
In Vivo and In Vitro Characterization of the RNA Binding Capacity of SETD1A (KMT2F)

 , and
, and 
Abstract
:1. Introduction
2. Results
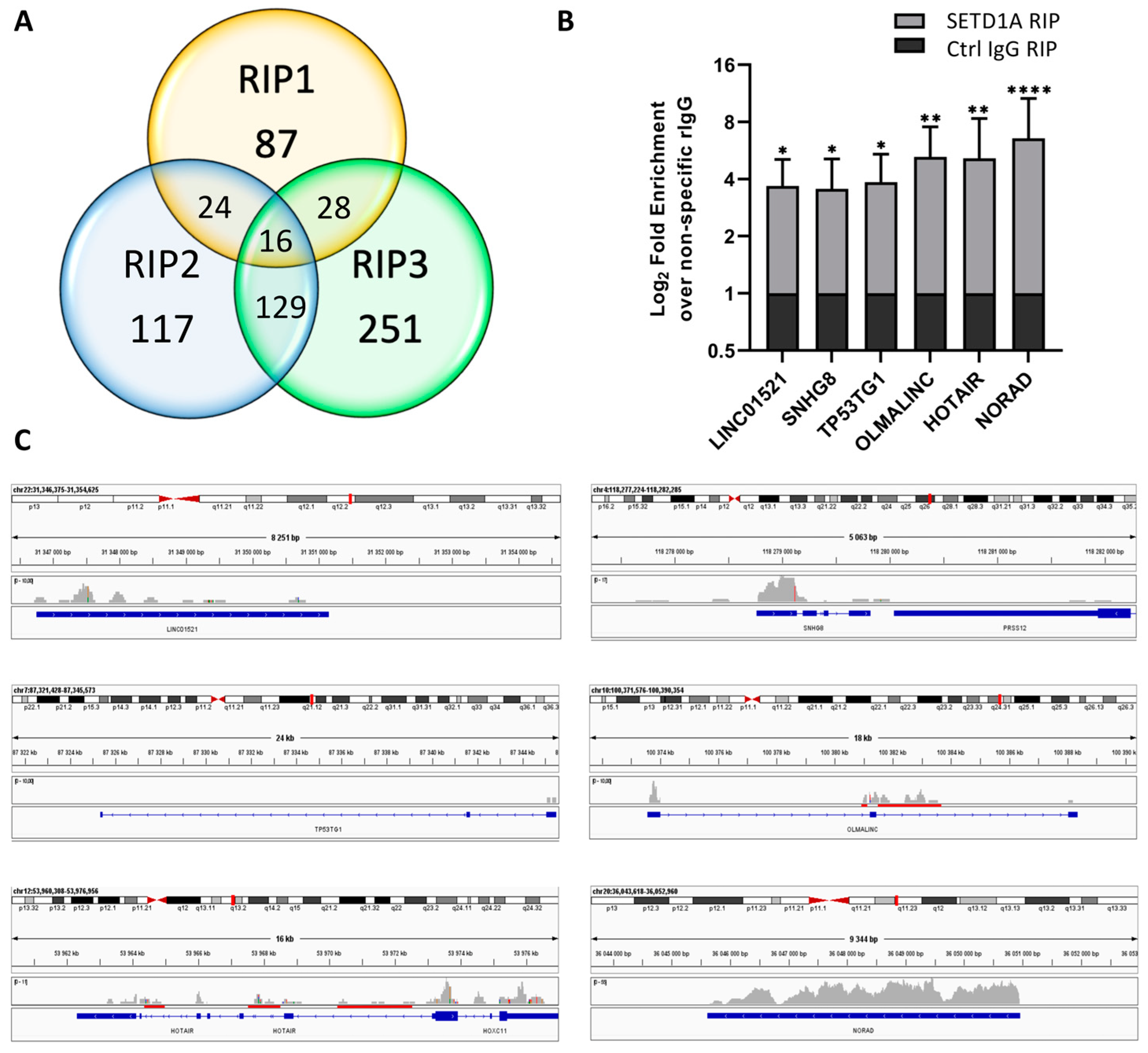
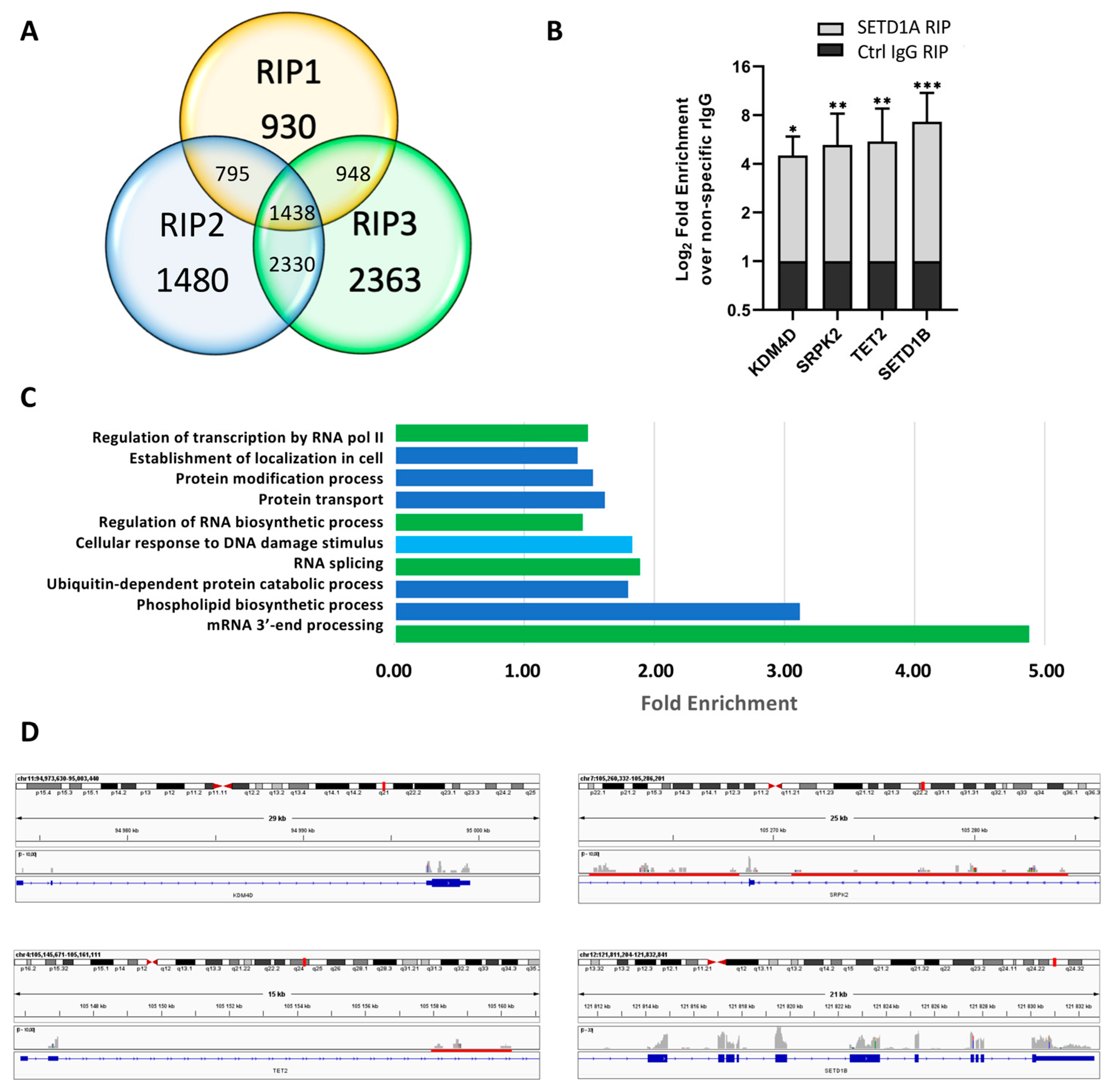
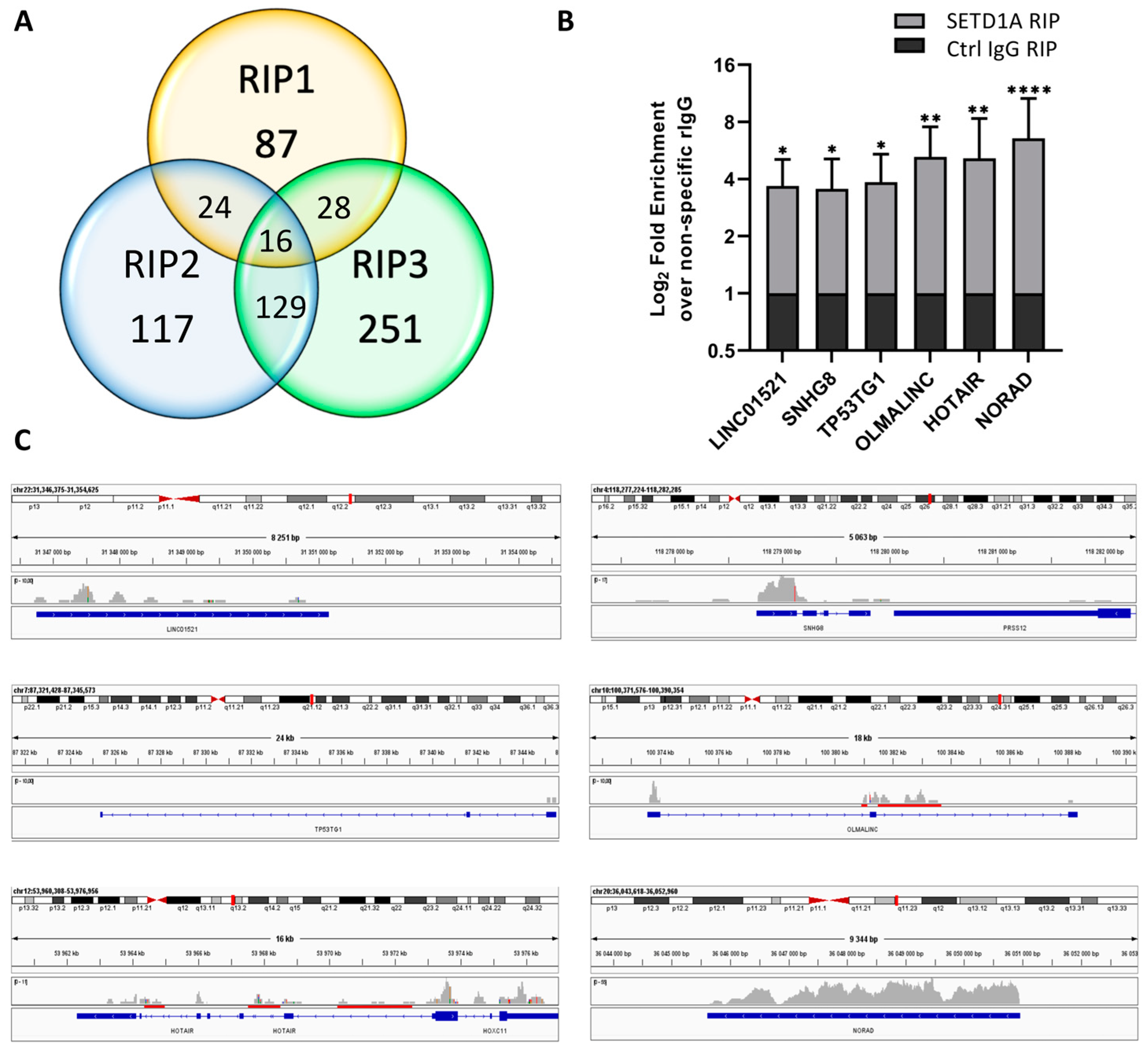
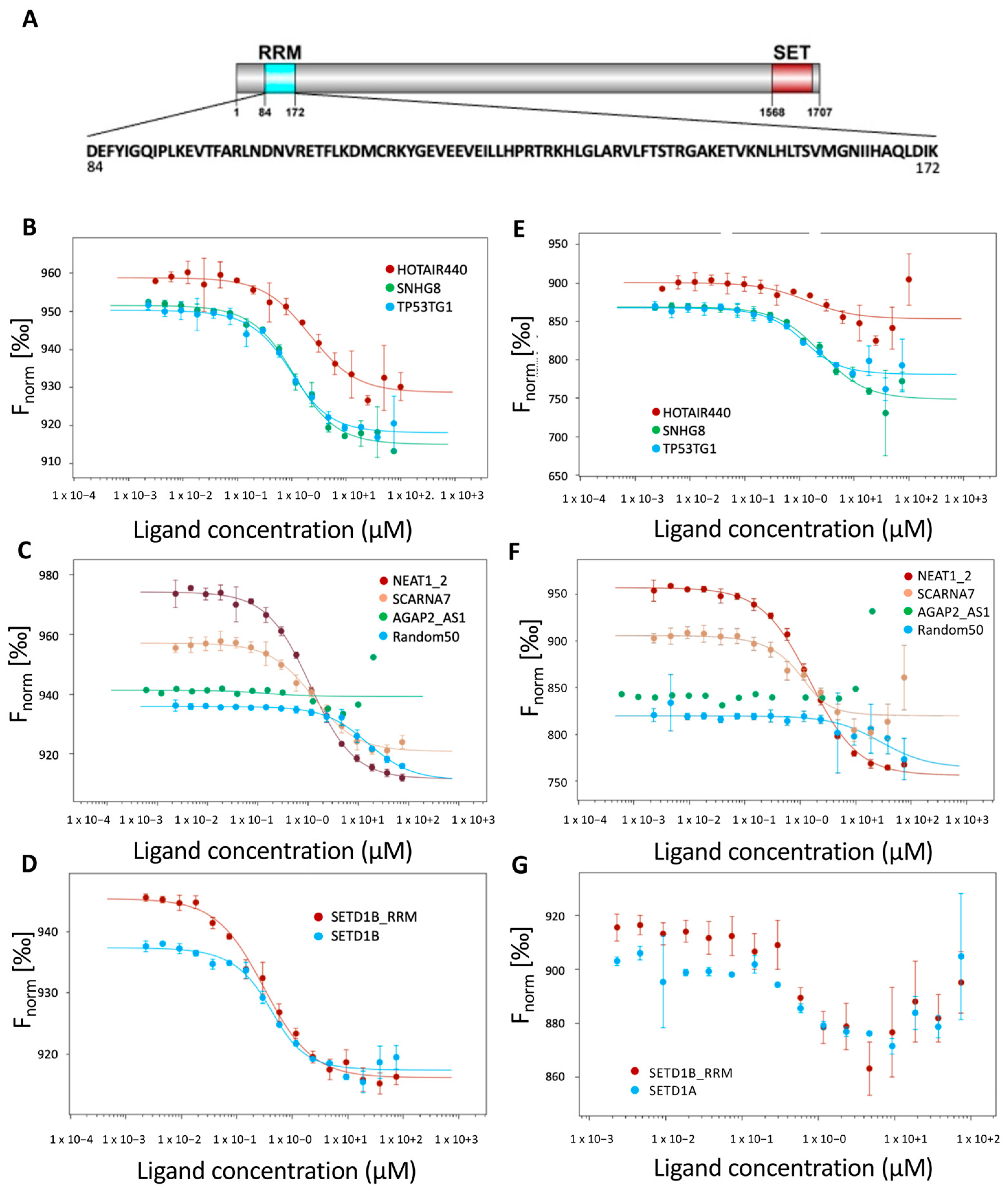
2.1. SETD1A Interacts with a Broad Range of Coding and Non-Coding RNAs
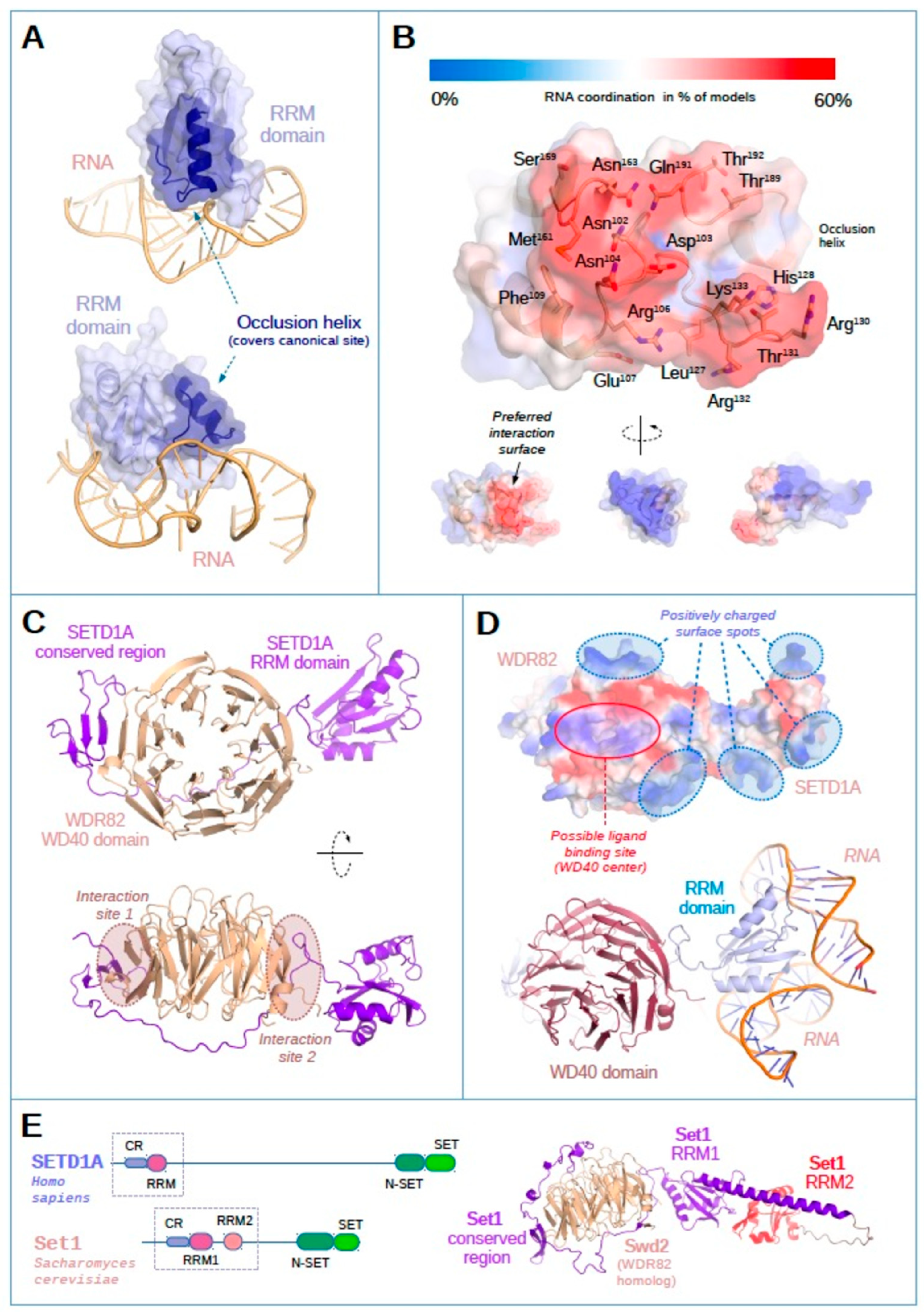
2.2. The SETD1A RRM Domain Is an Independent RNA Recognition Unit
2.3. Possible RNA-Interacting Interface of SED1A RRM
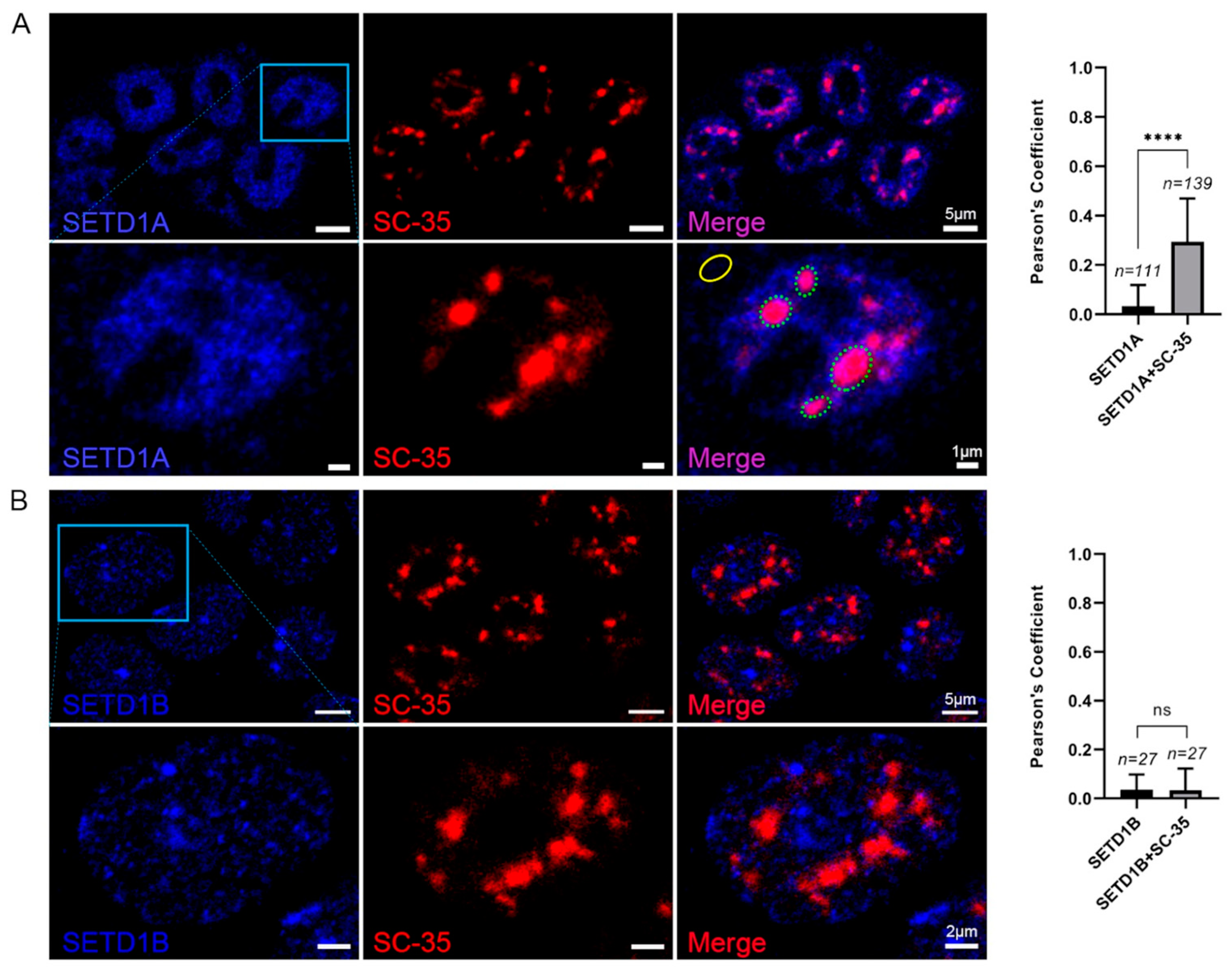
2.4. SETD1A Localizes to Splicing Speckles in the Nucleus
3. Discussion
4. Materials and Methods
4.1. Experimental Models
4.2. Cell Culture Conditions
4.3. Nuclear Extraction for RNA Immunoprecipitation
4.4. RNA Immunoprecipitation (RIP)
4.5. RNA Purification
4.6. RNA Sequencing Data Analysis
4.7. Bioinformatics Analyses
4.8. cDNA Synthesis
4.9. RT-qPCR
4.10. SETD1A_RRM Expression and Purification
4.11. Far-UV Circular Dichroism
4.12. RNA In Vitro Transcription, Purification, and Labeling
4.13. Electrophoretic Mobility Shift Assay (EMSA)
4.14. Microscale Thermophoresis (MST)
4.15. In Silico Structural Modeling
4.16. Co-Immunoprecipitation (Co-IP)
4.17. Western Blot
4.18. Immunoflourescence Staining (IFS)
4.19. Confocal Microscopy
4.20. Co-Localization Intensity Analysis
4.21. Statistical Analysis
4.21.1. qPCR Experiments
4.21.2. Colocalization Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- Small Cajal body-specific RNA 7 (SCARNA7), 330 nt

- AGAP2 antisense RNA 1 (AGAP2-AS1), 1567 nt

- Nuclear Paraspeckle Assembly Transcript 1_2 (12_13k segment), NEAT1_2, 976 nt
- HOX transcript antisense RNA (1-440), HOTAIR440, 440nt
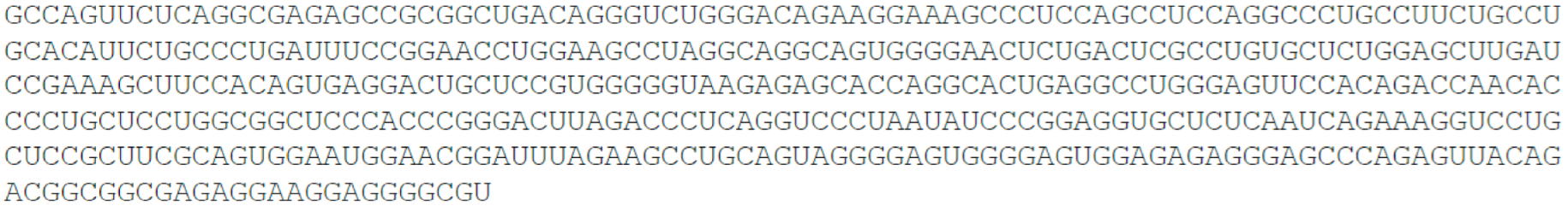
- TP53 target long non-coding RNA, TP53TG1, 707 nt
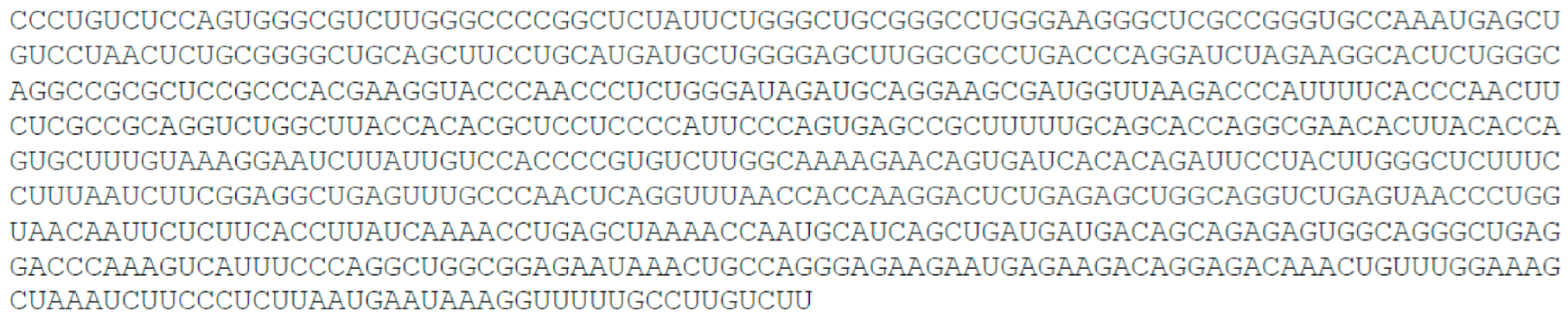
- Small nucleolar RNA host gene 8, SNHG8, 570 nt
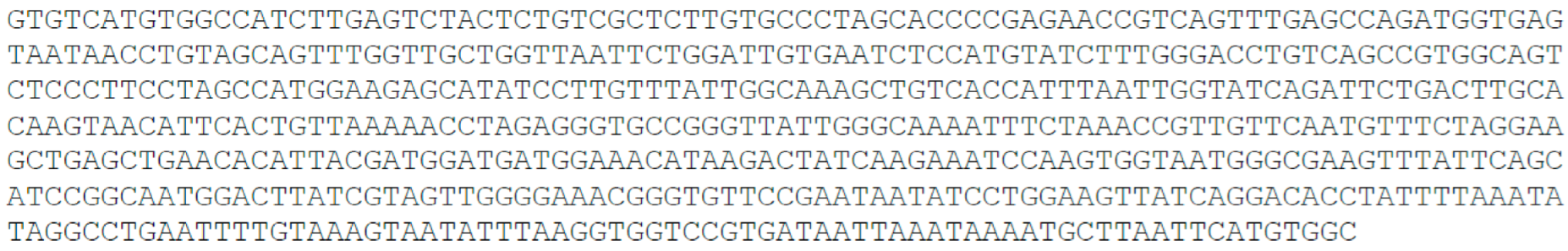
- Random 50 nucleotide, 50 nt
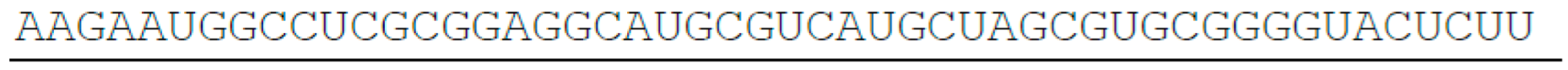
- SETD1B, 511 nt

- SETD1A, 2643 nt

References
- Herz, H.-M.; Mohan, M.; Garruss, A.S.; Liang, K.; Takahashi, Y.-H.; Mickey, K.; Voets, O.; Verrijzer, C.P.; Shilatifard, A. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012, 26, 2604–2620. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Herz, H.-M.; Smith, E.R.; Zhang, Y.; Jackson, J.; Washburn, M.P.; Washburn, M.P.; Florens, L.; Eissenberg, C.J.; Shilatifard, A. The COMPASS family of H3K4 methylases in Drosophila. Mol. Cell. Biol. 2011, 31, 4310–4318. [Google Scholar] [CrossRef]
- Sugeedha, J.; Gautam, J.; Tyagi, S. SET1/MLL family of proteins: Functions beyond histone methylation. Epigenetics 2021, 16, 469–487. [Google Scholar] [CrossRef] [PubMed]
- van Nuland, R.; Smits, A.H.; Pallaki, P.; Jansen, P.W.T.C.; Vermeulen, M.; Timmers, H.T.M. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol. Cell. Biol. 2013, 33, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.J.; Dingwall, A.K. The cancer COMPASS: Navigating the functions of MLL complexes in cancer. Cancer Genet. 2015, 208, 178–191. [Google Scholar] [CrossRef]
- Mishra, B.P.; Zaffuto, K.M.; Artinger, E.L.; Org, T.; Mikkola, H.K.A.; Cheng, C.; Djabali, M.; Ernst, P. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 2014, 7, 1239–1247. [Google Scholar] [CrossRef]
- Lee, J.; Saha, P.K.; Yang, Q.-H.; Lee, S.; Park, J.Y.; Suh, Y.; Lee, S.-K.; Chan, L.; Roeder, R.G.; Lee, J.W. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 19229–19234. [Google Scholar] [CrossRef]
- Lee, J.-E.; Wang, C.; Xu, S.; Cho, Y.-W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2013, 2, e01503. [Google Scholar] [CrossRef]
- Bledau, A.S.; Schmidt, K.; Neumann, K.; Hill, U.; Ciotta, G.; Gupta, A.; Torres, D.C.; Fu, J.; Kranz, A.; Stewart, A.F.; et al. The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development 2014, 141, 1022–1035. [Google Scholar] [CrossRef]
- Lee, J.-H.; Tate, C.M.; You, J.-S.; Skalnik, D.G. Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J. Biol. Chem. 2007, 282, 13419–13428. [Google Scholar] [CrossRef]
- Cao, K.; Collings, C.K.; Marshall, S.A.; Morgan, M.A.; Rendleman, E.J.; Wang, L.; Sze, C.C.; Sun, T.; Bartom, E.T.; Shilatifard, A. SET1A/COMPASS and shadow enhancers in the regulation of homeotic gene expression. Genes Dev. 2017, 31, 787–801. [Google Scholar] [CrossRef]
- Kranz, A.; Anastassiadis, K. The role of SETD1A and SETD1B in development and disease. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194578. [Google Scholar] [CrossRef]
- Singh, T.; Kurki, M.I.; Curtis, D.; Purcell, S.M.; Crooks, L.; McRae, J.; Suvisaari, J.; Chheda, H.; Blackwood, D.; Breen, G.; et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 2016, 19, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Krzyzewska, I.M.; Maas, S.M.; Henneman, P.; Lip, K.V.D.; Venema, A.; Baranano, K.; Chassevent, A.; Aref-Eshghi, E.; van Essen, A.J.; Fukuda, T.; et al. A genome-wide DNA methylation signature for SETD1B-related syndrome. Clin. Epigenetics 2019, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Hiraide, T.; Nakashima, M.; Yamoto, K.; Fukuda, T.; Kato, M.; Ikeda, H.; Sugie, Y.; Aoto, K.; Kaname, T.; Nakabayashi, K.; et al. De novo variants in SETD1B are associated with intellectual disability, epilepsy and autism. Hum. Genet. 2018, 137, 95–104. [Google Scholar] [CrossRef]
- Hiraide, T.; Hattori, A.; Ieda, D.; Hori, I.; Saitoh, S.; Nakashima, M.; Saitsu, H. De novo variants in SETD1B cause intellectual disability, autism spectrum disorder, and epilepsy with myoclonic absences. Epilepsia Open 2019, 4, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Arndt, K.; Kranz, A.; Fohgrub, J.; Jolly, A.; Bledau, A.S.; Di Virgilio, M.; Dahl, A.; Höfer, T.; Stewart, A.F.; Waskow, C. SETD1A protects HSCs from activation-induced functional decline in vivo. Blood 2018, 131, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Trésaugues, L.; Dehé, P.-M.; Guérois, R.; Rodriguez-Gil, A.; Varlet, I.; Salah, P.; Pamblanco, M.; Luciano, P.; Quevillon-Cheruel, S.; Sollier, J.; et al. Structural characterization of Set1 RNA recognition motifs and their role in histone H3 lysine 4 methylation. J. Mol. Biol. 2006, 359, 1170–1181. [Google Scholar] [CrossRef]
- Luciano, P.; Jeon, J.; El-Kaoutari, A.; Challal, D.; Bonnet, A.; Barucco, M.; Candelli, T.; Jourquin, F.; Lesage, P.; Kim, J.; et al. Binding to RNA regulates Set1 function. Cell Discov. 2017, 3, 17040. [Google Scholar] [CrossRef]
- Hoshii, T.; Cifani, P.; Feng, Z.; Huang, C.-H.; Koche, R.; Chen, C.-W.; Delaney, C.D.; Lowe, S.W.; Kentsis, A.; Armstrong, S.A. A Non-catalytic Function of SETD1A Regulates Cyclin K and the DNA Damage Response. Cell 2018, 172, 1007–1021.e17. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R.; Xu, W.; Delcuve, G.P. Histone H3K4 trimethylation: Dynamic interplay with pre-mRNA splicing. Biochem. Cell Biol. 2016, 94, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Rojas, C.; Hernandez, A.J.; Sarma, K.; Lee, J.T. Regulatory interactions between RNA and polycomb repressive complex 2. Mol. Cell 2014, 55, 171–185. [Google Scholar] [CrossRef]
- Antonov, I.; Medvedeva, Y. Direct Interactions with Nascent Transcripts Is Potentially a Common Targeting Mechanism of Long Non-Coding RNAs. Genes 2020, 11, 1483. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zhu, X.; Chen, Y.; Wei, H.; Chen, Q.; Chi, X.; Qi, B.; Zhang, L.; Zhao, Y.; Gao, G.F.; et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 2014, 16, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tang, Y.; Ge, Y.; Zhang, S.; Zheng, M. Long non-coding RNA NRAV enhances proliferation and invasion of hepatocellular carcinoma cells by modulating the Wnt/β-catenin signaling pathway. Bioengineered 2022, 13, 10026–10037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, K.; Li, J.; Chen, F.; Li, Y.; Lin, Q. Long Noncoding RNA HCG11 Acts as a Tumor Suppressor in Gastric Cancer by Regulating miR-942-5p/BRMS1 Axis. J. Oncol. 2021, 2021, 9961189. [Google Scholar] [CrossRef]
- Xu, J.; Xu, W.; Yang, X.; Liu, Z.; Sun, Q. LncRNA HCG11/miR-579-3p/MDM2 axis modulates malignant biological properties in pancreatic carcinoma via Notch/Hes1 signaling pathway. Aging 2021, 13, 16471–16484. [Google Scholar] [CrossRef]
- Poltronieri, P.; Ahmad, A.; Uddin, S. LncRNAs in Cancer Metastasis Therapy Resistance Volume, I.I; Frontiers Media SA: Lausanne, Switzerland, 2023. [Google Scholar]
- Wang, L.; Du, L.; Duan, W.; Yan, S.; Xie, Y.; Wang, C. Overexpression of long noncoding RNA NORAD in colorectal cancer associates with tumor progression. Onco Targets Ther. 2018, 11, 6757–6766. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef]
- Micsonai, A.; Moussong, É.; Wien, F.; Boros, E.; Vadászi, H.; Murvai, N.; Lee, Y.H.; Molnár, T.; Réfrégiers, M.; Goto, Y.; et al. BeStSel: Webserver for secondary structure and fold prediction for protein CD spectroscopy. Nucleic Acids Res. 2022, 50, W90–W98. [Google Scholar] [CrossRef]
- Jiang, H.; Cole, P.A. N-Terminal Protein Labeling with N-Hydroxysuccinimide Esters and Microscale Thermophoresis Measurements of Protein-Protein Interactions Using Labeled Protein. Curr. Protoc. 2021, 1, e14. [Google Scholar] [CrossRef]
- Bao, S.; Xu, C. Molecular insight into the SETD1A/B N-terminal region and its interaction with WDR82. Biochem. Biophys. Res. Commun. 2023, 658, 136–140. [Google Scholar] [CrossRef]
- Zeke, A.; Schád, É.; Horváth, T.; Abukhairan, R.; Szabó, B.; Tantos, A. Deep Structural Insights into RNA-Binding Disordered Protein Regions. Wiley Interdiscip. Rev. RNA 2022, 13, e1714. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Skalnik, D.G. Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A Histone H3-Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol. Cell. Biol. 2008, 28, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Galganski, L.; Urbanek, M.O.; Krzyzosiak, W.J. Nuclear speckles: Molecular organization, biological function and role in disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef]
- Ilik, İ.A.; Malszycki, M.; Lübke, A.K.; Schade, C.; Meierhofer, D.; Aktaş, T. SON and SRRM2 are essential for nuclear speckle formation. Elife 2020, 9, e60579. [Google Scholar] [CrossRef]
- Hirose, T.; Nakagawa, S. Paraspeckles: Possible nuclear hubs by the RNA for the RNA. Biomol. Concepts 2012, 3, 415–428. [Google Scholar] [CrossRef]
- GHendrickson, D.; Kelley, D.R.; Tenen, D.; Bernstein, B.; Rinn, J.L. Widespread RNA binding by chromatin-associated proteins. Genome Biol. 2016, 17, 28. [Google Scholar]
- Sayou, C.; Millán-Zambrano, G.; Santos-Rosa, H.; Petfalski, E.; Robson, S.; Houseley, J.; Kouzarides, T.; Tollervey, D. RNA Binding by Histone Methyltransferases Set1 and Set2. Mol. Cell. Biol. 2017, 37, e00165-17. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Xiao, Y.; Tang, J.; Wu, J.; Ma, J.; Tian, B.; Zhou, Y.; Wang, H.; Yang, D.; Liao, Q.J.; et al. Role of lncRNA and EZH2 Interaction/Regulatory Network in Lung Cancer. J. Cancer 2018, 9, 4156–4165. [Google Scholar] [CrossRef] [PubMed]
- Hao, A.; Wang, Y.; Stovall, D.B.; Wang, Y.; Sui, G. Emerging Roles of LncRNAs in the EZH2-regulated Oncogenic Network. Int. J. Biol. Sci. 2021, 17, 3268–3280. [Google Scholar] [CrossRef]
- Mirzaei, S.; Gholami, M.H.; Hushmandi, K.; Hashemi, F.; Zabolian, A.; Canadas, I.; Zarrabi, A.; Nabavi, N.; Aref, A.R.; Crea, F.; et al. The long and short non-coding RNAs modulating EZH2 signaling in cancer. J. Hematol. Oncol. 2022, 15, 18. [Google Scholar] [CrossRef]
- Hu, H.; Guo, X.; Mu, T.; Song, H. Long non-coding RNA telomerase RNA elements improve glucocorticoid-induced osteoporosis by EZH2 to regulate DKK1. Int. J. Rheum. Dis. 2023, 26, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Li, G.; Son, J.; Xu, C.-F.; Margueron, R.; Neubert, T.A.; Reinberg, D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated up-regulates its binding to ncRNA. Genes Dev. 2010, 24, 2615–2620. [Google Scholar] [CrossRef] [PubMed]
- Carlson, S.M.; Moore, K.E.; Sankaran, S.M.; Reynoird, N.; Elias, J.E.; Gozani, O. A Proteomic Strategy Identifies Lysine Methylation of Splicing Factor snRNP70 by the SETMAR Enzyme. J. Biol. Chem. 2015, 290, 12040–12047. [Google Scholar] [CrossRef]
- Carlson, S.M.; Soulette, C.M.; Yang, Z.; Elias, J.E.; Brooks, A.N.; Gozani, O. RBM25 is a global splicing factor promoting inclusion of alternatively spliced exons and is itself regulated by lysine mono-methylation. J. Biol. Chem. 2017, 292, 13381–13390. [Google Scholar] [CrossRef]
- Moore, K.E.; Carlson, S.M.; Camp, N.D.; Cheung, P.; James, R.G.; Chua, K.F.; Wolf-Yadlin, A.; Gozani, O. A general molecular affinity strategy for global detection and proteomic analysis of lysine methylation. Mol. Cell 2013, 50, 444–456. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Shavrukov, Y. Plant Genotyping: Methods and Protocols; Springer Nature: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.-P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Marmisolle, F.E.; García, M.L.; Reyes, C.A. RNA-binding protein immunoprecipitation as a tool to investigate plant miRNA processing interference by regulatory proteins of diverse origin. Plant Methods 2018, 14, 9. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Sarzynska, J.; Popenda, M.; Antczak, M.; Szachniuk, M. RNA tertiary structure prediction using RNAComposer in CASP15. Proteins 2023, 91, 1461–1534. [Google Scholar] [CrossRef]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. A knowledge-based scoring function for protein-RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Res. 2014, 42, e55. [Google Scholar] [CrossRef] [PubMed]
- Kamina, A.D.; Williams, N. Non-canonical binding interactions of the RNA recognition motif (RRM) domains of P34 protein modulate binding within the 5S ribonucleoprotein particle (5S RNP). PLoS ONE 2017, 12, e0177890. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Chamorro, J.; Piñeiro, D.; Gordon, J.M.B.; Ramajo, J.; Francisco-Velilla, R.; Macias, M.J.; Martinez-Salas, E. Identification of novel non-canonical RNA-binding sites in Gemin5 involved in internal initiation of translation. Nucleic Acids Res. 2014, 42, 5742–5754. [Google Scholar] [CrossRef] [PubMed]
- Szabó, B.; Murvai, N.; Abukhairan, R.; Schád, É.; Kardos, J.; Szeder, B.; Buday, L.; Tantos, Á. Disordered Regions of Mixed Lineage Leukemia 4 (MLL4) Protein Are Capable of RNA Binding. Int. J. Mol. Sci. 2018, 19, 3478. [Google Scholar] [CrossRef]
- Halbach, A.; Zhang, H.; Wengi, A.; Jablonska, Z.; Gruber, I.M.L.; Halbeisen, R.E.; Dehé, P.M.; Kemmeren, P.; Holstege, F.; Géli, V.; et al. Cotranslational assembly of the yeast SET1C histone methyltransferase complex. EMBO J. 2009, 28, 2959–2970. [Google Scholar] [CrossRef]
- Hollmann, N.M.; Jagtap, P.K.A.; Masiewicz, P.; Guitart, T.; Simon, B.; Provaznik, J.; Stein, F.; Haberkant, P.; Sweetapple, L.J.; Villacorta, L.; et al. Pseudo-RNA-Binding Domains Mediate RNA Structure Specificity in Upstream of N-Ras. Cell Rep. 2020, 32, 107930. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA | Kd T-Jump (μM) | Kd Thermophoresis (μM) | Kd Back Diffusion (μM) |
|---|---|---|---|
| SNHG8 (570 nt) | 0.87 | 2.19 | 1.19 |
| TP53TG1 (707 nt) | 0.67 | 0.69 | 1.05 |
| HOTAIR440 (440 nt) | 2.09 | 1.39 | 1.45 |
| NEAT1_2 (2976 nt) | 1.11 | 1.52 | 1.89 |
| AGAP2_AS1 (1567 nt) | n.d. | n.d. | n.d. |
| SCARNA7 (330 nt) | 1.28 | 0.31 | 1.17 |
| SETD1B (511 nt) | 0.31 | n.d. | 0,31 |
| SETD1A (2643 nt) | 0.21 | n.d. | 0.06 |
| Random50 (50 nt) | 15.50 | 28.14 | 11.77 |
| LED Power (%) | MST Power (%) | Before MST (s) | MST on (s) | After MST (s) | Delay (s) |
|---|---|---|---|---|---|
| 20–40 | 40 | 5 | 30 | 5 | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amin, H.M.; Szabo, B.; Abukhairan, R.; Zeke, A.; Kardos, J.; Schad, E.; Tantos, A. In Vivo and In Vitro Characterization of the RNA Binding Capacity of SETD1A (KMT2F). Int. J. Mol. Sci. 2023, 24, 16032. https://doi.org/10.3390/ijms242216032
Amin HM, Szabo B, Abukhairan R, Zeke A, Kardos J, Schad E, Tantos A. In Vivo and In Vitro Characterization of the RNA Binding Capacity of SETD1A (KMT2F). International Journal of Molecular Sciences. 2023; 24(22):16032. https://doi.org/10.3390/ijms242216032
Chicago/Turabian StyleAmin, Harem Muhamad, Beata Szabo, Rawan Abukhairan, Andras Zeke, József Kardos, Eva Schad, and Agnes Tantos. 2023. "In Vivo and In Vitro Characterization of the RNA Binding Capacity of SETD1A (KMT2F)" International Journal of Molecular Sciences 24, no. 22: 16032. https://doi.org/10.3390/ijms242216032
APA StyleAmin, H. M., Szabo, B., Abukhairan, R., Zeke, A., Kardos, J., Schad, E., & Tantos, A. (2023). In Vivo and In Vitro Characterization of the RNA Binding Capacity of SETD1A (KMT2F). International Journal of Molecular Sciences, 24(22), 16032. https://doi.org/10.3390/ijms242216032