
Changing the Electron Acceptor Specificity of Rhodobacter capsulatus Formate Dehydrogenase from NAD+ to NADP+

Abstract
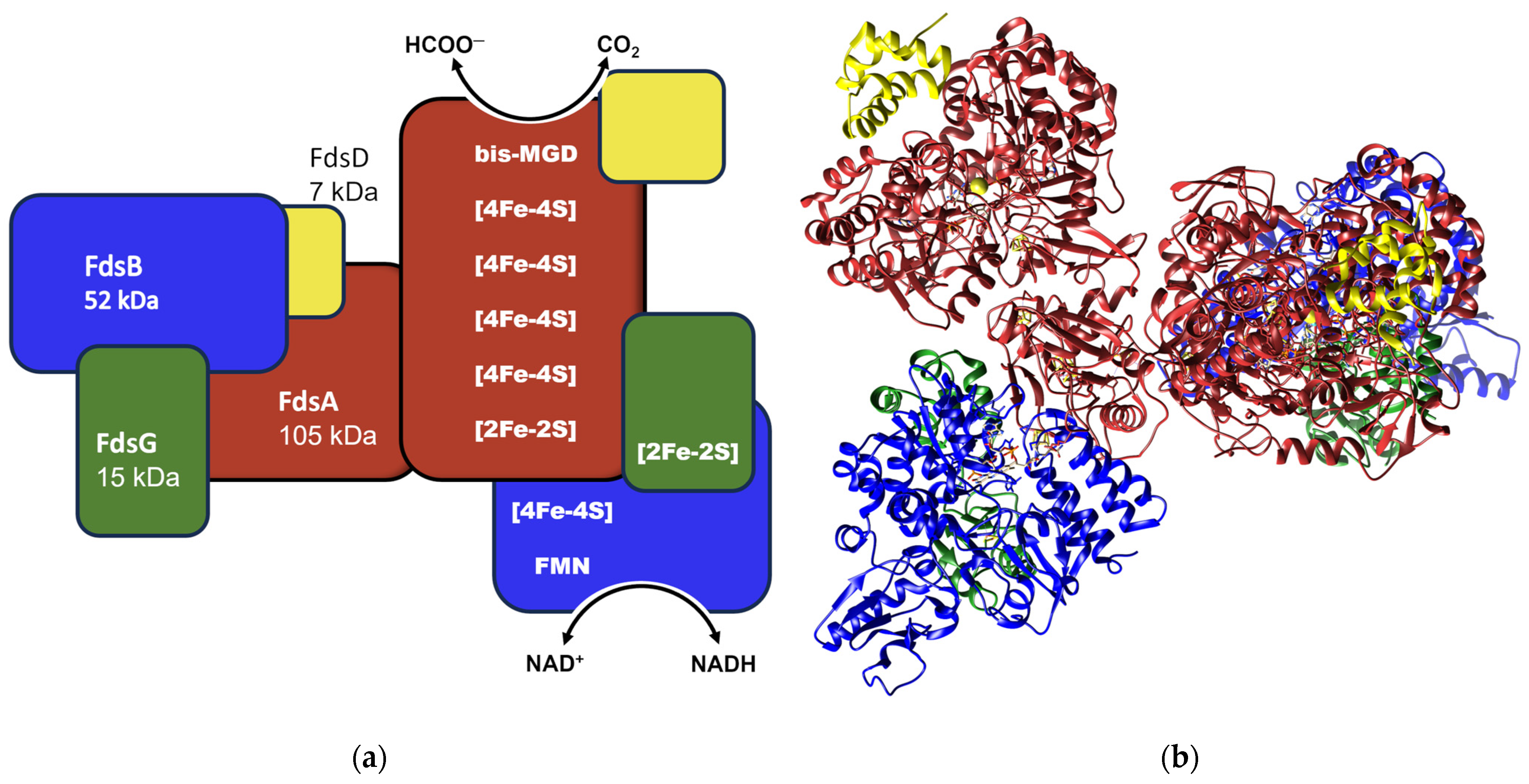
:1. Introduction
2. Results
2.1. Production of RcFDH Variants at the NAD+ Site
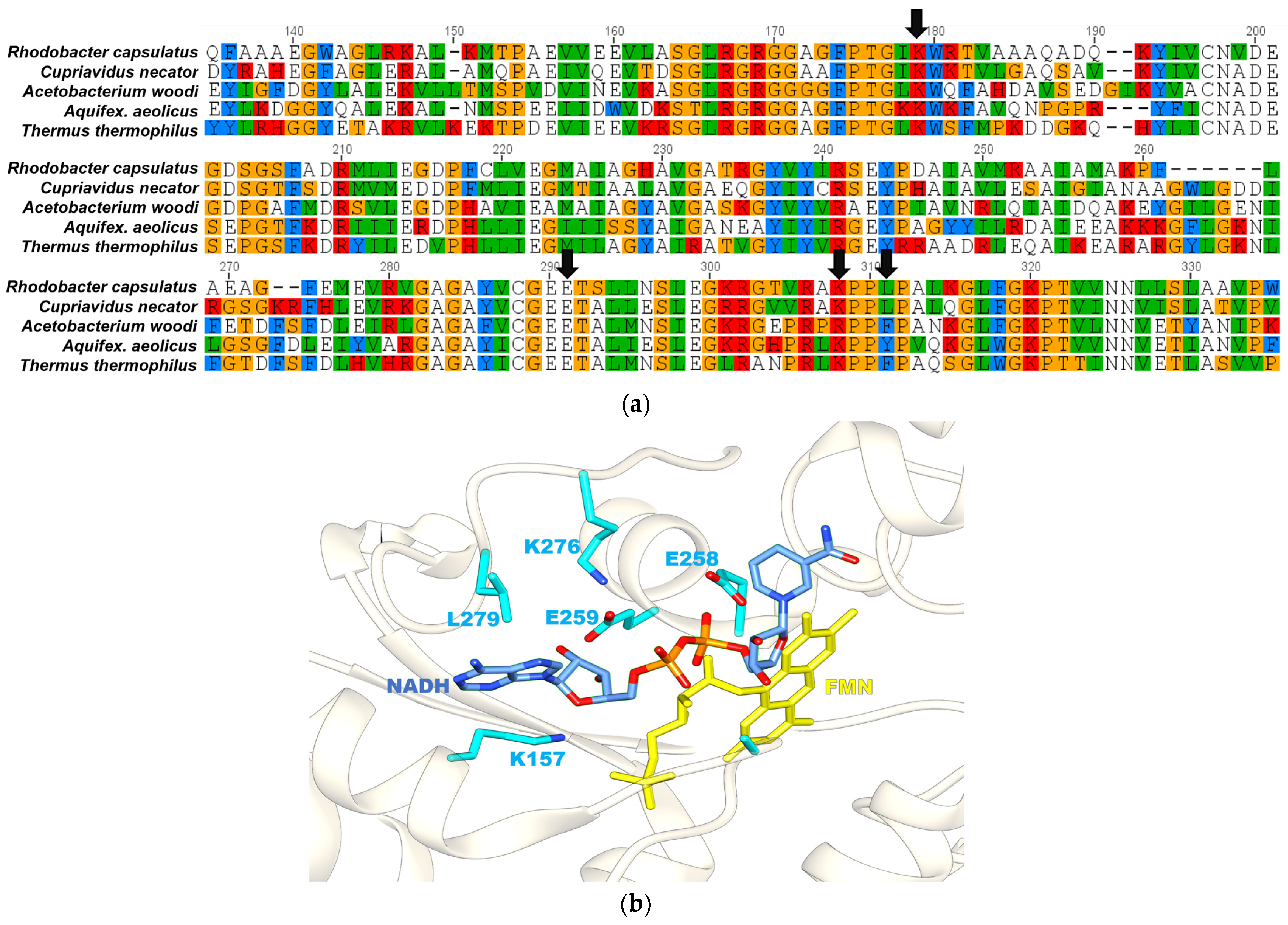
2.2. Lys157 of FdsB Subunit Is Essential for the Binding of the Nicotinamide Cofactor
2.3. Cofactor Specificity of RcFDH Variants Testing Formate Oxidation
2.4. CO2 Reduction Catalyzed by RcFDH Variants Using NADPH
3. Discussion
4. Materials and Methods
4.1. Site-Directed Mutagenesis
4.2. Protein Purification
4.3. Enzymatic Assays
4.4. CO2 Reduction Reactions
4.5. Determination of Cofactor Saturation
4.6. Formate Detection Using Gas Chromatography-Mass Spectrometry (GC-MS)
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olivier, J.G.; Schure, K.M.; Peters, J.A.H.W. Trends in Global CO2 and Total Greenhouse Gas Emissions; PBL Netherlands Environmental Assessment Agency: The Hague, The Netherlands, 2020; p. 4331. [Google Scholar]
- Yishai, O.; Lindner, S.N.; Gonzalez de la Cruz, J.; Tenenboim, H.; Bar-Even, A. The formate bio-economy. Curr. Opin. Chem. Biol. 2016, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bierbaumer, S.; Nattermann, M.; Schulz, L.; Zschoche, R.; Erb, T.J.; Winkler, C.K.; Tinzl, M.; Glueck, S.M. Enzymatic Conversion of CO2: From Natural to Artificial Utilization. Chem. Rev. 2023, 123, 5702–5754. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Hall, J.; Basu, P. The mononuclear molybdenum enzymes. Chem. Rev. 2014, 96, 2757–2816. [Google Scholar] [CrossRef] [PubMed]
- Schute, H.; Flossdorf, J.; Sahm, H.; Kula, M.R. Purification and properties of formaldehyde dehydrogenase and formate dehydrogenase from Candida boidinii. Eur. J. Biochem. 1976, 62, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Rusching, U.; Müller, U.; Willnow, P.; Höpner, T. CO2 reduction to formate by NADH catalysed by formate dehydrogenase from Pseudomonas oxalaticus. Eur. J. Biochem. 1976, 70, 325–330. [Google Scholar] [CrossRef]
- Popov, V.O.; Lamzin, V.S. NAD (+)-dependent formate dehydrogenase. Biochem. J. 1994, 301, 625. [Google Scholar] [CrossRef]
- Alqarni, M.H.; Foudah, A.I.; Muharram, M.M.; Budurian, H.; Labrou, N.E. Probing the Role of the Conserved Arg174 in Formate Dehydrogenase by Chemical Modification and Site-Directed Mutagenesis. Molecules 2021, 26, 1222. [Google Scholar] [CrossRef]
- Pagano, P.; Guo, Q.; Ranasinghe, C.; Schroeder, E.; Robben, K.; Hase, F.; Ye, H.; Wickersham, K.; Aspuru-Guzik, A.; Major, D.T. Oscillatory active-site motions correlate with kinetic isotope effects in formate dehydrogenase. ACS Catal. 2019, 9, 11199–11206. [Google Scholar] [CrossRef]
- Bommarius, A.; Drauz, K.; Hummel, W.; Kula, M.; Wandrey, C. Some new developments in reductive amtnation with cofactor regeneration. Biocatalysis 1994, 10, 37–47. [Google Scholar] [CrossRef]
- Shaked, Z.e.; Whitesides, G.M. Enzyme-catalyzed organic synthesis: NADH regeneration by using formate dehydrogenase. J. Am. Chem. Soc. 1980, 102, 7104–7105. [Google Scholar] [CrossRef]
- Gröger, H.; Rollmann, C.; Hüsken, H.; Werner, H.; Chamouleau, F.; Hagedorn, C.; Drauz, K.; Hummel, W. Coupled Cofactor-Dependent Enzymatic Reaction Systems in Aqueous Media. U.S. Patent 7,632,665, 15 December 2009. [Google Scholar]
- Bachosz, K.; Zdarta, J.; Bilal, M.; Meyer, A.S.; Jesionowski, T. Enzymatic cofactor regeneration systems: A new perspective on efficiency assessment. Sci. Total Environ. 2023, 868, 161630. [Google Scholar] [CrossRef] [PubMed]
- Iobbi-Nivol, C.; Leimkühler, S. Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Calzadiaz-Ramirez, L.; Meyer, A.S. Formate dehydrogenases for CO2 utilization. Curr. Opin. Biotechnol. 2022, 73, 95–100. [Google Scholar] [CrossRef]
- Alpdagtas, S.; Binay, B. NADP+-dependent formate dehydrogenase: A review. Biocatal. Biotransformation 2021, 39, 260–268. [Google Scholar] [CrossRef]
- Andreesen, J.R.; Ljungdahl, L.G. Nicotinamide adenine dinucleotide phosphate-dependent formate dehydrogenase from Clostridium thermoaceticum: Purification and properties. J. Bacteriol. 1974, 120, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.B.; Stadtman, T. Reconstitution of a formate-NADP+ oxidoreductase from formate dehydrogenase and a 5-deazaflavin-linked NADP+ reductase isolated from Methanococcus vannielii. J. Biol. Chem. 1980, 255, 1049–1053. [Google Scholar] [CrossRef]
- Ihara, M.; Kawano, Y.; Urano, M.; Okabe, A. Light driven CO2 fixation by using cyanobacterial photosystem I and NADPH-dependent formate dehydrogenase. PLoS ONE 2013, 8, e71581. [Google Scholar] [CrossRef]
- Hartmann, T.; Schrapers, P.; Utesch, T.; Nimtz, M.; Rippers, Y.; Dau, H.; Mroginski, M.A.; Haumann, M.; Leimkuhler, S. The Molybdenum Active Site of Formate Dehydrogenase Is Capable of Catalyzing C-H Bond Cleavage and Oxygen Atom Transfer Reactions. Biochemistry 2016, 55, 2381–2389. [Google Scholar] [CrossRef]
- Hartmann, T.; Leimkühler, S. The oxygen-tolerant and NAD-dependent formate dehydrogenase from Rhodobacter capsulatus is able to catalyze the reduction of CO2 to formate. FEBS J. 2013, 280, 6083–6096. [Google Scholar] [CrossRef]
- Kumar, H.; Khosraneh, M.; Bandaru, S.S.; Schulzke, C.; Leimkühler, S. The Mechanism of Metal-Containing Formate Dehydrogenases Revisited: The Formation of Bicarbonate as Product Intermediate Provides Evidence for an Oxygen Atom Transfer Mechanism. Molecules 2023, 28, 1537. [Google Scholar] [CrossRef]
- Laun, K.; Duffus, B.R.; Kumar, H.; Oudsen, J.P.H.; Karafoulidi-Retsou, C.; Tadjoung Waffo, A.; Hildebrandt, P.; Hoang Ly, K.; Leimkühler, S.; Katz, S.; et al. A Minimal Light-Driven System to Study the Enzymatic CO2 Reduction of Formate Dehydrogenase. ChemCatChem 2022, 14, e202201067. [Google Scholar] [CrossRef]
- Duffus, B.R.; Schrapers, P.; Schuth, N.; Mebs, S.; Dau, H.; Leimkuhler, S.; Haumann, M. Anion Binding and Oxidative Modification at the Molybdenum Cofactor of Formate Dehydrogenase from Rhodobacter capsulatus Studied by X-ray Absorption Spectroscopy. Inorg. Chem. 2020, 59, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Radon, C.; Mittelstadt, G.; Duffus, B.R.; Burger, J.; Hartmann, T.; Mielke, T.; Teutloff, C.; Leimkuhler, S.; Wendler, P. Cryo-EM structures reveal intricate Fe-S cluster arrangement and charging in Rhodobacter capsulatus formate dehydrogenase. Nat. Commun. 2020, 11, 1912. [Google Scholar] [CrossRef]
- Sazanov, L.A.; Hinchliffe, P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 2006, 311, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Niks, D.; Hakopian, S.; Tam, T.K.; Yu, X.; Hille, R.; Blaha, G.M. Crystallographic and kinetic analyses of the FdsBG subcomplex of the cytosolic formate dehydrogenase FdsABG from Cupriavidus necator. J. Biol. Chem. 2020, 295, 6570–6585. [Google Scholar] [CrossRef]
- Katsyv, A.; Kumar, A.; Saura, P.; Pöverlein, M.C.; Freibert, S.A.; Stripp, T.S.; Jain, S.; Gamiz-Hernandez, A.P.; Kaila, V.R.; Müller, V. Molecular basis of the electron bifurcation mechanism in the [FeFe]-hydrogenase complex HydABC. J. Am. Chem. Soc. 2023, 145, 5696–5709. [Google Scholar] [CrossRef]
- Schulte, M.; Frick, K.; Gnandt, E.; Jurkovic, S.; Burschel, S.; Labatzke, R.; Aierstock, K.; Fiegen, D.; Wohlwend, D.; Gerhardt, S. A mechanism to prevent production of reactive oxygen species by Escherichia coli respiratory complex I. Nat. Commun. 2019, 10, 2551. [Google Scholar] [CrossRef]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef]
- Cahn, J.K.; Werlang, C.A.; Baumschlager, A.; Brinkmann-Chen, S.; Mayo, S.L.; Arnold, F.H. A General Tool for Engineering the NAD/NADP Cofactor Preference of Oxidoreductases. ACS Synth. Biol. 2017, 6, 326–333. [Google Scholar] [CrossRef]
- Hartmann, T.; Schwanhold, N.; Leimkuhler, S. Assembly and catalysis of molybdenum or tungsten-containing formate dehydrogenases from bacteria. Biochim. Biophys. Acta 2015, 1854, 1090–1100. [Google Scholar] [CrossRef]
- Morina, K.; Schulte, M.; Hubrich, F.; Dörner, K.; Steimle, S.; Stolpe, S.; Friedrich, T. Engineering the respiratory complex I to energy-converting NADPH: Ubiquinone oxidoreductase. J. Biol. Chem. 2011, 286, 34627–34634. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Geng, Q.; Chen, C.; Zheng, Y.C.; Yu, H.L.; Xu, J.H. Engineering a Formate Dehydrogenase for NADPH Regeneration. Chembiochem 2023, e202300390. [Google Scholar] [CrossRef] [PubMed]
- Chanique, A.M.; Parra, L.P. Protein Engineering for Nicotinamide Coenzyme Specificity in Oxidoreductases: Attempts and Challenges. Front. Microbiol. 2018, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Baccour, M.; Lamotte, A.; Sakai, K.; Dubreucq, E.; Mehdi, A.; Kano, K.; Galarneau, A.; Drone, J.; Brun, N. Production of formate from CO2 gas under ambient conditions: Towards flow-through enzyme reactors. Green. Chem. 2020, 22, 3727–3733. [Google Scholar] [CrossRef]
- Medipally, H.; Guarneri, A.; Pospisil, L.; Franssen, M.C.R.; van Berkel, W.J.H.; Paul, C.E.; Nowaczyk, M.M. Light-Driven NADPH Cofactor Recycling by Photosystem I for Biocatalytic Reactions. ChemCatChem 2023, 15, e202300821. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NADP+ | NAD+ | |||||
|---|---|---|---|---|---|---|
| KM a (mM) | kcat a (min−1) | kcat/KM (min−1 mM−1) | KM (Mm) | kcat (min−1) | kcat/KM (min−1 mM−1) | |
| RcFDHWT | ND b | ND | 0.12 ± 0.001 | 3565 ± 105 | 29,708 | |
| FdsBE259R | 3.31 ± 1.4 | 367 ± 45 | 105.7 | 7.48 ± 2.1 | 863.6 ± 56 | 115.06 |
| FdsBE259K | 5.2 ± 1.0 | 342 ± 40 | 68.4 | 4.36 ± 1.8 | 354 ± 55 | 81.2 |
| FdsBE259G | 5.5 ± 0.8 | 551 ± 78 | 112 | 3.0 ± 1.2 | 551 ± 95 | 183.7 |
| FdsBE259Q | 6.3 ± 0.7 | 496 ± 70 | 78.7 | 3.34 ± 1.7 | 1508 ± 120 | 451.5 |
| FdsBE259D | ND | ND | 2.75 ± 0.8 | 1545 ± 140 | 515 | |
| FdsBK157R | ND | ND | ND | ND | ||
| FdsBK157S | ND | ND | ND | ND | ||
| FdsBL279R | 5.4 ± 1.7 | 405 ± 56 | 75 | 6.4 ± 1.7 | 736 ± 84 | 115 |
| FdsBK276A | 4.2 ± 1.0 | 350 ± 39 | 83.3 | 6.8 ± 1.5 | 943 ± 114 | 138.6 |
| FdsBK276A/L279R | 5.2 ± 0.7 | 480 ± 29 | 92.3 | 7.0 ± 2.0 | 687 ± 78 | 98.1 |
| FdsBE259R/L279R | 3.9 ± 0.9 | 347 ± 42 | 89 | 5.9 ± 2.2 | 766 ± 98 | 129.8 |
| FdsBE259G/L279R | 4.1 ± 0.9 | 381 ± 51 | 92.9 | 6.2 ± 1.8 | 629 ± 74 | 101.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, H.; Leimkühler, S. Changing the Electron Acceptor Specificity of Rhodobacter capsulatus Formate Dehydrogenase from NAD+ to NADP+. Int. J. Mol. Sci. 2023, 24, 16067. https://doi.org/10.3390/ijms242216067
Kumar H, Leimkühler S. Changing the Electron Acceptor Specificity of Rhodobacter capsulatus Formate Dehydrogenase from NAD+ to NADP+. International Journal of Molecular Sciences. 2023; 24(22):16067. https://doi.org/10.3390/ijms242216067
Chicago/Turabian StyleKumar, Hemant, and Silke Leimkühler. 2023. "Changing the Electron Acceptor Specificity of Rhodobacter capsulatus Formate Dehydrogenase from NAD+ to NADP+" International Journal of Molecular Sciences 24, no. 22: 16067. https://doi.org/10.3390/ijms242216067
APA StyleKumar, H., & Leimkühler, S. (2023). Changing the Electron Acceptor Specificity of Rhodobacter capsulatus Formate Dehydrogenase from NAD+ to NADP+. International Journal of Molecular Sciences, 24(22), 16067. https://doi.org/10.3390/ijms242216067