1 Fusion as Resistance Mechanism to EGFR TKI in EGFR+NSCLC: Results from a Large-Scale Survey of NTRK1 Fusions in Chinese Patients with Lung Cancer. Clin. Lung Cancer 2020, 21, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yan, S.; Liu, Y.; Ma, L.; Liu, X.; Liu, Y.; Cheng, Y. Analysis of NTRK mutation and clinicopathologic factors in lung cancer patients in northeast China. Int. J. Biol. Markers 2020, 35, 36–40. [Google Scholar] [CrossRef]
- Severson, E.; Achyut, B.R.; Nesline, M.; Pabla, S.; Previs, R.A.; Kannan, G.; Chenn, A.; Zhang, S.; Klein, R.; Conroy, J.; et al. RNA sequencing identifies novel NRG1-fusions in solid tumors that lack co-occurring oncogenic drivers. J. Mol. Diagn 2023, 25, 454–466. [Google Scholar] [CrossRef]
- Drilon, A.; Somwar, R.; Mangatt, B.P.; Edgren, H.; Desmeules, P.; Ruusulehto, A.; Smith, R.S.; Delasos, L.; Vojnic, M.; Plodkowski, A.J.; et al. Response to ERBB3-Directed Targeted Therapy in NRG1-Rearranged Cancers. Cancer Discov. 2018, 8, 686–695. [Google Scholar] [CrossRef]
- Birchmeier, C.; Sharma, S.; Wigler, M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 9270–9274. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.H.; Chang, Y.J.; Huang, W.C.; Jang, T.H.; Kung, H.J.; Wang, W.C.; Yang, M.H.; Lin, M.C.; Huang, S.F.; Chou, S.W.; et al. EZH2-mediated upregulation of ROS1 oncogene promotes oral cancer metastasis. Oncogene 2017, 36, 6542–6554. [Google Scholar] [CrossRef]
- Grenier, K.; Rivière, J.B.; Bencheikh, B.O.A.; Corredor, A.L.G.; Shieh, B.C.; Wang, H.; Fiset, P.O.; Camilleri-Broët, S. Routine Clinically Detected Increased ROS1 Transcripts Are Related with ROS1 Expression by Immunohistochemistry and Associated with EGFR Mutations in Lung Adenocarcinoma. JTO Clin. Res. Rep. 2023, 4, 100530. [Google Scholar] [CrossRef]
- Lee, H.J.; Seol, H.S.; Kim, J.Y.; Chun, S.M.; Suh, Y.A.; Park, Y.S.; Kim, S.W.; Choi, C.M.; Park, S.I.; Kim, D.K.; et al. ROS1 receptor tyrosine kinase, a druggable target, is frequently overexpressed in non-small cell lung carcinomas via genetic and epigenetic mechanisms. Ann. Surg. Oncol. 2013, 20, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; O’Neill, K.; Riggs, M.; Wigler, M. Characterization of ROS1 cDNA from a human glioblastoma cell line. Proc. Natl. Acad. Sci. USA 1990, 87, 4799–4803. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, J.; Wong, R.; Charest, A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim. Biophys. Acta 2009, 1795, 37–52. [Google Scholar] [CrossRef]
- Drilon, A.; Jenkins, C.; Iyer, S.; Schoenfeld, A.; Keddy, C.; Davare, M.A. ROS1-dependent cancers—Biology, diagnostics and therapeutics. Nat. Rev. Clin. Oncol. 2021, 18, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; San Román-Gil, M.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef]
- Shaw, A.T.; Ou, S.H.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef]
- Barbacid, M.; Lamballe, F.; Pulido, D.; Klein, R. The trk family of tyrosine protein kinase receptors. Biochim. Biophys. Acta 1991, 1072, 115–127. [Google Scholar] [CrossRef]
- Barker, P.A.; Lomen-Hoerth, C.; Gensch, E.M.; Meakin, S.O.; Glass, D.J.; Shooter, E.M. Tissue-specific alternative splicing generates two isoforms of the trkA receptor. J. Biol. Chem. 1993, 268, 15150–15157. [Google Scholar] [CrossRef]
- Martin-Zanca, D.; Oskam, R.; Mitra, G.; Copeland, T.; Barbacid, M. Molecular and biochemical characterization of the human trk proto-oncogene. Mol. Cell Biol. 1989, 9, 24–33. [Google Scholar]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Hempstead, B.L.; Martin-Zanca, D.; Chao, M.V.; Parada, L.F. The trk proto-oncogene product: A signal transducing receptor for nerve growth factor. Science 1991, 252, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Jing, S.Q.; Nanduri, V.; O’Rourke, E.; Barbacid, M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 1991, 65, 189–197. [Google Scholar] [CrossRef]
- Klesse, L.J.; Parada, L.F. Trks: Signal transduction and intracellular pathways. Microsc. Res. Technol. 1999, 45, 210–216. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed]
- Indo, Y.; Mardy, S.; Tsuruta, M.; Karim, M.A.; Matsuda, I. Structure and organization of the humanTRKA gene encoding a high affinity receptor for nerve growth factor. Jpn. J. Hum. Genet. 1997, 42, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Reuther, G.W.; Lambert, Q.T.; Caligiuri, M.A.; Der, C.J. Identification and characterization of an activating TrkA deletion mutation in acute myeloid leukemia. Mol. Cell Biol. 2000, 20, 8655–8666. [Google Scholar] [CrossRef]
- Gao, F.; Griffin, N.; Faulkner, S.; Rowe, C.W.; Williams, L.; Roselli, S.; Thorne, R.F.; Ferdoushi, A.; Jobling, P.; Walker, M.M.; et al. The neurotrophic tyrosine kinase receptor TrkA and its ligand NGF are increased in squamous cell carcinomas of the lung. Sci. Rep. 2018, 8, 8135. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; Rowe, C.W.; Rodrigues Oliveira, S.M.; Roselli, S.; Thorne, R.F.; Oldmeadow, C.; Attia, J.; Jiang, C.C.; Zhang, X.D.; et al. Neurotrophin Receptors TrkA, p75(NTR), and Sortilin Are Increased and Targetable in Thyroid Cancer. Am. J. Pathol. 2018, 188, 229–241. [Google Scholar] [CrossRef]
- Lagadec, C.; Meignan, S.; Adriaenssens, E.; Foveau, B.; Vanhecke, E.; Romon, R.; Toillon, R.A.; Oxombre, B.; Hondermarck, H.; Le Bourhis, X. TrkA overexpression enhances growth and metastasis of breast cancer cells. Oncogene 2009, 28, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, S.; Griffin, N.; Rowe, C.W.; Jobling, P.; Lombard, J.M.; Oliveira, S.M.; Walker, M.M.; Hondermarck, H. Nerve growth factor and its receptor tyrosine kinase TrkA are overexpressed in cervical squamous cell carcinoma. FASEB Bioadv. 2020, 2, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Bauer, T.M.; Lee, J.J.; Dowlati, A.; Brose, M.S.; Farago, A.F.; Taylor, M.; Shaw, A.T.; Montez, S.; Meric-Bernstam, F.; et al. Larotrectinib in adult patients with solid tumours: A multi-centre, open-label, phase I dose-escalation study. Ann. Oncol. 2019, 30, 325–331. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Steinthorsdottir, V.; Stefansson, H.; Ghosh, S.; Birgisdottir, B.; Bjornsdottir, S.; Fasquel, A.C.; Olafsson, O.; Stefansson, K.; Gulcher, J.R. Multiple novel transcription initiation sites for NRG1. Gene 2004, 342, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Falls, D.L. Neuregulins: Functions, forms, and signaling strategies. Exp. Cell Res. 2003, 284, 14–30. [Google Scholar] [CrossRef]
- Meyer, D.; Birchmeier, C. Multiple essential functions of neuregulin in development. Nature 1995, 378, 386–390. [Google Scholar] [CrossRef]
- Kramer, R.; Bucay, N.; Kane, D.J.; Martin, L.E.; Tarpley, J.E.; Theill, L.E. Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proc. Natl. Acad. Sci. USA 1996, 93, 4833–4838. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F., 3rd; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef]
- Nagasaka, M.; Ou, S.I. NRG1 and NRG2 fusion positive solid tumor malignancies: A paradigm of ligand-fusion oncogenesis. Trends Cancer 2022, 8, 242–258. [Google Scholar] [CrossRef]
- Busfield, S.J.; Michnick, D.A.; Chickering, T.W.; Revett, T.L.; Ma, J.; Woolf, E.A.; Comrack, C.A.; Dussault, B.J.; Woolf, J.; Goodearl, A.D.; et al. Characterization of a neuregulin-related gene, Don-1, that is highly expressed in restricted regions of the cerebellum and hippocampus. Mol. Cell Biol. 1997, 17, 4007–4014. [Google Scholar] [CrossRef] [PubMed]
- Schoeberl, B.; Kudla, A.; Masson, K.; Kalra, A.; Curley, M.; Finn, G.; Pace, E.; Harms, B.; Kim, J.; Kearns, J.; et al. Systems biology driving drug development: From design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121). npj Syst. Biol. Appl. 2017, 3, 16034. [Google Scholar] [CrossRef] [PubMed]
- Carrizosa, D.R.; Burkard, M.E.; Elamin, Y.Y.; Desai, J.; Gadgeel, S.M.; Lin, J.J.; Waqar, S.N.; Spigel, D.R.; Chae, Y.K.; Cheema, P.K.; et al. CRESTONE: Initial efficacy and safety of seribantumab in solid tumors harboring NRG1 fusions. J. Clin. Oncol. 2022, 40, 3006. [Google Scholar] [CrossRef]
- Spigel, D.; Waqar, S.N.; Burkard, M.E.; Lin, J.J.; Chae, Y.K.; Socinski, M.A.; Gadgeel, S.; Reckamp, K.L.; Leland, S.M.; Plessinger, D.; et al. MO01.33 CRESTONE—Clinical Study of REsponse to Seribantumab in Tumors with NEuregulin-1 (NRG1) Fusions—A Phase 2 Study of the anti-HER3 mAb for Advanced or Metastatic Solid Tumors (NCT04383210). J. Thorac. Oncol. 2021, 16, S29–S30. [Google Scholar] [CrossRef]
- Schram, A.M.; Goto, K.; Kim, D.-W.; Martin-Romano, P.; Ou, S.-H.I.; O’Kane, G.M.; O’Reilly, E.M.; Umemoto, K.; Duruisseaux, M.; Neuzillet, C.; et al. Efficacy and safety of zenocutuzumab, a HER2 x HER3 bispecific antibody, across advanced NRG1 fusion (NRG1+) cancers. J. Clin. Oncol. 2022, 40, 105. [Google Scholar] [CrossRef]
- Arar, M.; Xu, Y.C.; Elshihabi, I.; Barnes, J.L.; Choudhury, G.G.; Abboud, H.E. Platelet-derived growth factor receptor beta regulates migration and DNA synthesis in metanephric mesenchymal cells. J. Biol. Chem. 2000, 275, 9527–9533. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Hoch, R.V.; Soriano, P. Roles of PDGF in animal development. Development 2003, 130, 4769–4784. [Google Scholar] [CrossRef] [PubMed]
- Appiah-Kubi, K.; Lan, T.; Wang, Y.; Qian, H.; Wu, M.; Yao, X.; Wu, Y.; Chen, Y. Platelet-derived growth factor receptors (PDGFRs) fusion genes involvement in hematological malignancies. Crit. Rev. Oncol. Hematol. 2017, 109, 20–34. [Google Scholar] [CrossRef]
- Heidaran, M.A.; Pierce, J.H.; Jensen, R.A.; Matsui, T.; Aaronson, S.A. Chimeric alpha- and beta-platelet-derived growth factor (PDGF) receptors define three immunoglobulin-like domains of the alpha-PDGF receptor that determine PDGF-AA binding specificity. J. Biol. Chem. 1990, 265, 18741–18744. [Google Scholar] [CrossRef] [PubMed]
- Lubinus, M.; Meier, K.E.; Smith, E.A.; Gause, K.C.; LeRoy, E.C.; Trojanowska, M. Independent effects of platelet-derived growth factor isoforms on mitogen-activated protein kinase activation and mitogenesis in human dermal fibroblasts. J. Biol. Chem. 1994, 269, 9822–9825. [Google Scholar] [CrossRef]
- Tsao, A.S.; Wei, W.; Kuhn, E.; Spencer, L.; Solis, L.M.; Suraokar, M.; Lee, J.J.; Hong, W.K.; Wistuba, I.I. Immunohistochemical overexpression of platelet-derived growth factor receptor-beta (PDGFR-β) is associated with PDGFRB gene copy number gain in sarcomatoid non-small-cell lung cancer. Clin. Lung Cancer 2011, 12, 369–374. [Google Scholar] [CrossRef]
- Kim, M.S.; Choi, H.S.; Wu, M.; Myung, J.; Kim, E.J.; Kim, Y.S.; Ro, S.; Ha, S.E.; Bartlett, A.; Wei, L.; et al. Potential Role of PDGFRβ-Associated THBS4 in Colorectal Cancer Development. Cancers 2020, 12, 2533. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Morandi, P.; Krishnamurthy, S.; Reuben, J.M.; Lee, B.N.; Francis, D.; Booser, D.J.; Green, M.C.; Arun, B.K.; Pusztai, L.; et al. Imatinib mesylate (Gleevec) in advanced breast cancer-expressing C-Kit or PDGFR-beta: Clinical activity and biological correlations. Ann. Oncol. 2008, 19, 1713–1719. [Google Scholar] [CrossRef]
- David, M.; Cross, N.C.; Burgstaller, S.; Chase, A.; Curtis, C.; Dang, R.; Gardembas, M.; Goldman, J.M.; Grand, F.; Hughes, G.; et al. Durable responses to imatinib in patients with PDGFRB fusion gene-positive and BCR-ABL-negative chronic myeloproliferative disorders. Blood 2007, 109, 61–64. [Google Scholar] [CrossRef]
- Apperley, J.F.; Gardembas, M.; Melo, J.V.; Russell-Jones, R.; Bain, B.J.; Baxter, E.J.; Chase, A.; Chessells, J.M.; Colombat, M.; Dearden, C.E.; et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N. Engl. J. Med. 2002, 347, 481–487. [Google Scholar] [CrossRef]
- Wang, P.; Song, L.; Ge, H.; Jin, P.; Jiang, Y.; Hu, W.; Geng, N. Crenolanib, a PDGFR inhibitor, suppresses lung cancer cell proliferation and inhibits tumor growth in vivo. OncoTargets Ther. 2014, 7, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Fujino, S.; Miyoshi, N.; Ito, A.; Yasui, M.; Ohue, M.; Ogino, T.; Takahashi, H.; Uemura, M.; Matsuda, C.; Mizushima, T.; et al. Crenolanib Regulates ERK and AKT/mTOR Signaling Pathways in RAS/BRAF-Mutated Colorectal Cancer Cells and Organoids. Mol. Cancer Res. 2021, 19, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.J.; Johnson, H.; Bronson, R.T.; de Feraudy, S.; White, F.; Charest, A. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res. 2012, 72, 3764–3774. [Google Scholar] [CrossRef]
- Neel, D.S.; Allegakoen, D.V.; Olivas, V.; Mayekar, M.K.; Hemmati, G.; Chatterjee, N.; Blakely, C.M.; McCoach, C.E.; Rotow, J.K.; Le, A.; et al. Differential Subcellular Localization Regulates Oncogenic Signaling by ROS1 Kinase Fusion Proteins. Cancer Res. 2019, 79, 546–556. [Google Scholar] [CrossRef]
- Cui, M.; Han, Y.; Li, P.; Zhang, J.; Ou, Q.; Tong, X.; Zhao, R.; Dong, N.; Wu, X.; Li, W.; et al. Molecular and clinicopathological characteristics of ROS1-rearranged non-small-cell lung cancers identified by next-generation sequencing. Mol. Oncol. 2020, 14, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chen, Z.; Huang, M.; Zhang, D.; Hu, M.; Jiao, F.; Quan, M. Detection of ROS1 gene fusions using next-generation sequencing for patients with malignancy in China. Front. Cell Dev. Biol. 2022, 10, 1035033. [Google Scholar] [CrossRef]
- Muminovic, M.; Uribe, C.R.C.; Alvarez-Pinzon, A.; Shan, K.; Raez, L.E. Importance of ROS1 gene fusions in non-small cell lung cancer. Cancer Drug Resist. 2023, 6, 332–344. [Google Scholar] [CrossRef]
- Bergethon, K.; Shaw, A.T.; Ou, S.-H.I.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 Rearrangements Define a Unique Molecular Class of Lung Cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef]
- Cai, W.; Li, X.; Su, C.; Fan, L.; Zheng, L.; Fei, K.; Zhou, C.; Manegold, C.; Schmid-Bindert, G. ROS1 fusions in Chinese patients with non-small-cell lung cancer. Ann. Oncol. 2013, 24, 1822–1827. [Google Scholar] [CrossRef]
- Lu, S.; Pan, H.; Wu, L.; Yao, Y.; He, J.; Wang, Y.; Wang, X.; Fang, Y.; Zhou, Z.; Wang, X.; et al. Efficacy, safety and pharmacokinetics of Unecritinib (TQ-B3101) for patients with ROS1 positive advanced non-small cell lung cancer: A Phase I/II Trial. Signal Transduct. Target. Ther. 2023, 8, 249. [Google Scholar] [CrossRef]
- Wang, V.; Bivona, T.; Ali, S.M.; Schrock, A.B.; Miller, V.A. CD74-ROS1 Fusion in NSCLC Detected by Hybrid Capture-Based Tissue Genomic Profiling and ctDNA Assays. J. Thorac. Oncol. 2017, 12, e19–e20. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Chen, P.; Zang, F.; Liu, Y.; Xu, X.; Su, Y.; Chen, J.; Lin, L.; Zhang, L.; Zhang, T. A patient with classic biphasic pulmonary blastoma harboring CD74-ROS1 fusion responds to crizotinib. Onco. Targets Ther. 2018, 11, 157–161. [Google Scholar] [CrossRef]
- Mizuno, T.; Fujiwara, Y.; Yoshida, K.; Kohno, T.; Ohe, Y. Next-Generation Sequencer Analysis of Pulmonary Pleomorphic Carcinoma with a CD74-ROS1 Fusion Successfully Treated with Crizotinib. J. Thorac. Oncol. 2019, 14, e106–e108. [Google Scholar] [CrossRef]
- Wang, G.; Gao, J.; Lv, J.; Chen, X.; Wu, J.; Wang, R.; Jiang, J. Effective Treatment with Cabozantinib in an Advanced Non-Small-Cell Lung Cancer Patient Harboring a CD74-ROS1 Fusion: A Case Report. Onco Targets Ther. 2020, 13, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Huang, S.; Ye, X.; Feng, L.; Lu, Y.; Zhou, C.; Zhao, J.; He, T.; Wang, J.; Li, B. Crizotinib resistance conferred by BRAF V600E mutation in non-small cell lung cancer harboring an oncogenic ROS1 fusion. Cancer Treat. Res. Commun. 2021, 27, 100377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Z.; Han, X.; Li, J.; Guo, H.; Shi, J. Acquired MET D1228N Mutations Mediate Crizotinib Resistance in Lung Adenocarcinoma with ROS1 Fusion: A Case Report. Oncologist 2021, 26, 178–181. [Google Scholar] [CrossRef]
- Cheng, Y.; Yang, J.; Wang, D.; Yan, D. ROS1 fusion lung adenosquamous carcinoma patient with short-term clinical benefit after crizotinib treatment: A case report. Ann. Transl. Med. 2022, 10, 157. [Google Scholar] [CrossRef]
- Tyler, L.C.; Le, A.T.; Chen, N.; Nijmeh, H.; Bao, L.; Wilson, T.R.; Chen, D.; Simmons, B.; Turner, K.M.; Perusse, D.; et al. MET gene amplification is a mechanism of resistance to entrectinib in ROS1+NSCLC. Thorac. Cancer 2022, 13, 3032–3041. [Google Scholar] [CrossRef]
- Tanaka, S.; Yoshimura, N.; Asakawa, R.; Tobita, S.; Yaga, M.; Ueno, K. A case of CD74-ROS1-positive lung adenocarcinoma diagnosed by next-generation sequencing achieved long-term survival with pemetrexed regimens. Thorac. Cancer 2023, 14, 2618–2621. [Google Scholar] [CrossRef]
- Pizzutilo, E.G.; Alberto Giuseppe, A.; Roazzi, L.; Romanò, R.; Motta, V.; Lauricella, C.; Marrapese, G.; Cerea, G.; Signorelli, D.; Veronese, S.M.; et al. Repotrectinib Overcomes F2004V Resistance Mutation in ROS1-Rearranged Non-Small Cell Lung Cancer: A Case Report. JTO Clin. Res. Rep. 2023. [Google Scholar] [CrossRef]
- Cha, Y.J.; Lee, C.; Joo, B.; Kim, K.A.; Lee, C.K.; Shim, H.S. Clinicopathological Characteristics of NRG1 Fusion-Positive Solid Tumors in Korean Patients. Cancer Res. Treat. 2023, 55, 1087–1095. [Google Scholar] [CrossRef]
- Chen, K.; Li, W.; Xi, X.; Zhong, J. A case of multiple primary lung adenocarcinoma with a CD74-NRG1 fusion protein and HER2 mutation benefit from combined target therapy. Thorac. Cancer 2022, 13, 3063–3067. [Google Scholar] [CrossRef]
- Abe, A.; Emi, N.; Tanimoto, M.; Terasaki, H.; Marunouchi, T.; Saito, H. Fusion of the platelet-derived growth factor receptor beta to a novel gene CEV14 in acute myelogenous leukemia after clonal evolution. Blood 1997, 90, 4271–4277. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Jang, J.H.; Koh, E.H. TRIP11-PDGFRB fusion in a patient with a therapy-related myeloid neoplasm with t(5;14)(q33;q32) after treatment for acute promyelocytic leukemia. Mol. Cytogenet. 2014, 7, 103. [Google Scholar] [CrossRef]
- Fayiga, F.F.; Reyes-Hadsall, S.C.; Moreno, B.A.; Oh, K.S.; Brathwaite, C.; Duarte, A.M. Novel ANKRD26 and PDGFRB gene mutations in pediatric case of non-Langerhans cell histiocytosis: Case report and literature review. J. Cutan. Pathol. 2023, 50, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Ou, S.I. Is NRG2α Fusion a “Doppelgänger” to NRG1α/β Fusions in Oncology? J. Thorac. Oncol. 2020, 15, 878–880. [Google Scholar] [CrossRef]
- Drilon, A.; Wang, L.; Arcila, M.E.; Balasubramanian, S.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.; Lipson, D.; Miller, V.A.; Kris, M.G.; et al. Broad, Hybrid Capture-Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin. Cancer Res. 2015, 21, 3631–3639. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Park, S.; Park, H.; Kim, S.; Lee, J.; Lee, J.; Youk, J.; Yi, K.; An, Y.; Park, I.K.; et al. Tracing Oncogene Rearrangements in the Mutational History of Lung Adenocarcinoma. Cell 2019, 177, 1842–1857.e21. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, Y.; Ning, H.; Chen, X.; Tan, X.; Ding, X. Clinic-pathologic features and gene fusion pattern of ALK and ROS1 in non-small cell lung cancer show association with household coal combustion. Transl. Cancer Res. 2019, 8, 2164–2174. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Zhang, R.; Xu, Q.; Yang, H.; Lizaso, A.; Xu, C.; Liu, J.; Wang, W.; Ou, S.I.; et al. Clinical and molecular factors that impact the efficacy of first-line crizotinib in ROS1-rearranged non-small-cell lung cancer: A large multicenter retrospective study. BMC Med. 2021, 19, 206. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Hsieh, M.S.; Wu, S.G.; Chang, Y.L.; Shih, J.Y.; Liu, Y.N.; Tsai, M.F.; Tsai, T.H.; Yu, C.J.; Yang, J.C.; et al. Clinical and the prognostic characteristics of lung adenocarcinoma patients with ROS1 fusion in comparison with other driver mutations in East Asian populations. J. Thorac. Oncol. 2014, 9, 1171–1179. [Google Scholar] [CrossRef]
- Kim, H.R.; Lim, S.M.; Kim, H.J.; Hwang, S.K.; Park, J.K.; Shin, E.; Bae, M.K.; Ou, S.H.; Wang, J.; Jewell, S.S.; et al. The frequency and impact of ROS1 rearrangement on clinical outcomes in never smokers with lung adenocarcinoma. Ann. Oncol. 2013, 24, 2364–2370. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.D.; Le, A.T.; Theodoro, M.F.; Skokan, M.C.; Aisner, D.L.; Berge, E.M.; Terracciano, L.M.; Cappuzzo, F.; Incarbone, M.; Roncalli, M.; et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 4570–4579. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Li, Y.; Xiao, L.; Xiong, Y.; Liu, L.; Jiang, W.; Heng, J.; Qu, J.; Yang, N.; Zhang, Y. Crizotinib presented with promising efficacy but for concomitant mutation in next- generation sequencing-identified ROS1-rearranged non-small-cell lung cancer. Onco Targets Ther. 2018, 11, 6937–6945. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Kohno, T.; Tsuta, K.; Wakai, S.; Arai, Y.; Shimada, Y.; Asamura, H.; Furuta, K.; Shibata, T.; Tsuda, H. ROS1-rearranged lung cancer: A clinicopathologic and molecular study of 15 surgical cases. Am. J. Surg. Pathol. 2013, 37, 554–562. [Google Scholar] [CrossRef]
- Koopman, B.; Kuijpers, C.; Groen, H.J.M.; Timens, W.; Schuuring, E.; Willems, S.M.; van Kempen, L.C. Detection of NTRK Fusions and TRK Expression and Performance of pan- TRK Immunohistochemistry in Routine Diagnostics: Results from a Nationwide Community-Based Cohort. Diagnostics 2022, 12, 668. [Google Scholar] [CrossRef]
- Ptáková, N.; Martínek, P.; Holubec, L.; Janovský, V.; Vančurová, J.; Grossmann, P.; Navarro, P.A.; Rodriguez Moreno, J.F.; Alaghehbandan, R.; Hes, O.; et al. Identification of tumors with NRG1 rearrangement, including a novel putative pathogenic UNC5D-NRG1 gene fusion in prostate cancer by data-drilling a de-identified tumor database. Genes Chromosomes Cancer 2021, 60, 474–481. [Google Scholar] [CrossRef] [PubMed]
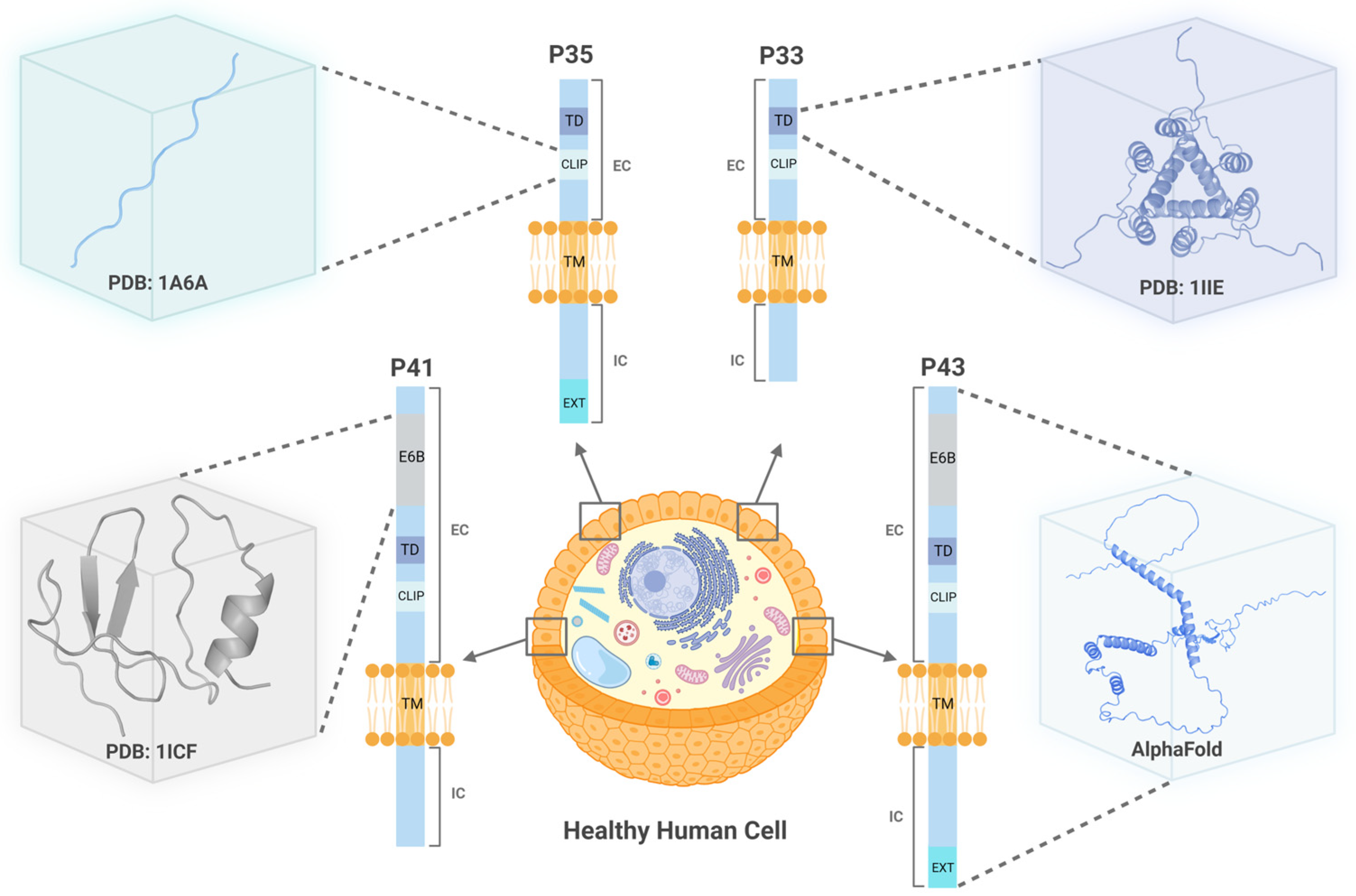
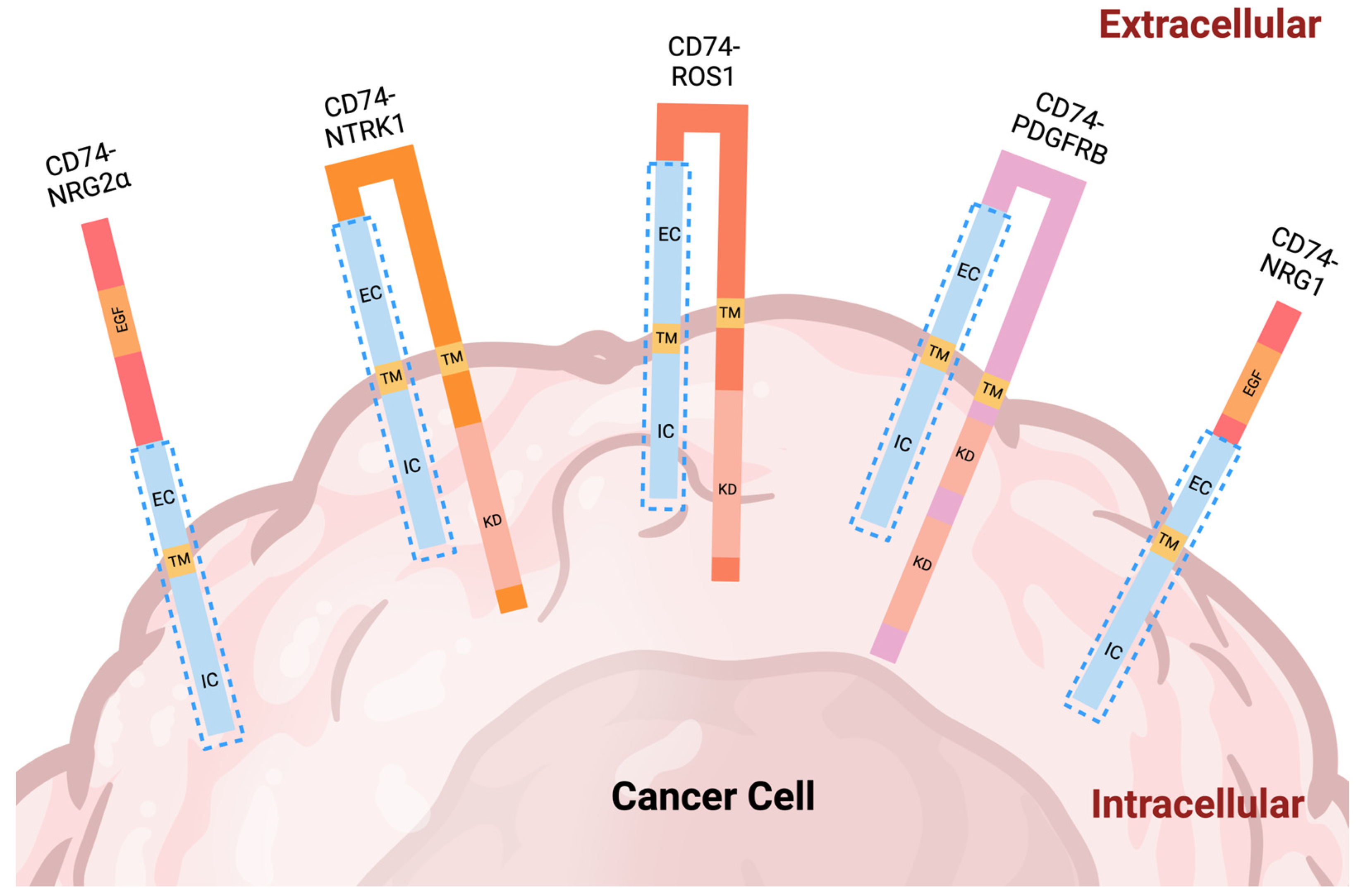
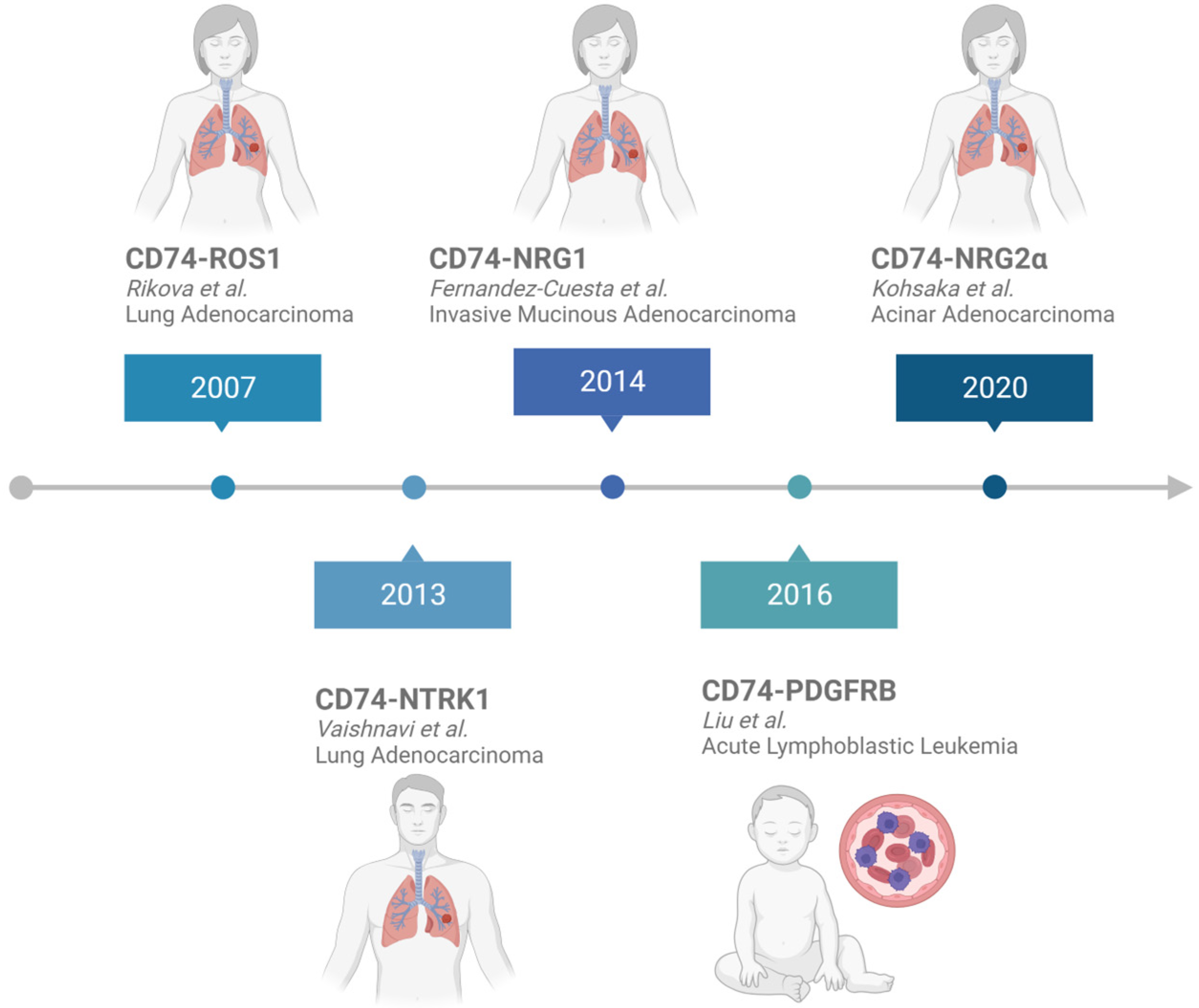

{kind=link}
{kind=link}
{kind=link}
| CD74 Fusion Protein | Exon Breakpoint | Cancer | Reference |
|---|---|---|---|
| CD74-ROS1 | C6-R34 | NSCLC | [30] |
| C6-R32 | NSCLC | [57] | |
| C7-R32 | NSCLC | [60] | |
| C7-R34 | IBC | [55] | |
| C3-R34 | NSCLC | [61] | |
| C6-R33 | NSCLC | [62] | |
| C6-R35 | NSCLC | [63] | |
| CD74-NTRK1 | C8-N12 | NSCLC | [31] |
| C7-N8 | NSCLC | [64] | |
| C6-N12 | NSCLC | [65] | |
| CD74-NRG1 | C6-N6 | IMA | [32] |
| C8-N6 | IMA | [59] | |
| C6-N4 | NSCLC | [66] | |
| C7-N6 | IMA | [67] | |
| CD74-PDGFRB | C6-P11 | B-ALL | [33] |
| CD74-NRG2α | C6-N2 | NSCLC | [34] |
| Diagnosis | Variant | Detection Method | Age | Gender | Smoker/Pack Year (PY) | Stage | Treatment | TKI Resistance | Lifespan OS | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| NSCLC | C7-R32 C6-R34 | RT-PCR | 61 | F | 0 | IV (T4N3M1b) | 1st: pemetrexed + cisplatin * refused crizotinib for economic reason | - | - | [60] |
| IBC | C7-R34 | NGS | 64 | F | - | cT4N3M1 | 1st: paclitaxel 2nd: capecitabine * refused crizotinib for economic reason | - | Died 4 mos. after dx | [55] |
| NSCLC | C3-R34 | NGS | 38 | F | 0 | IVA (T1aN3M1A) | 1st: cisplatin, pemetrexed, + bevacizumab 2nd: docetaxel + ramucirumab * 3rd: crizotinib * 4th: entrectinib | Crizotinib resistance: brain metastasis | 1 mo. PFS | [61] |
| Metastatic NSCLC | - | CGP (NGS) ctDNA assay | 41 | F | 0 | - | 1st: carboplatin, pemetrexed + bevacizumab * 2nd: crizotinib | - | PFS 4 mos. after crizotinib | [132] |
| CBPB | - | NGS | 44 | F | 0 | IV (cT3N1M1b) | * 1st: crizotinib | Crizotinib resistance: ROS1 G2032R mutation | PFS for 3 mos. PD after crizotinib resistance | [133] |
| PPC | - | NGS RT-PCR | 56 | F | 0 | IIIA (T2aN2M0) | 1st: resection 2nd: paclitaxel, carboplatin + bevacizumab * 3rd: crizotinib | - | CR after 6 mos. | [134] |
| NSCLC | C6-R34 | NGS | 40 | F | 0 | - | 1st: gemcitabine + cisplatin 2nd: docetaxel 3rd: gefitinib * 4th: cabozantinib * 5th: crizotinib | Cabozantinib resistance | SD after 2 mos. crizotinib | [135] |
| NSCLC | C6-R33 | NGS RT-qPCR | 51 | F | - | IVB (pT4N3M1c) | 1st: pemetrexed + cisplatin * 2nd: crizotinib | - | PFS 15.7 mos. after crizotinib | [58] |
| Metastatic NSCLC | C6-R33 | NGS | 60 | F | 0 | - | 1st: radiotherapy * 2nd: entrectinib | - | 5 mos. SD | [62] |
| NSCLC | C6-R33 | NGS | 53 | F | - | IV | * 1st: crizotinib * 2nd: dabrefenib | Crizotinib resistance: BRAF V600E mutation | 2 mos. PR after crizotinib. Dead 15 days after dabrefenib | [136] |
| Metastatic NSCLC | - | NGS | 30 | F | - | IIIC (T3N3M0) | 1st: cisplatin + pemetrexed 2nd: nedaplatin + pemetrexed * 3rd: crizotinib * 4th: cabozantinib | Crizotinib resistance: MET D1228N mutation | Dead 21 months from dx | [137] |
| NSCLC- ASC | C6-R34 | NGS | 43 | F | 0 | IIIA (pT2aN2M0) | 1st: albumin-bound paclitaxel + camrelizumab * 2nd: crizotinib 3rd: pemetrexed + carboplatin + bevacizumab 4th: cisplatin + gemcitabine + bevacizumab + * crizotinib | - | Dead ~15 mos. after dx | [138] |
| NSCLC | C6-R32 C6-R35 | DNA NGS RNA NGS FISH Sanger Seq | 44 | F | 0 | IVB | * 1st: crizotinib * 2nd: lorlatinib 3rd: chemotherapy | Crizotinib resistance: bone metastasis; lorlatinib resistance: ROS1 G2032R mutation | Dead ~19 mos. after dx | [63] |
| NSCLC | - | Molecular testing—not specified | 54 | M | 0 | IV | * 1st: crizotinib * 2nd: entrectinib 3rd: carboplatin + pemetrexed 4th: pemetrexed + pembrolizumab * 5th: repotrectinib * 6th: cabozantinib | Possible entrectinib resistance: MET amplification | Deceased ~50 months after dx | [139] |
| NSCLC | - | NGS | 54 | F | 30 | IVA (T2aN3M1a) | 1st: cisplatin + pemetrexed 2nd: carboplatin + paclitaxel 3rd: pemetrexed 4th: nivolumab 5th: docetaxel 6th: pemetrexed 7th: S-1 8th: gemcitabine * 9th: entrectinib | - | 1st: PR 2nd: SD 3rd: PR 4th: SD 5th: PR 6th: PD 7th: PD 8th: PD 9th: PR | [140] |
| NSCLC | - | NGS | 49 | F | 0 | IV | * 1st: entrectinib * 2nd: crizotinib 3rd: carboplatin * 4th: repotrectinib | Possible entrectinib resistance: ROS1 F2004V mutation | 1st: PR 2nd: SD 3rd: PD 4th: PR | [141] |
| Diagnosis | Variant | Detection Method | Age | Gender | Smoker/Pack Year (PY) | Stage | Treatment | Lifespan OS | Reference |
|---|---|---|---|---|---|---|---|---|---|
| NSCLC | - | Genetic Testing | 62 | F | 0 | IV | 1st: pemetrexed + cisplatin 2nd: atezolizumab + albumin-bound paclitaxel + carboplatin * 3rd: afatinib + pyrotinib | PFS 5 mos. | [143] |
| IMA | C6-N6 | NGS | 86 | M | 0 | IIA (pT2bN0M0) | 1st: carboplatin + pemetrexed 2nd: paclitaxel + bevacizumab 3rd: nivolumab 4th: GSK2849330 (anti-ERBB3 mAb) * 5th: afatinib | PR 1 year and 7 mos. On GSK2849330 | [67] |
| IMA | C8-N6 | Multiplex PCR | 81 | M | Former, smoked cigars for 1 year | IIB (pT3N0M0) | * 1st: afatinib | SD at 6 wks. and PD at 13 wks. | [67] |
| IMA | C7-N6 | NGS | 51 | M | <1 | IIIB (T4N2M0) | 1st: cisplatin + pemetrexed * 2nd: afatinib | PD at 8 wks. after afatinib started | [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vargas, J.; Pantouris, G. Analysis of CD74 Occurrence in Oncogenic Fusion Proteins. Int. J. Mol. Sci. 2023, 24, 15981. https://doi.org/10.3390/ijms242115981
Vargas J, Pantouris G. Analysis of CD74 Occurrence in Oncogenic Fusion Proteins. International Journal of Molecular Sciences. 2023; 24(21):15981. https://doi.org/10.3390/ijms242115981
Chicago/Turabian StyleVargas, Jasmine, and Georgios Pantouris. 2023. "Analysis of CD74 Occurrence in Oncogenic Fusion Proteins" International Journal of Molecular Sciences 24, no. 21: 15981. https://doi.org/10.3390/ijms242115981
APA StyleVargas, J., & Pantouris, G. (2023). Analysis of CD74 Occurrence in Oncogenic Fusion Proteins. International Journal of Molecular Sciences, 24(21), 15981. https://doi.org/10.3390/ijms242115981