Abstract
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) causes coronavirus disease 2019 (COVID-19), which has killed ~7 million persons worldwide. Chronic kidney disease (CKD) is the most common risk factor for severe COVID-19 and one that most increases the risk of COVID-19-related death. Moreover, CKD increases the risk of acute kidney injury (AKI), and COVID-19 patients with AKI are at an increased risk of death. However, the molecular basis underlying this risk has not been well characterized. CKD patients are at increased risk of death from multiple infections, to which immune deficiency in non-specific host defenses may contribute. However, COVID-19-associated AKI has specific molecular features and CKD modulates the local (kidney) and systemic (lung, aorta) expression of host genes encoding coronavirus-associated receptors and factors (SCARFs), which SARS-CoV-2 hijacks to enter cells and replicate. We review the interaction between kidney disease and COVID-19, including the over 200 host genes that may influence the severity of COVID-19, and provide evidence suggesting that kidney disease may modulate the expression of SCARF genes and other key host genes involved in an effective adaptive defense against coronaviruses. Given the poor response of certain CKD populations (e.g., kidney transplant recipients) to SARS-CoV-2 vaccines and their suboptimal outcomes when infected, we propose a research agenda focusing on CKD to develop the concept of comorbidity-specific targeted therapeutic approaches to SARS-CoV-2 infection or to future coronavirus infections.
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused over 769 million confirmed cases of coronavirus disease 2019 (COVID-19) and over 6.95 million deaths worldwide, as reported by the World Health Organization (WHO), but the real numbers are believed to be much higher [1]. The WHO declared the COVID-19 outbreak a pandemic on 11 March 2020, and on 5 May 2023, declared that it is no longer a public health emergency of international concern [2]. The disease is now considered endemic. In this regard, it is still topical to advance our understanding of the pathogenesis of COVID-19 and the molecular mechanisms that determine the susceptibility to severe disease. Thus, the peak number of globally diagnosed cases was observed as recently as December 2022, and an uptick of cases driven by novel varieties was observed in July–August 2023 [1]. Additionally, any acquired knowledge will be helpful in managing the next viral pandemic outbreak. Moreover, reinfection contributed to additional risks of death (HR: 2.17, 95% CI 1.93–2.45), hospitalization (HR = 3.32, 95% CI 3.13–3.51), and sequelae among different organ systems, compared with no reinfection, despite vaccination status [3]. Kidney disorders showed the highest risk after reinfection (HR = 3.55, 95% CI = 3.18–3.97; burden = 38.31, 95% CI = 32.86–44.37), and this risk remained elevated independently of the status of vaccination [3] (Figure 1).
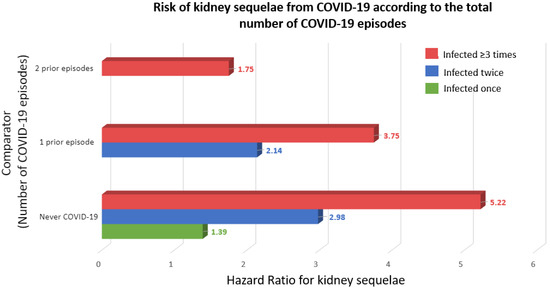
Figure 1.
Risk of kidney sequelae from COVID-19 according to the total number of COVID-19 episodes [3]. Kidney sequelae were defined as acute kidney injury or chronic kidney disease. Outcomes were defined at time of first incidence of their component individual sequela.
A plethora of manuscripts have identified host genes hijacked by SARS-CoV-2 and have explored the genetic predisposition to COVID-19 severity, and they have improved our understanding of the role of host genes in facilitating or limiting disease severity [4,5]. Key host genes include those encoding SARS-CoV-2 and coronavirus-associated receptors and factors (SCARFs), that is, molecules involved in SARS-CoV-2 cell entry, replication, and assembly, as well as those encoding host defense mechanisms. In a recent example, a common HLA-B allele (HLA-B*15:0) explains why around 20% of persons infected by SARS-CoV-2 remain asymptomatic, as it is associated with memory T-cell cross-reactivity to a peptide from seasonal coronaviruses [6]. However, an understanding of the molecular mechanisms of susceptibility to severe COVID-19 in patients with kidney disease has lagged, despite chronic kidney disease (CKD) being the most common risk factor for severe COVID-19 worldwide and one resulting in the most increase in the risk of COVID-19-related death [7,8,9]. We now first review the concepts of CKD and acute kidney injury (AKI) and the clinical interaction between COVID-19 and CKD or AKI. This is followed by an assessment of persistent unmet needs, ranging from the suboptimal response to SARS-CoV-2 vaccines in some populations of patients with CKD to the persistent risks represented by novel coronaviruses or SARS-CoV-2 variants. We then proceed to assess and extract data regarding the recent information on the specific molecular pathogenesis of COVID-19-associated AKI and the molecular mechanisms that may underlie the increased severity of COVID-19 in patients with CKD, which illustrate the feasibility and need to better understand the molecular pathogenesis of COVID-19 in the context of kidney disease as a means to develop kidney disease-specific targeted therapeutic approaches that improve the COVID-19 outcomes in patients with kidney disease. Finally, we propose a research agenda.
2. Acute Kidney Injury and Chronic Kidney Disease
According to Kidney Disease: Improving Global Outcomes (KDIGO), kidney disease may be acute or chronic, and the diagnostic criteria are used to identify patients with adverse health outcomes [10,11]. Since 2002, CKD has been defined as the presence of structural and/or functional alterations in the kidneys persisting for longer than 3 months with an impact on health [12]. However, the current, more precise definition dates from 2012 [10,13]. This timing should be considered, as when the SARS-CoV-2 pandemic hit in 2020, most healthcare professionals worldwide had not studied the current concept of CKD in medical schools and neither had the health authorities. Consequently, the striking negative impact of COVID-19 in persons with CKD flew initially under the radar, unlike other comorbidities that were soon identified as risk factors for severe COVID-19, such as diabetes, hypertension, and cardiovascular disease [14]. The diagnostic criteria that by themselves allow for the detection of CKD include an estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73 m2 or urinary albumin–creatinine ratio ≥ 30 mg/g, or pathological changes in the urinary sediment, kidney histology or imaging, or kidney transplantation [13]. eGFR and albuminuria allow for the categorization of CKD into mild, moderate, or severe categories, which are associated with increasing risks of CKD progression, premature all-cause death, or AKI, among others [15] (Figure 2).
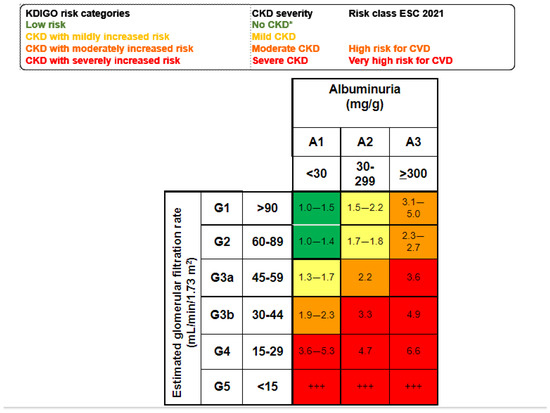
Figure 2.
Mild, moderate, and severe CKD according to KDIGO risk categories based on simultaneous assessment of eGFR and albuminuria. Albuminuria is usually assessed as the urinary albumin–creatinine ratio (UACR in a spot urine sample. Figure shows the risk of all-cause death for each cell) [10,15]. * No CKD if ther is no other evidence of CKD as imaging, hematuria or others. +++ Kidney failure: patients needing kidney replacement therapy and cardiovascular risk is extremely high.
Globally, there are 850 million people with CKD, and the number is expected to increase as the world population ages [16,17,18]. Prior to the SARS-CoV-2 pandemic, it was estimated that, by 2040, CKD would be the fifth global cause of death, as it is growing faster than most other common causes of death [16,17]. Indeed, the loss of key kidney functions, such as the production of the anti-aging protein Klotho, or the decrease in glomerular filtration and tubular secretion of waste molecules leading to the accumulation of uremic toxins, is believed to accelerate biological aging, leading to premature death, mainly from cardiovascular causes, infection, malignancy, and the lack of access to kidney replacement therapy (KRT) [16].
The progression of CKD is frequently non-linear, as CKD predisposes to AKI, and AKI may accelerate the progression of CKD [19,20]. As for CKD, the cut-off points selected to define AKI (an increase in serum creatinine by ≥0.3 mg/dL within 48 h) are associated with an increased risk of death that increases to 45–50% in those requiring KRT and may persist for up to a year [21,22,23].
3. CKD as a Risk Factor for COVID-19
Identifying the factors associated with severe COVID-19 is important to protect those in high-risk groups and, eventually, develop specific therapeutic approaches if the molecular mechanisms underlying the increased susceptibility are identified. CKD is the most common and the most influential comorbidity associated with COVID-19 severity. According to the prevalence data from the Global Burden of Diseases, Injuries, and Risk Factors Study (GBD) together with the UN population estimates for 2020, CKD is deemed the most prevalent risk factor for severe COVID-19 in adults [9]. In addition, CKD alone explains the increased risk of severe COVID-19 in approximately 5% of the global population. Another study conducted in the UK using primary care electronic records of 17,278,392 persons examined the factors associated with COVID-19-related death [8]. While older age, expectedly, showed the strongest association with poor outcomes, CKD was among the top five comorbidities conferring the highest risk of COVID-19-related death. The adjusted HR was 2.52 (2.33–2.72) for subjects with G4–5 CKD (eGFR < 30 mL/min/1.73 m2), 3.53 (2.77–4.49) for transplant recipients, and 3.69 (3.09–4.39) for patients with kidney failure, including those receiving dialysis. The Madrid REMER Registry study also showed that, in 2020, COVID-19 was the most common cause of death among patients on kidney replacement therapy (KRT), with overall mortality increasing by 34% compared with the average in the previous decade [24]. In this regard, patients on dialysis are at high risk of death from infection as a category encompassing different microbes. While the rate of mortality from infection decreased from 224 to 163 per 10,000 person-years for those commencing dialysis in 1980–2005 and 2006–2018, respectively, it remains over 20-fold higher than in the general population and disproportionally affects women and minorities [25]. Thus, the increased severity of COVID-19 should not come as a surprise, and the key question is to what extent the predisposition of CKD patients to severe COVID-19 is a further manifestation of the non-specific immune suppression in this population, or whether it is driven by specific factors that merit specific management.
4. COVID-19, AKI, and Mortality
COVID-19 may be complicated by AKI. This may result from systemic cytokine release syndrome due to the SARS-CoV-2 infection of kidney cells, specific disease entities such as collapsing focal segmental glomerulosclerosis or thrombotic microangiopathy, or a combination of these and other factors [26,27,28,29,30]. As is the case for AKI in other contexts, AKI is associated with an increased risk of death in patients with COVID-19 [31,32,33]. In the regions and countries hit earliest and hardest by the pandemic, the lack of enough resources to provide both ventilation support and KRT to all those in need may have contributed to a further increase in the mortality of COVID-19 AKI [34]. The fact that there is evidence of the infection of kidney cells during AKI and that AKI in COVID-19 is associated with adverse outcomes means that it is also of interest to understand the susceptibility of kidney cells to SARS-CoV-2 infection. Indeed, the SARS-CoV-2 infection of kidney cells was associated with an increased risk of death in patients with COVID-19 [35].
5. Suboptimal Response to SARS-CoV-2 Vaccines in Some Patient Populations with CKD
The most obvious preventive measure for those at high risk of severe COVID-19 is vaccination [36]. However, patients on dialysis and even more kidney transplant recipients have suboptimal responses to vaccines. As an example, patients on dialysis require specially designed regimens to optimize the response to vaccination against hepatitis B virus, and despite that, some patients are not immunized or are immunized only transiently [37]. There are multiple anti-SARS-CoV-2 vaccines, and reviewing the CKD literature on all of them is beyond the scope of this review. However, the mRNA-based vaccines (BNT162b2 and mRNA-1273) have been prospectively assessed using the same methods in patients with CKD not on dialysis, on hemodialysis/peritoneal dialysis, and those who underwent transplantation, and this may provide a flavor of the issues. After the initial vaccination schedule, the BNT162b2 (30 μcg) vaccine was associated with a six-fold higher risk for negative humoral response than mRNA-1273 (100 μcg), likely due to the different dose, as is the case for hepatitis B virus vaccines [38]. During the first year after vaccination, patients with non-dialysis CKD and those on dialysis presented good anti-spike antibody responses. However, anti-spike antibodies decreased over time. A third dose of vaccine induced seroconversion in a high percentage of antibody-negative patients after two doses, although responses were poorer in kidney transplant recipients [39]. The fourth dose seroconverted 72% of previously negative patients. Higher anti-spike antibody titers at 12 months were independently associated with repeated exposure to antigen (fourth vaccine dose and/or previous breakthrough infections). Breakthrough COVID-19 requiring admission was observed in CKD patients with lower antibody titers, and the least benefit from the fourth dose was observed in patients with the highest need for a vaccine booster (i.e., those with lower pre-booster antibody titers or kidney transplant recipients) [40]. Overall, kidney transplant recipients presented suboptimal responses after any vaccination schedule (initial, third, and fourth dose). Some patients had a persistently negative humoral response despite boosters [41], and the mortality of patients on KRT remains high despite increased access to critical care [42].
7. Molecular Pathogenesis of COVID-19-Associated AKI
Recently, the gene expression of kidneys from patients with COVID-19 was characterized, providing novel insight into the pathogenesis of kidney injury and, specifically, kidney injury when kidney cells are infected with SARS-CoV-2 [35]. Interestingly, two main components drove the kidney gene expression difference between COVID-19 status and the presence of renal SARS-CoV-2 RNA. Inflammation pathways were prominent among differentially expressed genes [35]. Additionally, IFN-α and -γ response pathways were specifically enriched in SARS-CoV-2-infected kidneys. The COVID-19 SARS-CoV-2-infected kidney transcriptomic signature differed from another viral nephropathy, hantavirus nephropathy [35]. Furthermore, the kidney transcriptomic signature of COVID-19 AKI was compared with that of AKI in the absence of COVID-19. Overall, there were 1191 differentially expressed genes (DEGs) in COVID-19 AKI, but none were detected in non-COVID-19 AKI, serving as potential specific players of COVID-19-induced AKI (647 upregulated over 1.5-fold and 347 downregulated to less than 0.5-fold [35] (Table 1).

Table 1.
Top 10 upregulated (A) and downregulated (B) genes in human COVID-19 AKI that are not significantly differentially expressed (adjusted p value < 0.05) in human non-COVID-19 AKI when either of them is compared with control. Data obtained from ref. [35].
When analyzing the DEGs using the bioinformatic platform Enrichr-KG [46], the most relevant transcription factors potentially driving the differences in kidney gene expression between COVID-19 AKI and non-COVID-19 AKI were POLR2L, EIF3K, BOLA3, RFXANK, and ZNF576 (Figure 3). POLR2L and EIF3K also contributed to explaining the full range of upregulated genes. The authors also identified an X-linked inhibitor of apoptosis-associated factor 1 (XAF1) as a critical target of SARS-CoV-2 infection of the kidneys [35].
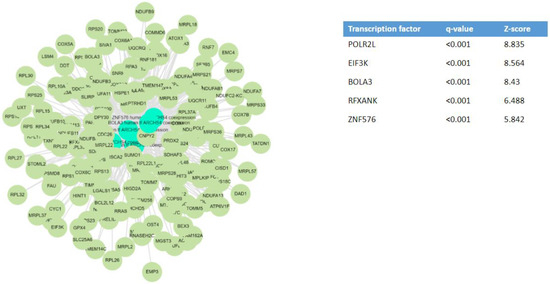
Figure 3.
Key transcription factors that may underlie the differential gene expression observed in human COVID-19-dependent AKI [35]. Transcription factor enrichment analysis from differentially expressed genes in COVID-19 AKI, but non-differentially expressed in non-COVID-19 AKI, was performed using the bioinformatic platform Enrichr-KG [46]. NCBI GEO under the accession number GSE210622.
8. SCARF Genes
SARS-CoV-2 and coronavirus-associated receptors and factors (SCARFs) are molecules involved in SARS-CoV-2 cell entry, replication, and assembly or that may be involved in these processes based on prior knowledge of the biology of other coronaviruses [4]. The entry mechanism of SARS-CoV-2, similar to other human coronaviruses, involves initial viral spike protein binding to a cell-surface receptor and its posterior cleavage by a host protease [46,47] (Figure 4). For both SARS-CoV and SARS-CoV-2, angiotensin-converting enzyme 2 (ACE2) in the cell membrane was identified as the primary receptor, and transmembrane serine protease type 2 (TMPRSS2) was indicated as the main cell-surface protease promoting virus entry [48]. However, the expression levels of ACE2 in the lung are relatively low and mostly limited to type 2 alveolar cells, while in other organs less affected by COVID-19, such as the small intestine, colon, or testis, ACE2 expression is much higher [49]. Thus, the evidence regarding ACE2 expression levels in different tissues and its correlation with COVID-19 clinical manifestations suggests that alternative factors are implicated in SARS-CoV-2 entry facilitation. This may include other molecules as well as the polarized distribution of ACE2. For example, proximal tubular cells express high amounts of ACE2 but on the tubular lumen [50]. Given the size of the SARS-CoV-2 virion, it is not expected to traverse a healthy glomerular filtration barrier, so proximal tubular cell entry via ACE2 would require either the disruption of the glomerular filtration barrier (e.g., glomerulopathy) or tubular injury (e.g., AKI) that disrupts the continuity of the tubular epithelial layer or alters the localization of ACE2 expression.
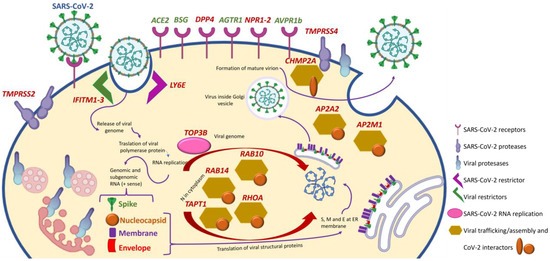
Figure 4.
Selected SCARF genes and impact of CKD. Those differentially expressed in at least one organ (kidney, lung, heart, or aorta) of mice with CKD and whose differential expression may be expected to increase the severity of COVID-19 are identified in red. The increased expression of all the genes shown will favor the biology of SARS-CoV-2 except for IFITM1–3 and LY6E, whose increased expression would impair SARS-CoV-2 entry into cells. Modified from [51] with permission.
Single-cell RNA sequencing identified cells expressing 28 SCARF genes in healthy human tissues [4]. SCARF genes include entry factors (ACE2 and TMPRSS2) and other potential cell-surface receptors validated in human cells that may facilitate cell entry as evaluated for SARS-CoV, SARS-CoV-2, hCoV-229E or MERS-CoV, such as basignin (Ok Blood Group, BSG), alanyl aminopeptidase (ANPEP), dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin/cluster of differentiation 209 (DC-SIGN, CD209), C-type lectin domain family 4 member G/M (CLEC4G/M), and dipeptidyl peptidase-4 (DPP4) [52,53,54,55]. Several cellular proteases were also included, such as TMPRSS4 which functions with a priming factor; TMPRSS11A/B, which activates the S peptide of other coronaviruses; furin, which activates MERS-CoV and possibly SARS-CoV-2 proteins; and cathepsins (CTSL/B), which can substitute TMPRSS2 to prime SARS-CoV [56,57]. Additionally, there are restriction factors known to protect cells against SARS-CoV-2 entry such as lymphocyte antigen 6 family member E (LY6E) and interferon-induced transmembrane proteins 1, 2, and 3 (IFITM1–3) [57,58].
At the post-entry level, DNA topoisomerase iii beta (TOP3B) and zinc-finger CCHC-type and RNA-binding motif containing 1 (ZCRB1) are essential for SARS-CoV-2 and SARS-CoV genome replication, respectively [59,60]. Proteins involved in the assembly and trafficking of RNA viruses that physically interact with SARS-CoV-2 structural proteins include members of the Rho-GTPase complex (RHOA, RAB10, RAB14, and RAB1A), members of the activating protein 2 (AP2) complex (AP2A2 and AP2M1), and charged multivesicular body protein 2A (CHMP2A) [61].
More recently, SARS-CoV-2 was reported to form complexes with self-proteins to exploit receptor-mediated endocytosis through the interaction of its spike with soluble ACE2 (sACE2) or sACE2–vasopressin via angiotensin II receptor type 1 (AT1), the target of angiotensin receptor blockers (ARBs), or arginine vasopressin receptor 1B (AVPR1B), respectively [62]. Notably, sACE2 was at some point contemplated as a therapeutic agent for COVID-19, as it could compete for cell membrane ACE2 and, thus, decrease viral entry into cells [63,64,65]. However, the only results reported in clinicaltrials.gov were disappointing (NCT04335136). The fact that sACE2 can actually facilitate virus entry through alternative receptors may contribute to explaining the disappointing results. Additionally, neuropilin 1 (NRP-1) served as an entry factor that potentiates SARS-CoV-2 infectivity in vitro [66].
9. Susceptibility to SARS-CoV-2 Infection and Severity of COVID-19
Environmental, clinical (e.g., CKD), and social factors play a key role in SARS-CoV-2 infection and the severity of COVID-19, but host genetic factors may also contribute, as indicated above for resistance to clinical infection [6]. A full understanding of the biological role of genetic factors in the pathogenesis of COVID-19 may help to identify mechanistic targets for therapeutic development [67]. However, susceptibility to SARS-CoV-2 infection is not easy to define, due to the different viral, host, and environmental factors, in addition to diverse phenotypes, vaccination response, and population differences, among others. Different designs have been adopted to evaluate the role of genetics on the susceptibility and severity of COVID-19, as recently summarized [5]. Information is derived mainly from studies designed to identify common single nucleotide polymorphisms (SNPs) and rare and ultra-rare variants associated with different phenotypes, obtained from analyses of single genes or candidate-pathway association studies, genome-wide association studies (GWAS), meta-analyses, or polygenic risk scores [67,68,69,70,71,72,73,74,75,76,77,78].
10. SCARF Genes and Kidney Disease
CKD is considered a systemic condition. Thus, CKD modifies the physiology and function of multiple organs, inducing a state of systemic inflammation and accelerating biological aging [79,80]. This is the consequence of the retention of uremic toxins or the loss of key kidney functions, such as the clearance of small protein proinflammatory mediators or the production of the anti-inflammatory and anti-aging protein Klotho [81,82,83]. This altered pathophysiological state may be predicted to alter the expression of multiple genes, potentially including SCARF and COVID-19 susceptibility genes. Additionally, both AKI and CKD are associated with cause-specific and shared changes in kidney gene expression, many of which are the consequence of local inflammation. As an example, kidney diseases studied up to now share the early loss of Klotho expression, which is driven by tubular cell stressors, including inflammatory cytokines [84,85]. Finally, AKI, COVID-19, and SARS-CoV-2 infection of kidney cells result in specific local gene expression patterns (Table 1).
We have used two approaches to address the impact of kidney disease on the expression of genes relevant to COVID-19 susceptibility or severity either locally in the kidney or systemically.
In an experimental interventional approach followed by the validation of kidney data in human kidney transcriptomic databases, we induced CKD in mice through the addition of adenine to food and assessed the kidney and systemic expression of 21 SCARF genes [85]. Mice with adenine-induced CKD retain uremic solutes, and, thus, allow us to explore the systemic impact of kidney disease, unlike other models of CKD, like unilateral ureteral obstruction, that allow for the study of the local molecular mechanisms of kidney injury but not the systemic consequences given the fact that one kidney is normal [86].
In mice with adenine-induced CKD, 20/21 (95%) SCARF genes studied were differentially expressed in at least one organ [51,87,88,89,90,91,92,93] (Table 2). This very high rate of differential gene expression already suggests that expanding the analysis to other SCARF or disease susceptibility genes may uncover further abnormalities that sensitize to more severe COVID-19. Indeed, for 15/22 (68%) SCARF genes, the differential expression would be expected to favor SARS-CoV-2 infection and/or severity (Figure 4). The largest impact of CKD on COVID-19-related gene expression was observed in the kidneys. Of the 15 differentially expressed genes whose differential expression would be expected to favor SARS-CoV-2 infection and/or severity, 13 were differentially expressed in the kidney, and 8 of them were validated in human CKD kidney transcriptomic datasets, including those for the most common cause of CKD, diabetic nephropathy.
Table 2.
SCARF genes that are differentially expressed in kidneys, lungs, heart, or aorta in mice with CKD [51].
Two genes were reported to protect from SARS-CoV-2 and were downregulated in at least one non-kidney target organ: Ifitm3 in the lung and Ly6e in the aorta [51]. This means that the downregulation of these genes during CKD may favor SARS-CoV-2 entry into lung or vascular cells, potentially increasing the severity of lung or vascular manifestations of COVID-19 in patients with CKD. The lungs and the vasculature are critical drivers of COVID-19 mortality. These findings should be validated in protein studies in the lungs or aorta of patients with CKD, ideally including immunohistochemistry studies that locate the cells where the differential expression is occurring. Additionally, the identification of the drivers of these changes in gene expression may provide tools to prevent or reverse them: Are they driven by uremic toxins, or by inflammatory cytokines or other factors? It may be argued that other SCARF genes in these organs are not differentially expressed in mice with CKD or even that the differential gene expression would be predicted to be protected from COVID-19. However, we should consider the context of this research: We already know that CKD patients are at increased risk of severe COVID-19, and the aim is to identify potential drivers of this increased risk. The differential expression of genes predicted to potentially result in protection from COVID-19 would not have a clinical translation, meaning that either they are not so influential on the overall pathophysiology of the disease, or they occur in non-relevant cell types, or they are not translated into functional protein changes.
The second approach was based on data extraction from the 237 SCARF genes and genes proposed to be involved in COVID-19 severity or SARS-CoV-2 susceptibility retrieved from the literature described above [5,53,54,55,57,59,60,62,67,75,77,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130] (Supplementary Table S1). They were assessed in murine and human transcriptomic databases.
First, we evaluated the expression in a normal murine kidney database (Table 3). The rationale would be that early proof-of-concept studies will be more feasible in murine models, and there are already humanized murine models of coronavirus infection [131]. A total of 178 COVID-19-related genes were found in the normal kidney database, with 143 genes considered to be expressed. Additionally, we evaluated the expression of COVID-19-related genes in a human AKI single-cell transcriptomic database [132]. In bulk kidney data, 94 out of 225 found genes (41.8%) were differentially expressed during human AKI (Table 3): Of those, 22 were upregulated ≥1.25-fold, and 34 were downregulated to less than 0.5-fold. All the analyzed cell types had some degree of differential expression of COVID-19-related genes, with the bulk of them being differentially expressed in the thick ascending limb of Henle and proximal tubular cells, while podocytes were on the other side of the spectrum (Table 3), likely because they are not primary targets of the most common cause of AKI, formerly known as acute tubular necrosis.
Table 3.
Expression of 237 genes proposed to be involved in COVID-19 severity or SARS-CoV-2 susceptibility (Supplementary Table S1) in the murine normal kidneys [133] or human AKI kidneys [132]. Data are expressed as the number of genes or number (% of differentially expressed genes among genes expressed by that cell).
Finally, differentially expressed genes in the kidneys of patients with COVID-19 and AKI included 19 of 237 COVID-19 susceptibility genes: RPL24, OAS1-3, ARHGAP27, PPP1R15A, GOLGA3, XCR1, MX1, RUSC1, TCF19, POLD1, NOTCH4, HLA-E, PROC, GC, MRPS21, PDE4A, and ATP5PO [35].
11. Proposal for a Research Agenda
Overall, there is convincing evidence that kidney disease is associated with an increased risk of COVID-19 death. Indeed, CKD is the most common risk factor for severe CKD and the one that most increases the risk of COVID-19 death, while the development of AKI during a COVID-19 episode also increases the risk of death. On top of this, vaccination in patients with CKD in response to the disease may be suboptimal in terms of the duration of protective anti-spike protein antibodies or even, for kidney transplant recipients, in terms of the development of protective antibodies. In recent months, a better understanding of genetic risk factors for severe COVID-19 has inaugurated a new era, in which the impact of comorbidities on the expression and activity of these key genes may be explored to eventually develop comorbidity-specific therapeutic approaches (Figure 5). Given its well-characterized impact on multiple organs and systems, CKD is an ideal candidate for proof-of-concept studies that address this hypothesis. Early preclinical evidence backed by data extracted from human transcriptomic databases suggests that kidney injury is associated with the local differential expression of multiple genes involved in SARS-CoV-2 pathogenicity or the host response against it. Moreover, the differential gene expression was also observed in target organs of COVID-19 in mice with CKD. Some of these changes in gene expression would be expected to favor a higher severity of COVID-19. Box 1 summarizes potential next steps in the quest for comorbidity-specific approaches to fight SARS-CoV-2 or future pandemics caused by coronaviruses or other viruses. The research agenda proposes to advance preclinical research to initial clinical translation (the observational validation of key preclinical findings) to advanced clinical translation (interventional studies), recognizing that preclinical studies may not be feasible for certain genes whose expression levels and/or functions significantly differ between humans and mice. Preclinical studies should start from a data-mining exercise that identifies all human and murine datasets that are informative on SCARF and COVID-19 host gene expression in different organs under conditions of CKD. This should be followed by cross-validation between human and murine data and the characterization of the regulators of differentially expressed genes. Ideally, the function of key genes should be explored in humanized models of COVID-19 in mice, as this will facilitate advances in the functional analysis of specific genes through gene-targeting approaches. The key murine observational data then should be validated in human samples. Finally, samples from interventional human studies should be assessed to understand how key COVID-19 host genes are regulated in human CKD. Finally, if safe drug interventions are identified (i.e., drug repurposing), a prospective study should assess the feasibility of modulating COVID-19 host genes in order to increase the resistance of CKD patients to COVID-19, eventually testing whether this is protective. The analysis should not be limited to gene expression, as protein levels and even protein function may be modified in CKD, for example, through post-translational modifications such as carbamylation and others.
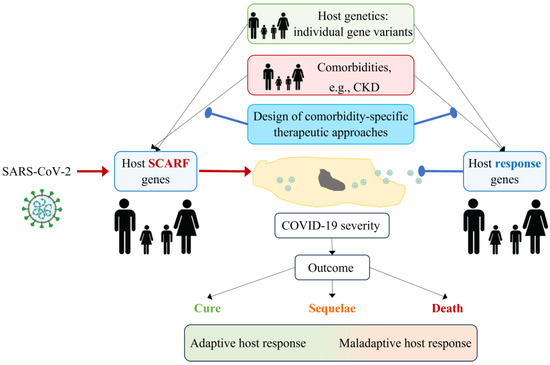
Figure 5.
Development of comorbidity-specific therapeutic approaches for COVID-19. The severity of COVID-19 depends on the interaction between SARS-CoV-2 and the host. SARS-CoV-2 hijacks host gene products (SCARFs) to enter cells and replicate, while host response genes try to eliminate the virus. Genetic variants in the host may determine whether SCARFs and host response genes are present and whether they are expressed and active at appropriate levels. Additionally, comorbidities (e.g., CKD) may modify the expression or activity of SCARFs and host response genes or their products. Proof-of-concept studies have demonstrated that CKD modifies the systemic expression of some SCARF genes in a manner that may facilitate viral entry and replication and, potentially, increase COVID-19 severity. The characterization of such interactions between comorbidities, SCARFs, and host response genes may help design comorbidity-specific therapeutic approaches to SARS-CoV-2 or future viral threats that minimize the risk of sequelae or death. SCARF: coronavirus-associated receptors and factors, CKD: chronic kidney disease.
Box 1. Toward developing comorbidity-specific therapeutic approaches. A research agenda.
Preclinical proof of concept *
- Use data-mining strategies to assemble all the available information from public databases and explore the expression for all known COVID-19 host genes in the kidneys and key target organs (e.g., lungs, vasculature, etc.) of mice with CKD;
- Develop ad hoc models of murine CKD to fill the gaps in knowledge. This will facilitate the development of preclinical studies using tools such as genetically modified animals;
- Gene expression, protein levels, and protein post-translational modifications and function may be explored;
- Once potential targets have been identified that may account for the increased severity of COVID-19 in patients with CKD:
- o
- Characterize the CKD-related modifiers that modulate gene or protein expression or function;
- o
- Confirm the role of the specific target in the severity of COVID-19 in humanized mouse models.
(Given the differences between humans and mice, for some genes, preclinical studies should be obviated as they will not be informative).
Initial clinical translation
- Use data-mining strategies to explore the expression for all known COVID-19 host genes in the kidneys and key target organs (e.g., lungs, vasculature, etc.) of persons with CKD;
- Use focused analysis to confirm findings in murine models;
- Use focused biobank searches to address persistent gaps in knowledge.
Advanced clinical translation
- Identify interventions that restore the expression and function of the CKD-influenced COVID-19 genes that appear key for the increased severity of COVID-19;
- Initiate clinical development of such therapeutic strategies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms242216078/s1.
Author Contributions
Conceptualization, A.O. and S.C.; writing—original draft preparation, S.C., A.O., D.A. and D.D.; writing—review and editing, S.C., A.O., D.A., D.D. and M.D.S.-N.; funding acquisition, A.O. and M.D.S.-N. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by Instituto de Salud Carlos III (ISCIII) FIS/Fondos FEDER (PI22/00050, and PI21/00251), ERA-PerMed-JTC2022 (SPAREKID AC22/00027), FRIAT, Comunidad de Madrid en Biomedicina P2022/BMD-7223, CIFRA_COR-CM. It also received funding from the ISCIII RICORS program RICORS2040 (RD21/0005/0001) funded by European Union–NextGenerationEU; Mecanismo para la Recuperación y la Resiliencia (MRR) and SPACKDc PMP21/00109; FEDER funds; COST Action PERMEDIK CA21165, supported by COST (European Cooperation in Science and Technology); PREVENTCKD Consortium (Project ID: 101101220) Programme EU4H DG/Agency: HADEA; KitNewCare (Project ID: 101137054); and HORIZON-HLTH-2023-CARE-04 Programme: HORIZON. DG/Agency: HADEA. MDS-N was supported by Spain’s Ministry of Science and Innovation (MICINN) Ramon y Cajal program RYC2018-024461-I. This research was funded by the AstraZeneca Foundation through the Program for the Promotion of Young Researchers “V Call for Young Researchers Awards of the AstraZeneca Foundation 2020”.
Conflicts of Interest
A.O. has received grants from Sanofi and consultancy or speaker fees or travel support from Adviccene, Alexion, Astellas, AstraZeneca, Amicus, Amgen, Boehringer Ingelheim, Fresenius Medical Care, GSK, Bayer, Sanofi-Genzyme, Menarini, Mundipharma, Kyowa Kirin, Lilly, Freeline, Idorsia, Chiesi, Otsuka, Novo-Nordisk, Sysmex and Vifor Fresenius Medical Care Renal Pharma and is the Director of the Catedra UAM-AstraZeneca of chronic kidney disease and electrolytes. He has stock in Telara Farma. S.C. has received honoraria for consultancy from Otsuka.
References
- WHO Coronavirus Dashboard. Available online: https://covid19.who.int/ (accessed on 27 July 2023).
- Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/interactive-timeline (accessed on 22 June 2019).
- Bowe, B.; Xie, Y.; Al-Aly, Z. Acute and postacute sequelae associated with SARS-CoV-2 reinfection. Nat. Med. 2022, 28, 2398–2405. [Google Scholar] [CrossRef]
- Singh, M.; Bansal, V.; Feschotte, C. A Single-Cell RNA Expression Map of Human Coronavirus Entry Factors. Cell Rep. 2020, 32, 108175. [Google Scholar] [CrossRef] [PubMed]
- Cappadona, C.; Rimoldi, V.; Paraboschi, E.M.; Asselta, R. Genetic susceptibility to severe COVID-19. Infect. Genet. Evol. 2023, 110, 105426. [Google Scholar] [CrossRef] [PubMed]
- Augusto, D.G.; Murdolo, L.D.; Chatzileontiadou, D.S.M.; Sabatino, J.J.; Yusufali, T.; Peyser, N.D.; Butcher, X.; Kizer, K.; Guthrie, K.; Murray, V.W.; et al. A common allele of HLA is associated with asymptomatic SARS-CoV-2 infection. Nature 2023, 620, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Council, E.-E.; Group, E.W. Chronic kidney disease is a key risk factor for severe COVID-19: A call to action by the ERA-EDTA. Nephrol. Dial. Transplant. 2021, 36, 87–94. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- Clark, A.; Jit, M.; Warren-Gash, C.; Guthrie, B.; Wang, H.H.X.; Mercer, S.W.; Sanderson, C.; McKee, M.; Troeger, C.; Ong, K.L.; et al. Global, regional, and national estimates of the population at increased risk of severe COVID-19 due to underlying health conditions in 2020: A modelling study. Lancet Glob. Health 2020, 8, e1003–e1017. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO); CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- KDIGO AKI Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int. Suppl. 2012, 2, 1–138. [Google Scholar]
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Am. J. Kidney Dis. 2002, 39 (Suppl. 1), i–ii+S1–S266. [Google Scholar]
- Perez-Gomez, M.V.; Bartsch, L.-A.; Castillo-Rodriguez, E.; Fernandez-Prado, R.; Fernandez-Fernandez, B.; Martin-Cleary, C.; Gracia-Iguacel, C.; Ortiz, A. Clarifying the concept of chronic kidney disease for non-nephrologists. Clin. Kidney J. 2019, 12, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Wanner, C.; Gansevoort, R.T.; Cozzolino, M.; Fliser, D.; Gambaro, G.; Ong, A.; Rosenkranz, A.R.; Rychlık, I.; Sarafidis, P.; et al. Chronic kidney disease as cardiovascular risk factor in routine clinical practice: A position statement by the Council of the European Renal Association. Clin. Kidney J. 2022, 16, 403–407. [Google Scholar] [CrossRef] [PubMed]
- AIRG-E; EKPF; ALCER; FRIAT; REDINREN; RICORS2040; SENEFRO; SET; ONT. CKD: The burden of disease invisible to re-search funders. Nefrologia 2022, 42, 65–84. [Google Scholar] [CrossRef]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.W.; et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication worldwide more than 850 million individuals have kidney diseases. Nephrol. Dial. Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef]
- Mehta, R.L.; Cerdá, J.; Burdmann, E.A.; Tonelli, M.; García-García, G.; Jha, V.; Susantitaphong, P.; Rocco, M.; Vanholder, R.; Sever, M.S.; et al. International Society of Nephrology’s 0by25 initi-ative for acute kidney injury (zero preventable deaths by 2025): A human rights case for nephrology. Lancet 2015, 385, 2616–2643. [Google Scholar] [CrossRef]
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.J. Acute kidney injury. Nat. Rev. Dis. Primers 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Carriazo, S.; Aparicio-Madre, M.I.; Tornero-Molina, F.; Fernández-Lucas, M.; Paraiso-Cuevas, V.; González-Parra, E.; del Río-Gallegos, F.; Marques-Vidas, M.; Alcázar-Arroyo, R.; Martins-Muñoz, J.; et al. Impact of different COVID-19 waves on kidney replacement therapy epidemiology and mortality: REMER 2020. Nephrol. Dial. Transplant. 2022, 37, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.H.; Au, E.H.; Davies, C.E.; Jaure, A.; Howell, M.; Lim, W.H.; Craig, J.C.; Teixeira-Pinto, A.; Wong, G. Long-term Trends in Infection-Related Mortality in Adults Treated With Maintenance Dialysis. Am. J. Kidney Dis. 2023, 82, 597–607. [Google Scholar] [CrossRef]
- Carriazo, S.; Kanbay, M.; Ortiz, A. Kidney disease and electrolytes in COVID-19: More than meets the eye. Clin. Kidney J. 2020, 13, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Radovic, S.; Meng, W.; Chen, L.; Paniz Mondolfi, A.E.; Bryce, C.; Grimes, Z.; Sordillo, E.M.; Cordon-Cardo, C.; Guo, H.; Huang, Y.; et al. SARS-CoV-2 infection of kidney tissues from se-vere COVID-19 patients. J. Med. Virol. 2023, 95, e28566. [Google Scholar] [CrossRef]
- Jansen, J.; Reimer, K.C.; Nagai, J.S.; Varghese, F.S.; Overheul, G.J.; de Beer, M.; Roverts, R.; Daviran, D.; Fermin, L.A.; Willemsen, B.; et al. SARS-CoV-2 infects the human kidney and drives fibrosis in kidney organoids. Cell Stem Cell 2022, 29, 217–231.e8. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human kidney is a target for novel severe acute respiratory syn-drome coronavirus 2 infection. Nat. Commun. 2021, 12, 2506. [Google Scholar] [CrossRef]
- Puelles, V.G.; Lütgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Rubin, S.; Orieux, A.; Prevel, R.; Garric, A.; Bats, M.L.; Dabernat, S.; Camou, F.; Guisset, O.; Issa, N.; Mourissoux, G.; et al. Characterization of acute kidney injury in critically ill pa-tients with severe coronavirus disease 2019. Clin Kidney J. 2020, 13, 354–361. [Google Scholar]
- Roushani, J.; Thomas, D.; Oliver, M.J.; Ip, J.; Tang, Y.; Yeung, A.; Taji, L.; Cooper, R.; Magner, P.O.; Garg, A.X.; et al. Acute kidney injury requiring renal replacement therapy in people with COVID-19 disease in Ontario, Canada: A prospective analysis of risk factors and outcomes. Clin. Kidney J. 2021, 15, 507–516. [Google Scholar] [CrossRef]
- Wan, Y.I.; Bien, Z.; Apea, V.J.; Orkin, C.M.; Dhairyawan, R.; Kirwan, C.J.; Pearse, R.M.; Puthucheary, Z.A.; Prowle, J.R. Acute kidney injury in COVID-19: Multicentre prospective analysis of registry data. Clin. Kidney J. 2021, 14, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Portolés, J.; López-Sánchez, P.; Martin-Rodríguez, L.; Serrano-Salazar, M.L.; Valdenebro-Recio, M.; Ramos, A.; Malo, R.M.; Zalamea, F.; Martin-Giner, J.M.; Marques, M.; et al. Acute and chronic kidney disease and risk of hospital mortality during COVID-19 pandemic waves in the pre-vaccination era. Clin. Kidney J. 2022, 16, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Isnard, P.; Vergnaud, P.; Garbay, S.; Jamme, M.; Eloudzeri, M.; Karras, A.; Anglicheau, D.; Galantine, V.; Eddine, A.J.; Gosset, C.; et al. A specific molecular signature in SARS-CoV-2-infected kidney biopsies. JCI Insight. 2023, 8, e165192. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H. COVID-19 Vaccines–Immunity, Variants, Boosters. N. Engl. J. Med. 2022, 387, 1011–1020. [Google Scholar] [CrossRef]
- Kong, N.C.T.; Beran, J.; Kee, S.A.; Miguel, J.L.; Sánchez, C.; Bayas, J.M.; Vilella, A.; Calbo-Torrecillas, F.; de Novales, E.L.; Srinivasa, K.; et al. A new adjuvant improves the immune response to hepati-tis B vaccine in hemodialysis patients. Kidney Int. 2008, 73, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, B.; Soler, M.J.; Ortiz, A.; Martínez Vaquera, S.; Jarava Mantecón, C.J.; Useche, G.; Sánchez Márquez, M.G.; Carnerero, M.; Jaldo Rodríguez, M.T.; Muñoz Ramos, P.; et al. Safety and immediate humoral re-sponse of COVID-19 vaccines in chronic kidney disease patients: The SENCOVAC study. Nephrol. Dial. Transplant. 2022, 37, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, B.; Soler, M.J.; Ortiz, A.; Orero, E.; Tejedor, S.; Mantecón, C.J.J.; Perez, V.O.G.; Franco, A.J.M.; Sánchez, C.A.; Carretero, M.P.; et al. Humoral Response to Third Dose of SARS-CoV-2 Vac-cines in the CKD Spectrum. Clin. J. Am. Soc. Nephrol. 2022, 17, 872–876. [Google Scholar] [CrossRef]
- Quiroga, B.; Soler, M.J.; Ortiz, A.; Jarava Mantecón, C.J.; Gomes Pérez, V.O.; Bordils, A.; Lacueva, J.; Marin Franco, A.J.; Delgado Conde, P.; Muñoz Ramos, P.; et al. Humoral response after the fourth dose of the SARS-CoV-2 vaccine in the CKD spectrum: A prespecified analysis of the SENCOVAC study. Nephrol. Dial. Transplant. 2023, 38, 969–981. [Google Scholar] [CrossRef]
- Quiroga, B.; Soler, M.J.; Ortiz, A.; de Sequera, P.; SENCOVAC Collaborative Network. Lessons from SENCOVAC: A prospective study evaluating the re-sponse to SARS-CoV-2 vaccination in the CKD spectrum. Nefrologia 2023, in press. [Google Scholar] [CrossRef]
- Quiroga, B.; Ortiz, A.; Cabezas-Reina, C.J.; Ruiz Fuentes, M.C.; López Jiménez, V.; Zárraga Larrondo, S.; Toapanta, N.; Molina Gómez, M.; de Sequera, P.; Sánchez-Álvarez, E.; et al. Evolving spectrum but persistent high mortality of COVID-19 among patients on kidney replacement therapy in the vaccine era: The Spanish COVID-19 KRT Registry. Clin. Kidney J. 2022, 15, 1685–1697. [Google Scholar] [CrossRef]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, G.M.; Ahuja, A.; Yung, M.Y.; Leung, C.; To, K.; et al. A Major Outbreak of Severe Acute Respiratory Syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- de Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Enrichr-KG Web-Server Application. Available online: https://maayanlab.cloud/enrichr-kg (accessed on 12 August 2023).
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- D’marco, L.; Puchades, M.J.; Romero-Parra, M.; Gimenez-Civera, E.; Soler, M.J.; Ortiz, A.; Gorriz, J.L. Coronavirus disease 2019 in chronic kidney disease. Clin. Kidney J. 2020, 13, 297–306. [Google Scholar] [CrossRef]
- Carriazo, S.; Ribagorda, M.; Pintor-Chocano, A.; Perez-Gomez, M.V.; Ortiz, A.; Sanchez-Niño, M.D. Increased expression of SCARF genes favoring SARS-CoV-2 infection in key target organs in CKD. Clin. Kidney J. 2023, sfad220. [Google Scholar] [CrossRef]
- Marzi, A.; Gramberg, T.; Simmons, G.; Möller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR Interact with the Glycoprotein of Marburg Virus and the S Protein of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Chen, Z.; Mi, L.; Xu, J.; Yu, J.; Wang, X.; Jiang, J.; Xing, J.; Shang, P.; Qian, A.; Li, Y.; et al. Function of HAb18G/CD147 in Invasion of Host Cells by Severe Acute Respiratory Syndrome Coronavirus. J. Infect. Dis. 2005, 191, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Vankadari, N.; Wilce, J.A. Emerging WuHan (COVID-19) coronavirus: Glycan shield and structure prediction of spike glyco-protein and its interaction with human CD26. Emerg. Microbes Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Zmora, P.; Hoffmann, M.; Kollmus, H.; Moldenhauer, A.-S.; Danov, O.; Braun, A.; Winkler, M.; Schughart, K.; Pöhlmann, S. TMPRSS11A activates the influenza A virus hemagglutinin and the MERS coronavirus spike protein and is insensitive against blockade by HAI-1. J. Biol. Chem. 2018, 293, 13863–13873. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Zmora, P.; Gierer, S.; Heurich, A.; Pöhlmann, S. Proteolytic activation of the SARS-coronavirus spike protein: Cutting enzymes at the cutting edge of antiviral research. Antivir. Res. 2013, 100, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zheng, S.; Chen, D.; Zheng, M.; Li, X.; Li, G.; Lin, H.; Chang, J.; Zeng, H.; Guo, J.T. LY6E Restricts Entry of Human Corona-viruses, Including Currently Pandemic SARS-CoV-2. J. Virol. 2020, 94, e00562-20. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, K.R.; Hirano, M.; Fagg, W.S.; McAnarney, E.T.; Shan, C.; Xie, X.; Hage, A.; Pietzsch, C.A.; Bukreyev, A.; Rajsbaum, R.; et al. Topoisomerase III-β is required for efficient replication of positive-sense RNA viruses. Antivir. Res. 2020, 182, 104874. [Google Scholar] [CrossRef]
- Tan, Y.W.; Hong, W.; Liu, D.X. Binding of the 5′-untranslated region of coronavirus RNA to zinc finger CCHC-type and RNA-binding motif 1 enhances viral replication and transcription. Nucleic Acids Res. 2012, 40, 5065–5077. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals tar-gets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.P.; Zhou, R.; Chan, K.-H.; Zhao, H.; Zhu, L.; et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228.e12. [Google Scholar] [CrossRef]
- Monteil, V.; Eaton, B.; Postnikova, E.; Murphy, M.; Braunsfeld, B.; Crozier, I.; Kricek, F.; Niederhöfer, J.; Schwarzböck, A.; Breid, H.; et al. Clinical grade ACE2 as a universal agent to block SARS-CoV-2 variants. EMBO Mol. Med. 2022, 14, e15230. [Google Scholar] [CrossRef]
- Shoemaker, R.H.; Panettieri, R.A.; Libutti, S.K.; Hochster, H.S.; Watts, N.R.; Wingfield, P.T.; Starkl, P.; Pimenov, L.; Gawish, R.; Hladik, A.; et al. Development of an aerosol intervention for COVID-19 disease: Tolerability of soluble ACE2 (APN01) administered via nebulizer. PLoS ONE 2022, 17, e0271066. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Q.; Yang, S.; Li, Y.; Dou, Y.; Deng, Y.Q.; Bi, J.; Tan, Y.; Wang, H.; Gong, W.; et al. hACE2 Fc-neutralization antibody cocktail provides synergistic protection against SARS-CoV-2 and its spike RBD variants. Cell Discov. 2021, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Mayi, B.S.; Leibowitz, J.A.; Woods, A.T.; Ammon, K.A.; Liu, A.E.; Raja, A. The role of Neuropilin-1 in COVID-19. PLoS Pathog. 2021, 17, e1009153. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef] [PubMed]
- The Severe COVID-19 GWAS Group. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Degenhardt, F.; Ellinghaus, D.; Juzenas, S.; Lerga-Jaso, J.; Wendorff, M.; Maya-Miles, D.; Uellendahl-Werth, F.; ElAbd, H.; Arora, J.; Lenning, O.B.; et al. Detailed stratified GWAS analysis for severe COVID-19 in four European populations. Hum. Mol. Genet. 2022, 31, 3945–3966. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. A first update on mapping the human genetic architecture of COVID-19. Nature 2022, 608, E1–E10. [Google Scholar] [CrossRef]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-Sonmez, T.; Coker, D.; Symons, A.; Esparza-Gordillo, J.; Aslibekyan, S.; Auton, A.; et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021, 53, 801–808. [Google Scholar] [CrossRef]
- Roberts, G.H.L.; Partha, R.; Rhead, B.; Knight, S.C.; Park, D.S.; Coignet, M.V.; Zhang, M.; Berkowitz, N.; Turrisini, D.A.; Gaddis, M.; et al. Expanded COVID-19 phenotype definitions reveal distinct patterns of genetic association and protective effects. Nat. Genet. 2022, 54, 374–381. [Google Scholar] [CrossRef]
- Horowitz, J.E.; Kosmicki, J.A.; Damask, A.; Sharma, D.; Roberts, G.H.; Justice, A.E.; Banerjee, N.; Coignet, M.V.; Yadav, A.; Leader, J.B.; et al. Genome-wide analysis provides genetic evi-dence that ACE2 influences COVID-19 risk and yields risk scores associated with severe disease. Nat. Genet. 2022, 54, 382–392. [Google Scholar] [CrossRef]
- Kousathanas, A.; Pairo-Castineira, E.; Rawlik, K.; Stuckey, A.; Odhams, C.A.; Walker, S.; Russell, C.D.; Malinauskas, T.; Wu, Y.; Millar, J.; et al. Whole-genome sequencing reveals host factors underlying critical COVID-19. Nature 2022, 607, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Ishak, A.; Mehendale, M.; AlRawashdeh, M.M.; Sestacovschi, C.; Sharath, M.; Pandav, K.; Marzban, S. The association of COVID-19 severity and susceptibility and genetic risk factors: A systematic review of the literature. Gene 2022, 836, 146674. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Rawlik, K.; Bretherick, A.D.; Qi, T.; Wu, Y.; Nassiri, I.; McConkey, G.A.; Zechner, M.; Klaric, L.; Griffiths, F.; et al. GWAS and meta-analysis identifies 49 genetic vari-ants underlying critical COVID-19. Nature 2023, 617, 764–768. [Google Scholar]
- Gupta, K.; Kaur, G.; Pathak, T.; Banerjee, I. Systematic review and meta-analysis of human genetic variants contributing to COVID-19 susceptibility and severity. Gene 2022, 844, 146790. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, V.; Cuccorese, M.; Trenti, T. Genetic polymorphisms of ACE1, ACE2, IFTM3, TMPRSS2 and TNFα genes associated with susceptibility and severity of SARS-CoV-2 infection: A systematic review and meta-analysis. Clin. Exp. Med. 2023, 23, 3251–3264. [Google Scholar] [PubMed]
- Tian, Y.E.; Cropley, V.; Maier, A.B.; Lautenschlager, N.T.; Breakspear, M.; Zalesky, A. Heterogeneous aging across multiple organ sys-tems and prediction of chronic disease and mortality. Nat. Med. 2023, 29, 1221–1231. [Google Scholar] [CrossRef]
- Zoccali, C.; Vanholder, R.; Massy, Z.A.; Ortiz, A.; Sarafidis, P.; Dekker, F.W.; Fliser, D.; Fouque, D.; Heine, G.H.; Jager, K.J.; et al. The systemic nature of CKD. Nat. Rev. Nephrol. 2017, 13, 344–358. [Google Scholar] [CrossRef]
- Fernández-Fernández, B.; Valiño-Rivas, L.; Sánchez-Niño, M.D.; Ortiz, A. Albuminuria Downregulation of the Anti-Aging Factor Klotho: The Missing Link Potentially Explaining the Association of Pathological Albuminuria with Premature Death. Adv. Ther. 2020, 37, 62–72. [Google Scholar] [CrossRef]
- Poveda, J.; Sanchez-Niño, M.D.; Glorieux, G.; Sanz, A.B.; Egido, J.; Vanholder, R.; Ortiz, A. p-Cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol. Dial. Transplant. 2013, 29, 56–64. [Google Scholar] [CrossRef]
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cy-tokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Nephrol. Dial. Transplant. 2018, 33, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The Inflammatory Cytokines TWEAK and TNFα Reduce Renal Klotho Expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Ucero, A.C.; Benito-Martin, A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Sanz, A.B.; Ramos, A.M.; Berzal, S.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Unilateral ureteral obstruction: Beyond obstruction. Int. Urol. Nephrol. 2013, 46, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Berthier, C.C.; Bethunaickan, R.; Gonzalez-Rivera, T.; Nair, V.; Ramanujam, M.; Zhang, W.; Bottinger, E.P.; Segerer, S.; Lindenmeyer, M.; Cohen, C.D.; et al. Cross-Species Transcriptional Network Analysis Defines Shared Inflammatory Responses in Murine and Human Lupus Nephritis. J. Immunol. 2012, 189, 988–1001. [Google Scholar] [CrossRef]
- Nakagawa, S.; Nishihara, K.; Miyata, H.; Shinke, H.; Tomita, E.; Kajiwara, M.; Matsubara, T.; Iehara, N.; Igarashi, Y.; Yamada, H.; et al. Molecular Markers of Tubulointerstitial Fibrosis and Tubular Cell Damage in Patients with Chronic Kidney Disease. PLoS ONE 2015, 10, e0136994. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Park, A.S.D.; Mohtat, D.; Thomas, D.B.; Pullman, J.M.; Susztak, K. Transcriptome Analysis of Human Diabetic Kidney Disease. Diabetes 2011, 60, 2354–2369. [Google Scholar] [CrossRef]
- Ju, W.; Greene, C.S.; Eichinger, F.; Nair, V.; Hodgin, J.B.; Bitzer, M.; Lee, Y.-S.; Zhu, Q.; Kehata, M.; Li, M.; et al. Defining cell-type specificity at the transcriptional level in human disease. Genome Res. 2013, 23, 1862–1873. [Google Scholar] [CrossRef]
- Schmid, H.; Boucherot, A.; Yasuda, Y.; Henger, A.; Brunner, B.; Eichinger, F.; Nitsche, A.; Kiss, E.; Bleich, M.; Gröne, H.J.; et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes. 2006, 55, 2993–3003. [Google Scholar]
- Hodgin, J.B.; Borczuk, A.C.; Nasr, S.H.; Markowitz, G.S.; Nair, V.; Martini, S.; Eichinger, F.; Vining, C.; Berthier, C.C.; Kretzler, M.; et al. A Molecular Profile of Focal Segmental Glomerulosclerosis from Formalin-Fixed, Paraffin-Embedded Tissue. Am. J. Pathol. 2010, 177, 1674–1686. [Google Scholar] [CrossRef]
- Neusser, M.A.; Lindenmeyer, M.T.; Moll, A.G.; Segerer, S.; Edenhofer, I.; Sen, K.; Stiehl, D.P.; Kretzler, M.; Gröne, H.J.; Schlöndorff, D.; et al. Human nephrosclerosis triggers a hypoxia-related glomerulopathy. Am. J. Pathol. 2010, 176, 594–607. [Google Scholar]
- Hofmann, H.; Pyrc, K.; Van Der Hoek, L.; Geier, M.; Berkhout, B.; Pöhlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.E.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, A.; Zelus, B.D.; Wentworth, D.E.; Talbot, P.J.; Holmes, K.V. Identification of a receptor-binding domain of the spike glyco-protein of human coronavirus HCoV-229E. J. Virol. 2003, 77, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Kolb, A.F.; Siddell, S.G.; Hegyi, A. Identification of residues critical for the human coronavirus 229E receptor function of human aminopeptidase N. J. Gen. Virol. 1997, 78, 2795–2802. [Google Scholar] [CrossRef]
- Kolb, A.F.; Maile, J.; Heister, A.; Siddell, S.G. Characterization of functional domains in the human coronavirus HCV 229E receptor. J. Gen. Virol. 1996, 77 Pt 10, 2515–2521. [Google Scholar] [CrossRef]
- Yeager, C.L.; Ashmun, R.A.; Williams, R.K.; Cardellichio, C.B.; Shapiro, L.H.; Look, A.T.; Holmes, K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992, 357, 420–422. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Huang, Y.; Ganesh, L.; Leung, K.; Kong, W.P.; Schwartz, O.; Subbarao, K.; Nabel, G.J. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004, 78, 5642–5650. [Google Scholar] [CrossRef]
- Gramberg, T.; Hofmann, H.; Möller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef] [PubMed]
- Kam, Y.-W.; Okumura, Y.; Kido, H.; Ng, L.F.P.; Bruzzone, R.; Altmeyer, R. Cleavage of the SARS Coronavirus Spike Glycoprotein by Airway Proteases Enhances Virus Entry into Human Bronchial Epithelial Cells In Vitro. PLoS ONE 2009, 4, e7870. [Google Scholar] [CrossRef]
- Wang, C.; Wang, J.; Shuai, L.; Ma, X.; Zhang, H.; Liu, R.; Chen, W.; Wang, X.; Ge, J.; Wen, Z.; et al. The Serine/Threonine Kinase AP2-Associated Kinase 1 Plays an Important Role in Rabies Virus Entry. Viruses 2019, 12, 45. [Google Scholar] [CrossRef]
- Bekerman, E.; Neveu, G.; Shulla, A.; Brannan, J.; Pu, S.-Y.; Wang, S.; Xiao, F.; Barouch-Bentov, R.; Bakken, R.R.; Mateo, R.; et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J. Clin. Investig. 2017, 127, 1338–1352. [Google Scholar] [CrossRef]
- Neveu, G.; Barouch-Bentov, R.; Ziv-Av, A.; Gerber, D.; Jacob, Y.; Einav, S. Identification and Targeting of an Interaction between a Tyrosine Motif within Hepatitis C Virus Core Protein and AP2M1 Essential for Viral Assembly. PLoS Pathog. 2012, 8, e1002845. [Google Scholar] [CrossRef]
- Neveu, G.; Ziv-Av, A.; Barouch-Bentov, R.; Berkerman, E.; Mulholland, J.; Einav, S.; DeWitt, W.S.; Emerson, R.O.; Lindau, P.; Vignali, M.; et al. AP-2-Associated Protein Kinase 1 and Cyclin G-Associated Kinase Regulate Hepatitis C Virus Entry and Are Potential Drug Targets. J. Virol. 2015, 89, 4387–4404. [Google Scholar] [CrossRef]
- von Schwedler, U.K.; Stuchell, M.; Müller, B.; Ward, D.M.; Chung, H.-Y.; Morita, E.; Wang, H.E.; Davis, T.; He, G.-P.; Cimbora, D.M.; et al. The Protein Network of HIV Budding. Cell 2003, 114, 701–713. [Google Scholar] [CrossRef]
- Cho, N.-J.; Lee, C.; Pang, P.S.; Pham, E.A.; Fram, B.; Nguyen, K.; Xiong, A.; Sklan, E.H.; Elazar, M.; Koytak, E.S.; et al. Phosphatidylinositol 4,5-Bisphosphate Is an HCV NS5A Ligand and Mediates Replication of the Viral Genome. Gastroenterology 2015, 148, 616–625. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sklan, E.H.; Serrano, R.L.; Einav, S.; Pfeffer, S.R.; Lambright, D.G.; Glenn, J.S. TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. J. Biol. Chem. 2007, 282, 36354–36361. [Google Scholar] [CrossRef] [PubMed]
- Jowers, T.P.; Featherstone, R.J.; Reynolds, D.K.; Brown, H.K.; James, J.; Prescott, A.; Haga, I.R.; Beard, P.M. RAB1A promotes Vaccinia virus replication by facilitating the production of intracellular enveloped virions. Virology 2015, 475, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Zenner, H.L.; Yoshimura, S.-I.; Barr, F.A.; Crump, C.M. Analysis of Rab GTPase-Activating Proteins Indicates that Rab1a/b and Rab43 Are Important for Herpes Simplex Virus 1 Secondary Envelopment. J. Virol. 2011, 85, 8012–8021. [Google Scholar] [CrossRef]
- Spearman, P. Viral interactions with host cell Rab GTPases. Small GTPases 2017, 9, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Pastey, M.K.; Crowe, J.E.; Graham, B.S. RhoA Interacts with the Fusion Glycoprotein of Respiratory Syncytial Virus and Facilitates Virus-Induced Syncytium Formation. J. Virol. 1999, 73, 7262–7270. [Google Scholar] [CrossRef]
- Stertz, S.; Shaw, M.L. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect. 2011, 13, 516–525. [Google Scholar] [CrossRef]
- Huang, I.C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef]
- Pfaender, S.; Mar, K.B.; Michailidis, E.; Kratzel, A.; Boys, I.N.; V’kovski, P.; Fan, W.; Kelly, J.N.; Hirt, D.; Ebert, N.; et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol. 2020, 5, 1330–1339. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Kavanagh Williamson, M.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Kim, Y.M.; Shin, E.C. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 2021, 53, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Dieter, C.; Brondani, L.d.A.; Leitão, C.B.; Gerchman, F.; Lemos, N.E.; Crispim, D. Genetic polymorphisms associated with susceptibility to COVID-19 disease and severity: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0270627. [Google Scholar] [CrossRef] [PubMed]
- Araújo, A.; Sgorlon, G.; Aguiar, L.E.; Cidrão, M.H.M.C.; Teixeira, K.S.; Salcedo, J.M.V.; Passos-Silva, A.M.; Vieira, D. Influence of polymorphic variations of IFNL, HLA, and IL-6 genes in severe cases of COVID-19. Exp. Biol. Med. 2023, 248, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Alsaedi, S.B.; Mineta, K.; Gao, X.; Gojobori, T. Computational network analysis of host genetic risk variants of severe COVID-19. Hum. Genom. 2023, 17, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Iniguez, M.; Pérez-Matute, P.; Villoslada-Blanco, P.; Recio-Fernandez, E.; Ezquerro-Pérez, D.; Alba, J.; Ferreira-Laso, M.L.; Oteo, J.A. ACE Gene Variants Rise the Risk of Severe COVID-19 in Patients With Hypertension, Dyslipidemia or Diabetes: A Spanish Pilot Study. Front. Endo-Crinol. 2021, 12, 688071. [Google Scholar] [CrossRef]
- Colona, V.L.; Vasiliou, V.; Watt, J.; Novelli, G.; Reichardt, J.K.V. Update on human genetic susceptibility to COVID-19: Susceptibility to virus and response. Hum. Genom. 2021, 15, 57. [Google Scholar] [CrossRef]
- Li, Y.; Ke, Y.; Xia, X.; Wang, Y.; Cheng, F.; Liu, X.; Jin, X.; Li, B.; Xie, C.; Liu, S.; et al. Genome-wide association study of COVID-19 severity among the Chinese population. Cell Discov. 2021, 7, 76. [Google Scholar] [CrossRef]
- Llewellyn, G.N.; Chen, H.Y.; Rogers, G.L.; Huang, X.; Sell, P.J.; Henley, J.E.; Cannon, P.M. Comparison of SARS-CoV-2 entry inhibitors based on ACE2 receptor or engineered Spike-binding peptides. J. Virol. 2023, 97, e0068423. [Google Scholar] [CrossRef]
- Hinze, C.; Kocks, C.; Leiz, J.; Karaiskos, N.; Boltengagen, A.; Cao, S.; Skopnik, C.M.; Klocke, J.; Hardenberg, J.H.; Stockmann, H.; et al. Single-cell transcriptomics reveals common epithelial re-sponse patterns in human acute kidney injury. Genome Med. 2022, 14, 103. [Google Scholar] [CrossRef]
- Valiño-Rivas, L.; Cuarental, L.; Agustin, M.; Husi, H.; Cannata-Ortiz, P.; Sanz, A.B.; Mischak, H.; Ortiz, A.; Sanchez-Niño, M.D. MAGE genes in the kidney: Identification of MAGED2 as upregulated during kidney injury and in stressed tubular cells. Nephrol. Dial. Transplant. 2018, 34, 1498–1507. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).