
Uncovering the Genetic and Molecular Features of Huntington’s Disease in Northern Colombia

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Demographic Findings
2.2. Molecular Diagnosis and Genotype Distribution
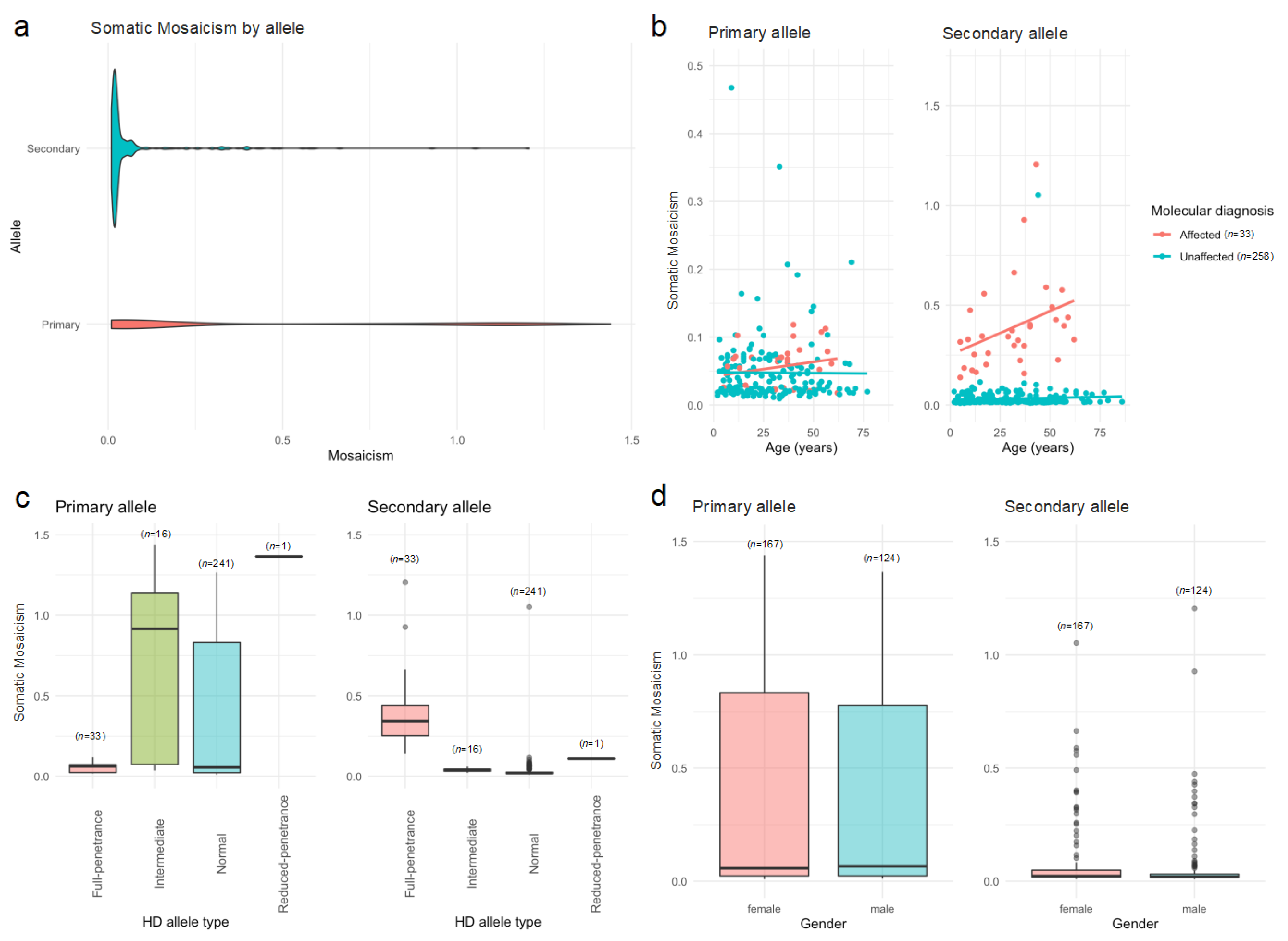
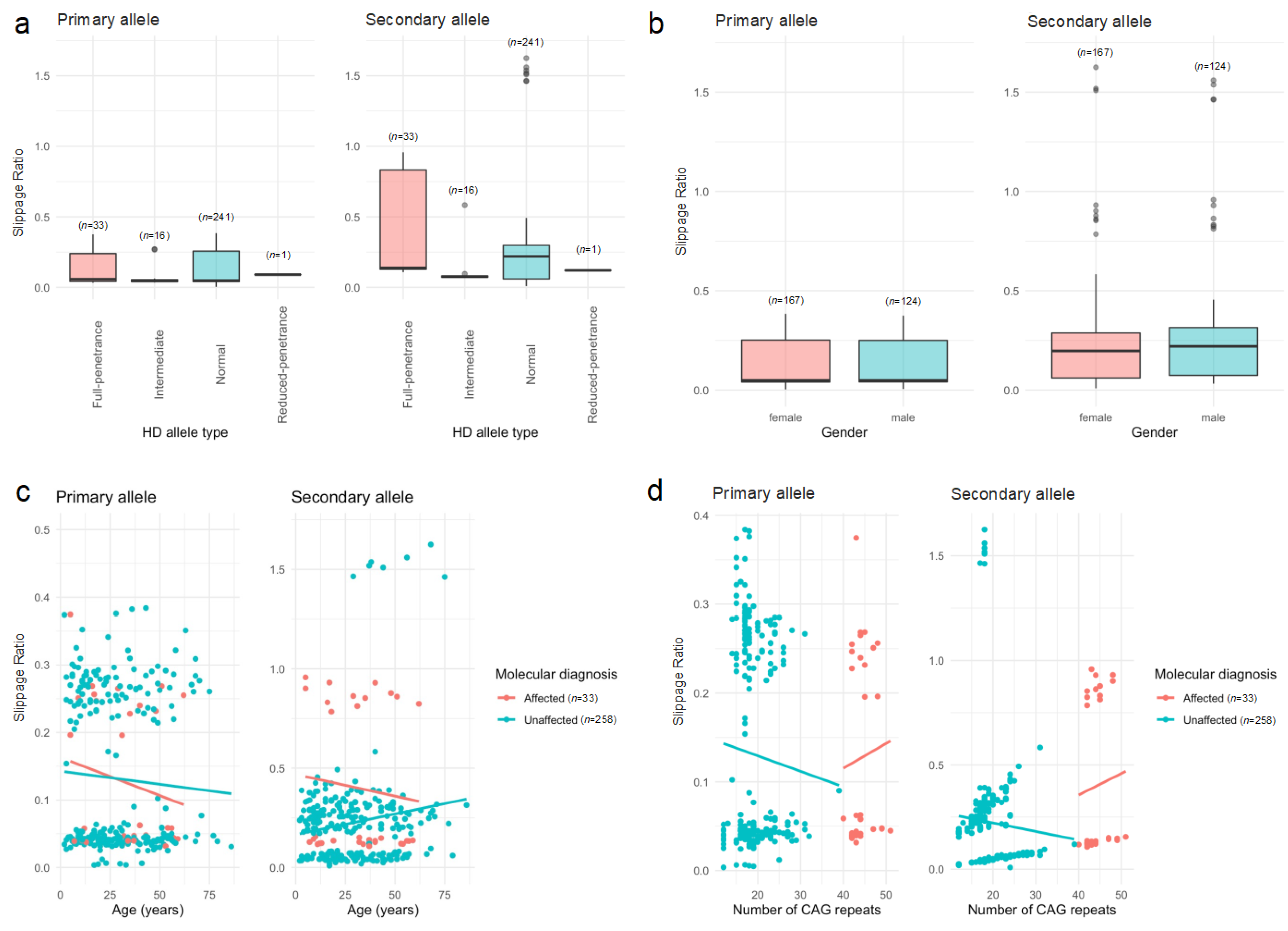
2.3. Heterogeneity Analysis
3. Discussion
4. Materials and Methods
4.1. Study Subjects
4.2. Blood Samples and Genomic DNA Extraction
4.3. Quantification of HTT CAG repeats
4.4. Bioinformatic and Statistical Analyses
4.4.1. Quantification of HTT CAG Repeats
4.4.2. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HD | Huntington’s disease. |
| HTT | Huntingtin, a protein coding gene. |
| CAG | Cytosine, adenine, guanine. |
| CCG | Cytosine, cytosine, guanine. |
References
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Marchina, E.; Misasi, S.; Bozzato, A.; Ferraboli, S.; Agosti, C.; Rozzini, L.; Borsani, G.; Barlati, S.; Padovani, A. Gene expression profile in fibroblasts of Huntington’s disease patients and controls. J. Neurol. Sci. 2014, 337, 42–46. [Google Scholar] [CrossRef]
- Potter, N.T.; Spector, E.B.; Prior, T.W. Technical Standards and Guidelines for Huntington Disease Testing. Genet. Med. 2004, 6, 61–65. [Google Scholar] [CrossRef]
- Gusella, J.F.; MacDonald, M.E. Huntington’s disease: Seeing the pathogenic process through a genetic lens. Trends Biochem. Sci. 2006, 31, 533–540. [Google Scholar] [CrossRef]
- Burton, A. Hope, humanity, and Huntington’s disease in Latin America. Lancet Neurol. 2013, 12, 133–134. [Google Scholar] [CrossRef]
- De Castro, M.; Restrepo, C.M. Genetics and genomic medicine in colombia. Mol. Genet. Genomic Med. 2015, 3, 84–91. [Google Scholar] [CrossRef]
- Daza, B.; Caiaffa, R.H.; Arteta, B.J.; Echeverría, R.V.; Ladrón de Guevara, Z.; Escamilla, M. Estudio neuroepidemiológico en Juande Acosta, Atlántico, Colombia. Acta Méd. Colomb. 1991, 17, 324. [Google Scholar]
- Sánchez-Castañeda, C.; Squitieri, F.; Di Paola, M.; Dayan, M.; Petrollini, M.; Sabatini, U. The role of iron in gray matter degeneration in huntington’s disease: A magnetic resonance imaging study. Hum. Brain Mapp. 2015, 36, 50–66. [Google Scholar] [CrossRef]
- Maxwell, A. ScaleHD Documentation. 2022. Available online: https://scalehd.readthedocs.io/_/downloads/en/latest/pdf/ (accessed on 20 March 2022).
- Pulkes, T.; Papsing, C.; Wattanapokayakit, S.; Mahasirimongkol, S. Cag-expansion haplotype analysis in a population with a low prevalence of huntington’s disease. J. Clin. Neurol. 2014, 10, 32–36. [Google Scholar] [CrossRef]
- Warby, S.C.; Visscher, H.; Collins, J.A.; Doty, C.N.; Carter, C.; Butland, S.L.; Hayden, A.R.; Kanazawa, I.; Ross, C.J.; Hayden, M.R. HTT haplotypes contribute to differences in Huntington disease prevalence between Europe and East Asia. Eur. J. Hum. Genet. 2011, 19, 561–566. [Google Scholar] [CrossRef]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E.; et al. A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 1994, 330, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov. Disord. 2012, 27, 1083–1091. [Google Scholar] [CrossRef]
- Hayden, M.R.; Berkowicz, A.L.; Beighton, P.H.; Yiptong, C. Huntington’s chorea on the island of Mauritius. S. Afr. Med. J. 1981, 60, 1001–1002. [Google Scholar] [PubMed]
- Apolinário, T.A.; Silva, I.d.S.d.; Agostinho, L.d.A.; Paiva, C.L.A. Investigation of intermediate CAG alleles of the HTT in the general population of Rio de Janeiro, Brazil, in comparison with a sample of Huntington disease-affected families. Mol. Genet. Genomic Med. 2020, 8, e1181. [Google Scholar] [CrossRef] [PubMed]
- Agostinho, L.D.A.; Rocha, C.F.; Medina-Acosta, E.; Barboza, H.N.; da Silva, A.F.A.; Pereira, S.P.; da Silva, I.D.S.; Paradela, E.R.; Figueiredo, A.L.D.S.; Nogueira, E.D.M.; et al. Haplotype analysis of the CAG and CCG repeats in 21 Brazilian families with Huntington’s disease. J. Hum. Genet. 2012, 57, 796–803. [Google Scholar] [CrossRef]
- Masuda, N.; Goto, J.; Murayama, N.; Watanabe, M.; Kondo, I. Kanazawa Analysis of triplet repeats in the huntingtin gene in Japanese families affected with Huntington’s disease. J. Med. Genet. 1995, 32, 701–705. [Google Scholar] [CrossRef]
- Ruiz de Sabando, A.; Urrutia Lafuente, E.; Galbete, A.; Ciosi, M.; García Amigot, F.; García Solaesa, V.; Spanish HD Collaborative Group; Monckton, D.G.; Ramos-Arroyo, M.A. Spanish HTT gene study reveals haplotype and allelic diversity with possible implications for germline expansion dynamics in Huntington disease. Hum. Mol. Genet. 2022, 32, 897–906. [Google Scholar] [CrossRef]
- Walker, R.H.; Gatto, E.M.; Bustamante, M.L.; Bernal-Pacheco, O.; Cardoso, F.; Castilhos, R.M.; Chana-Cuevas, P.; Cornejo-Olivas, M.; Estrada-Bellmann, I.; Jardim, L.B.; et al. Huntington’s disease-like disorders in Latin America and the Caribbean. Park. Relat. Disord. 2018, 53, 10–20. [Google Scholar] [CrossRef]
- Campbell, I.M.; Shaw, C.A.; Stankiewicz, P.; Lupski, J.R. Somatic mosaicism: Implications for disease and transmission genetics. Trends Genet. 2015, 31, 382–392. [Google Scholar] [CrossRef]
- Clever, F.; Cho, I.K.; Yang, J.; Chan, A.W.S. Progressive Polyglutamine Repeat Expansion in Peripheral Blood Cells and Sperm of Transgenic Huntington’s Disease Monkeys. J. Huntingt. Dis. 2019, 8, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Semaka, A.; Kay, C.; Doty, C.N.; Collins, J.A.; Tam, N.; Hayden, M.R. High frequency of intermediate alleles on Huntington disease-associated haplotypes in British Columbia’s general population. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2013, 162B, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Palareti, G.; Legnani, C.; Cosmi, B.; Antonucci, E.; Erba, N.; Poli, D.; Testa, S.; Tosetto, A.; DULCIS (D-dimer-ULtrasonography in Combination Italian Study) Investigators; De Micheli, V.; et al. Comparison between different D-Dimer cutoff values to assess the individual risk of recurrent venous thromboembolism: Analysis of results obtained in the DULCIS study. Int. J. Lab. Hematol. 2016, 38, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Kacher, R.; Lejeune, F.X.; Noel, S.; Cazeneuve, C.; Brice, A.; Humbert, S.; Durr, A. Propensity for somatic expansion increases over the course of life in huntington disease. Elife 2021, 10, e64674. [Google Scholar] [CrossRef] [PubMed]
- Viguera, E.; Canceill, D.; Ehrlich, S.D. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 2001, 20, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Sathe, S.; Ware, J.; Levey, J.; Neacy, E.; Blumenstein, R.; Noble, S.; Mühlbäck, A.; Rosser, A.; Landwehrmeyer, G.B.; Sampaio, C. Enroll-HD: An Integrated Clinical Research Platform and Worldwide Observational Study for Huntington’s Disease. Front. Neurol. 2021, 12, 667420. [Google Scholar] [CrossRef]
- Espinoza, F.A.; Turner, J.A.; Vergara, V.M.; Miller, R.L.; Mennigen, E.; Liu, J.; Misiura, M.B.; Ciarochi, J.; Johnson, H.J.; Long, J.D.; et al. Whole-Brain Connectivity in a Large Study of Huntington’s Disease Gene Mutation Carriers and Healthy Controls. Brain Connect. 2018, 8, 166–178. [Google Scholar] [CrossRef]
- Vélez, J.I. Machine Learning based Psychology: Advocating for A Data-Driven Approach. Int. J. Psychol. Res. 2021, 14, 6–11. [Google Scholar] [CrossRef]
- Mohan, A.; Sun, Z.; Ghosh, S.; Li, Y.; Sathe, S.; Hu, J.; Sampaio, C. A Machine-Learning Derived Huntington’s Disease Progression Model: Insights for Clinical Trial Design. Mov. Disord. 2022, 37, 553–562. [Google Scholar] [CrossRef]
- Riad, R.; Lunven, M.; Titeux, H.; Cao, X.N.; Hamet Bagnou, J.; Lemoine, L.; Montillot, J.; Sliwinski, A.; Youssov, K.; Cleret de Langavant, L.; et al. Predicting clinical scores in Huntington’s disease: A lightweight speech test. J. Neurol. 2022, 269, 5008–5021. [Google Scholar] [CrossRef]
- Odish, O.F.F.; Johnsen, K.; van Someren, P.; Roos, R.A.C.; van Dijk, J.G. EEG may serve as a biomarker in Huntington’s disease using machine learning automatic classification. Sci. Rep. 2018, 8, 16090. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.; Pinto, A.J.; Guest, J.S. Gene-Specific Primers for Improved Characterization of Mixed Phototrophic Communities. Appl. Environ. Microbiol. 2016, 82, 5878–5891. [Google Scholar] [CrossRef] [PubMed]
- Ciosi, M.; Cumming, S.A.; Alshammari, A.M.; Symeonidi, E.; Herzyk, P.; McGuinness, D.; Galbraith, J.; Hamilton, G.; Monckton, D.G. Library Preparation and MiSeq Sequencing for the Genotyping-by-Sequencing of the Huntington Disease HTT Exon One Trinucleotide Repeat and the Quantification of Somatic Mosaicism; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J.; Fadrosh, D.W.; Ma, B.; Gajer, P.; et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Affected n = 33 (11.3%) | Unaffected n = 258 (88.7%) | Statistic a | p-Value |
|---|---|---|---|---|
| Gender | Frequency (%) | 0.03 (1) | 0.867 | |
| Female | 18 (10.8) | 149 (89.2) | ||
| Male | 15 (12.1) | 109 (87.9) | ||
| Age | Frequency (%) | 5.34 (3) | 0.148 | |
| Children (<15 y) | 8 (11.4) | 62 (88.6) | ||
| AYA (15–29 y) | 4 (5) | 76 (95) | ||
| Adults (30–59 y) | 19 (15.6) | 103 (84.4) | ||
| Elderly (>59 y) | 2 (12.5) | 14 (87.5) | ||
| Schooling (years) | Frequency (%) | 1 (4) | 0.909 | |
| 0 | 0 (0) | 5 (100) | ||
| 1 | 2 (9.5) | 19 (90.5) | ||
| 2 | 8 (11.8) | 60 (88.2) | ||
| 3 | 13 (12.6) | 90 (87.4) | ||
| 4 | 8 (13.8) | 50 (86.2) | ||
| HD type | Frequency (%) | 525.87 (3) | <0.00001 | |
| Normal | - | 241 (82.8) | ||
| Intermediate | - | 16 (5.5) | ||
| Reduced penetrance | - | 1 (0.3) | ||
| Full penetrance | 33 (11.3) | - | ||
| Population | Number of CAG Repeats | Reference | ||||
|---|---|---|---|---|---|---|
| Mean | SD | Range | n | p-Value | ||
| Thai | 16.5 | 1.9 | 8–28 | 449 | Not reported | [10] |
| European | 18.4 | 3.7 | 8–35 | 479 | <0.0001 | [12] |
| American | 19.7 | 3.2 | 11–34 | 545 | <0.0001 | [13] |
| Finnish | 17.1 | 1.8 | 14–23 | 48 | 0.255 | [12] |
| Black | 16.2 | 2.5 | 8–24 | 113 | 0.55 | [12] |
| Chinese | 16.4 | 1.5 | 8–20 | 90 | 1 | [12] |
| Japanese | 16.6 | 1.3 | 13–23 | 166 | 1 | [12] |
| Colombian | 18.2 | 3 | 12–35 | 257 | 0.00001 | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, M.; Ríos-Anillo, M.R.; Acosta-López, J.E.; Cervantes-Henríquez, M.L.; Martínez-Banfi, M.; Pineda-Alhucema, W.; Puentes-Rozo, P.; Sánchez-Barros, C.; Pinzón, A.; Patel, H.R.; et al. Uncovering the Genetic and Molecular Features of Huntington’s Disease in Northern Colombia. Int. J. Mol. Sci. 2023, 24, 16154. https://doi.org/10.3390/ijms242216154
Ahmad M, Ríos-Anillo MR, Acosta-López JE, Cervantes-Henríquez ML, Martínez-Banfi M, Pineda-Alhucema W, Puentes-Rozo P, Sánchez-Barros C, Pinzón A, Patel HR, et al. Uncovering the Genetic and Molecular Features of Huntington’s Disease in Northern Colombia. International Journal of Molecular Sciences. 2023; 24(22):16154. https://doi.org/10.3390/ijms242216154
Chicago/Turabian StyleAhmad, Mostapha, Margarita R. Ríos-Anillo, Johan E. Acosta-López, Martha L. Cervantes-Henríquez, Martha Martínez-Banfi, Wilmar Pineda-Alhucema, Pedro Puentes-Rozo, Cristian Sánchez-Barros, Andrés Pinzón, Hardip R. Patel, and et al. 2023. "Uncovering the Genetic and Molecular Features of Huntington’s Disease in Northern Colombia" International Journal of Molecular Sciences 24, no. 22: 16154. https://doi.org/10.3390/ijms242216154