

Naphthyl-Substituted Indole and Pyrrole Carboxylic Acids as Effective Antibiotic Potentiators—Inhibitors of Bacterial Cystathionine γ-Lyase

 ,
,  , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
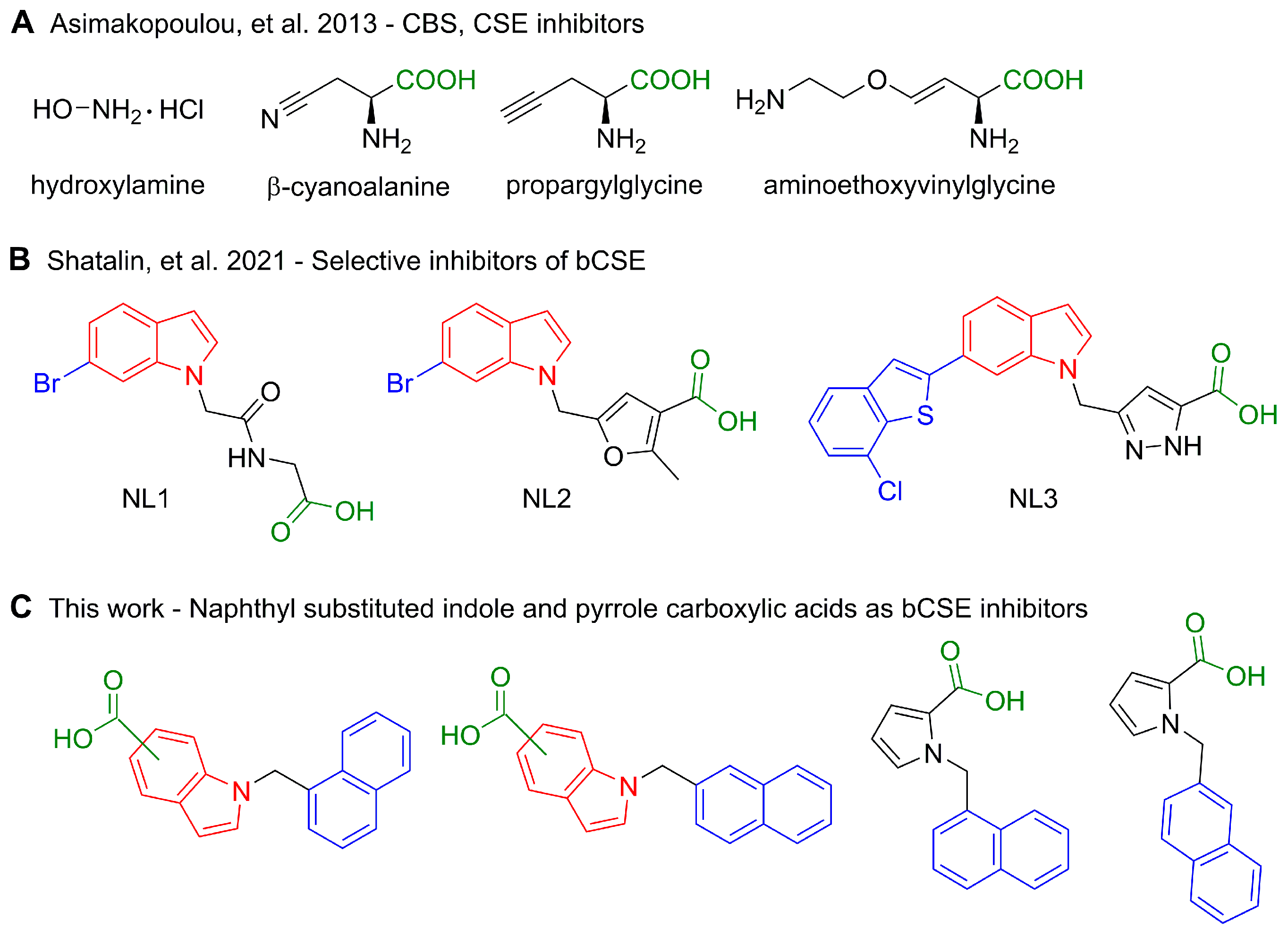
2.1. Design Strategy and Synthesis
2.2. Enzymatic (bCSE and Human CSE (hCSE)) Assays
2.3. Antibacterial Activity
2.4. ADMET Experiments
2.5. Molecular Dynamics Simulations
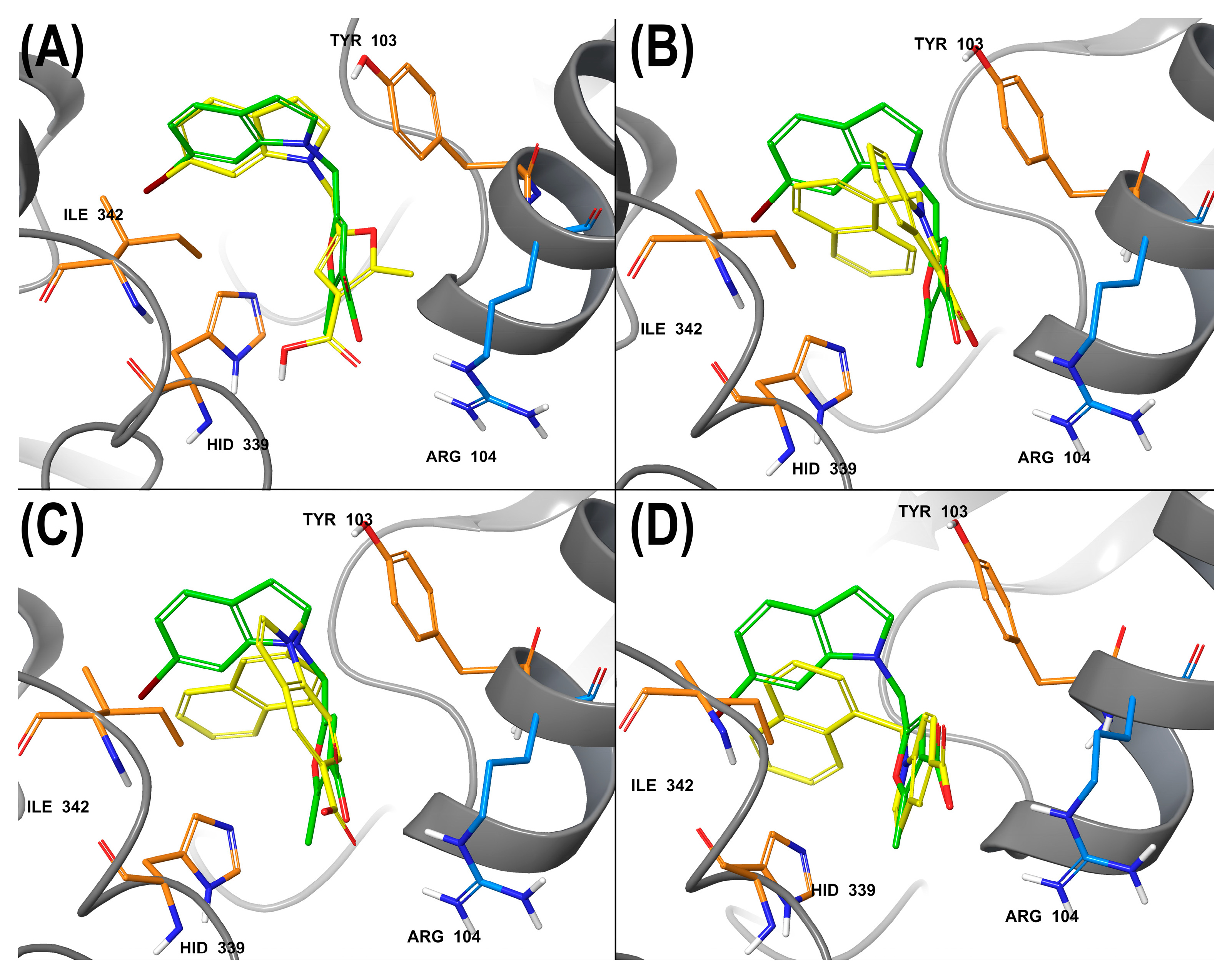
2.6. Molecular Docking
3. Materials and Methods
3.1. General Procedures, Materials, and Equipment
3.1.1. Glassware and Reaction Techniques
3.1.2. Solvents, Reagents, and Chemicals
3.1.3. Column Chromatography and Thin Layer Chromatography (TLC)
3.1.4. NMR Spectroscopy (Engelhardt Institute of Molecular Biology RAS, Moscow, Russia)
3.1.5. Mass Spectrometry (MS) (Sirius University of Science and Technology, Sochi, Russia)
3.1.6. Purity Analysis (Sirius University of Science and Technology, Sochi, Russia)
3.2. Synthesis of Target Compounds
3.2.1. General Procedure 1: N-alkylation of Indole Carboxylic Acids to Obtain 1a, 1b, 1c, 3a, 4a, and 4b

3.2.2. General Procedure 2: N-alkylation of Pyrrole and Indole Carboxylates to Obtain 2c, 2d, 3c, 5c, and 5d

3.2.3. General Procedure 3: Saponification of N-alkylated Pyrrole and Indole Carboxylates to Obtain Target Compounds 2a, 2b, 3b, 5a, and 5b

3.3. IC50 Assay
3.4. Microbiological Assays
3.4.1. Tested Bacterial Strains
3.4.2. Assessment of MICs
3.5. ADMET Experiments
3.5.1. Caco-2 Cell Penetration
3.5.2. Stability in SGF, SIF, and Human Plasma
3.5.3. Plasma Protein Binding
3.5.4. Microsomal Stability Assay
3.5.5. Solubility
3.5.6. LogD
3.5.7. LC–MS/MS Analysis
3.5.8. Cytotoxicity Assay
3.6. Lab Scale Preparation, Purification, and Validation of Proteins bCSE and hCSE
3.7. Other Methods and Funnel Metadynamics
3.7.1. Parameter Sets
3.7.2. System Preparation
3.7.3. Molecular Dynamics Simulations
3.7.4. Funnel Metadynamics
3.8. Molecular Docking
3.8.1. Protein and Ligand Structure Preparation
3.8.2. GridBox Generation and the Docking Protocol
3.8.3. The MM–GBSA Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Kraker, M.E.A.; Stewardson, A.J.; Harbarth, S. Will 10 Million People Die a Year Due to Antimicrobial Resistance by 2050? PLoS Med. 2016, 13, e1002184. [Google Scholar] [CrossRef] [PubMed]
- Gould, K. Antibiotics: From Prehistory to the Present Day. J. Antimicrob. Chemother. 2016, 71, 572–575. [Google Scholar] [CrossRef] [PubMed]
- WHO List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/en/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 14 October 2023).
- Donadio, S.; Maffioli, S.; Monciardini, P.; Sosio, M.; Jabes, D. Antibiotic Discovery in the Twenty-First Century: Current Trends and Future Perspectives. J. Antibiot. 2010, 63, 423–430. [Google Scholar] [CrossRef]
- Fernandes, P.; Martens, E.; Bertrand, D.; Pereira, D. The Solithromycin Journey—It Is All in the Chemistry. Bioorg. Med. Chem. 2016, 24, 6420–6428. [Google Scholar] [CrossRef] [PubMed]
- Årdal, C.; Balasegaram, M.; Laxminarayan, R.; McAdams, D.; Outterson, K.; Rex, J.H.; Sumpradit, N. Antibiotic Development—Economic, Regulatory and Societal Challenges. Nat. Rev. Microbiol. 2020, 18, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Wang, S. Antibiotic Modification Addressing Resistance. In Antibiotic Drug Resistance; Capelo-Martínez, J., Igrejas, G., Eds.; Wiley: Hoboken, NJ, USA, 2019; pp. 407–428. [Google Scholar]
- Douafer, H.; Andrieu, V.; Phanstiel, O.; Brunel, J.M. Antibiotic Adjuvants: Make Antibiotics Great Again! J. Med. Chem. 2019, 62, 8665–8681. [Google Scholar] [CrossRef]
- Wright, G.D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef]
- Fasnacht, M.; Polacek, N. Oxidative Stress in Bacteria and the Central Dogma of Molecular Biology. Front. Mol. Biosci. 2021, 8, 671037. [Google Scholar] [CrossRef]
- Mironov, A.; Seregina, T.; Nagornykh, M.; Luhachack, L.G.; Korolkova, N.; Lopes, L.E.; Kotova, V.; Zavilgelsky, G.; Shakulov, R.; Shatalin, K.; et al. Mechanism of H2S-Mediated Protection against Oxidative Stress in Escherichia Coli. Proc. Natl. Acad. Sci. USA 2017, 114, 6022–6027. [Google Scholar] [CrossRef]
- Shatalin, K.; Shatalina, E.; Mironov, A.; Nudler, E. H2S: A Universal Defense Against Antibiotics in Bacteria. Science 2011, 334, 986–990. [Google Scholar] [CrossRef]
- Shatalin, K.; Nuthanakanti, A.; Kaushik, A.; Shishov, D.; Peselis, A.; Shamovsky, I.; Pani, B.; Lechpammer, M.; Vasilyev, N.; Shatalina, E.; et al. Inhibitors of Bacterial H2S Biogenesis Targeting Antibiotic Resistance and Tolerance. Science 2021, 372, 1169–1175. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E. Impact of Hydrogen Sulfide on Mitochondrial and Bacterial Bioenergetics. Int. J. Mol. Sci. 2021, 22, 12688. [Google Scholar] [CrossRef] [PubMed]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of Commonly Used Pharmacological Inhibitors for Cystathionine β-Synthase (CBS) and Cystathionine γ-Lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Kudličková, Z.; Michalková, R.; Salayová, A.; Ksiažek, M.; Vilková, M.; Bekešová, S.; Mojžiš, J. Design, Synthesis, and Evaluation of Novel Indole Hybrid Chalcones and Their Antiproliferative and Antioxidant Activity. Molecules 2023, 28, 6583. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Moi, D.; Pedrini, M.; Pérez-Peña, H.; Pieraccini, S.; Dimasi, A.; Stagno, C.; Micale, N.; Schirmeister, T.; Sibille, G.; et al. Synthesis of SARS-CoV-2 M pro Inhibitors Bearing a Cinnamic Ester Warhead with in Vitro Activity against Human Coronaviruses. Org. Biomol. Chem. 2023, 21, 3811–3824. [Google Scholar] [CrossRef]
- Jagadeesan, S.; Karpagam, S. Novel Series of N-Acyl Substituted Indole Based Piperazine, Thiazole and Tetrazoles as Potential Antibacterial, Antifungal, Antioxidant and Cytotoxic Agents, and Their Docking Investigation as Potential Mcl-1 Inhibitors. J. Mol. Struct. 2023, 1271, 134013. [Google Scholar] [CrossRef]
- Peng, B.; Chen, W.; Liu, C.; Rosser, E.W.; Pacheco, A.; Zhao, Y.; Aguilar, H.C.; Xian, M. Fluorescent Probes Based on Nucleophilic Substitution-Cyclization for Hydrogen Sulfide Detection and Bioimaging. Chem. Eur. J. 2014, 20, 1010–1016. [Google Scholar] [CrossRef]
- Dwyer, D.J.; Kohanski, M.A.; Hayete, B.; Collins, J.J. Gyrase Inhibitors Induce an Oxidative Damage Cellular Death Pathway in Escherichia Coli. Mol. Syst. Biol. 2007, 3, 91. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A Common Mechanism of Cellular Death Induced by Bactericidal Antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef]
- Qin, H.-L.; Liu, J.; Fang, W.-Y.; Ravindar, L.; Rakesh, K.P. Indole-Based Derivatives as Potential Antibacterial Activity against Methicillin-Resistance Staphylococcus Aureus (MRSA). Eur. J. Med. Chem. 2020, 194, 112245. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Steegborn, C.; Clausen, T.; Sondermann, P.; Jacob, U.; Worbs, M.; Marinkovic, S.; Huber, R.; Wahl, M.C. Kinetics and Inhibition of Recombinant Human Cystathionine γ-Lyase. J. Biol. Chem. 1999, 274, 12675–12684. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J. Lipophilicity in Drug Discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Słoczyńska, K.; Gunia-Krzyżak, A.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Żelaszczyk, D.; Popiół, J.; Pękala, E. Metabolic Stability and Its Role in the Discovery of New Chemical Entities. Acta Pharm. 2019, 69, 345–361. [Google Scholar] [CrossRef]
- Gardiner, P.; Cox, R.J.; Grime, K. Plasma Protein Binding as an Optimizable Parameter for Acidic Drugs. Drug Metab. Dispos. 2019, 47, 865–873. [Google Scholar] [CrossRef]
- Smith, D.A.; Di, L.; Kerns, E.H. The Effect of Plasma Protein Binding on in Vivo Efficacy: Misconceptions in Drug Discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939. [Google Scholar] [CrossRef]
- Messerschmidt, A.; Worbs, M.; Steegborn, C.; Wahl, M.C.; Huber, R.; Laber, B.; Clausen, T. Determinants of Enzymatic Specificity in the Cys-Met-Metabolism PLP-Dependent Enzyme Family: Crystal Structure of Cystathionine γ-Lyase from Yeast and Intrafamiliar Structure Comparison. Biol. Chem. 2003, 384, 373–386. [Google Scholar] [CrossRef]
- Kaplan, M.M.; Flavin, M. Cystathionine γ-Synthetase of Salmonella. J. Biol. Chem. 1966, 241, 4463–4471. [Google Scholar] [CrossRef]
- Limongelli, V.; Bonomi, M.; Parrinello, M. Funnel Metadynamics as Accurate Binding Free-Energy Method. Proc. Natl. Acad. Sci. USA 2013, 110, 6358–6363. [Google Scholar] [CrossRef]
- Raniolo, S.; Limongelli, V. Ligand Binding Free-Energy Calculations with Funnel Metadynamics. Nat. Protoc. 2020, 15, 2837–2866. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- The PLUMED consortium. Promoting Transparency and Reproducibility in Enhanced Molecular Simulations. Nat. Methods 2019, 16, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Alferova, V.A.; Maviza, T.P.; Biryukov, M.V.; Zakalyukina, Y.V.; Lukianov, D.A.; Skvortsov, D.A.; Vasilyeva, L.A.; Tashlitsky, V.N.; Polshakov, V.I.; Sergiev, P.V.; et al. Biological Evaluation and Spectral Characterization of a Novel Tetracenomycin X Congener. Biochimie 2022, 192, 63–71. [Google Scholar] [CrossRef]
- Osterman, I.A.; Wieland, M.; Maviza, T.P.; Lashkevich, K.A.; Lukianov, D.A.; Komarova, E.S.; Zakalyukina, Y.V.; Buschauer, R.; Shiriaev, D.I.; Leyn, S.A.; et al. Tetracenomycin X Inhibits Translation by Binding within the Ribosomal Exit Tunnel. Nat. Chem. Biol. 2020, 16, 1071–1077. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y.; Yu, A.; Sun, D.; Yu, L.X. Oral Drug Absorption. In Developing Solid Oral Dosage Forms; Qui, Y., Chen, Y., Zhang, G., Yu, L., Mantri, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 331–354. [Google Scholar]
- Volpe, D.A. Variability in Caco-2 and MDCK Cell-Based Intestinal Permeability Assays. J. Pharm. Sci. 2008, 97, 712–725. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (Resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Connolly, M.L. Analytical Molecular Surface Calculation. J. Appl. Crystallogr. 1983, 16, 548–558. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Bernetti, M.; Bussi, G. Pressure Control Using Stochastic Cell Rescaling. J. Chem. Phys. 2020, 153, 114107. [Google Scholar] [CrossRef] [PubMed]
- Bhakat, S.; Söderhjelm, P. Resolving the Problem of Trapped Water in Binding Cavities: Prediction of Host–Guest Binding Free Energies in the SAMPL5 Challenge by Funnel Metadynamics. J. Comput. Aided Mol. Des. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for pKa Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible Ligand Docking with Glide. Curr. Protoc. Bioinform. 2007, 18. [Google Scholar] [CrossRef]
- Suenaga, A.; Okimoto, N.; Hirano, Y.; Fukui, K. An Efficient Computational Method for Calculating Ligand Binding Affinities. PLoS ONE 2012, 7, e42846. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Subhas Bose, D.; Idrees, M.; Todewale, I.K.; Jakka, N.M.; Venkateswara Rao, J. Hybrids of Privileged Structures Benzothiazoles and Pyrrolo[2,1-c] [1,4]Benzodiazepin-5-One, and Diversity-Oriented Synthesis of Benzothiazoles. Eur. J. Med. Chem. 2012, 50, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Li, X.; Deng, S.; Qi, X.; Cong, H.; Cheng, H.-G.; Shi, L.; Zhou, Q.; Zhuang, L. From N–H Nitration to Controllable Aromatic Mononitration and Dinitration─The Discovery of a Versatile and Powerful N-Nitropyrazole Nitrating Reagent. JACS 2022, 2, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Brown, F.J.; Cronk, L.A.; Aharony, D.; Snyder, D.W. 1,3,6-Trisubstituted Indoles as Peptidoleukotriene Antagonists: Benefits of a Second, Polar, Pyrrole Substituent. J. Med. Chem. 1992, 35, 2419–2439. [Google Scholar] [CrossRef] [PubMed]
- Houck, H.A.; Blasco, E.; Du Prez, F.E.; Barner-Kowollik, C. Light-Stabilized Dynamic Materials. J. Am. Chem. Soc. 2019, 141, 12329–12337. [Google Scholar] [CrossRef] [PubMed]
- Young, B.M.; Rossi, P.; Slavish, P.J.; Cui, Y.; Sowaileh, M.; Das, J.; Kalodimos, C.G.; Rankovic, Z. Synthesis of Isotopically Labeled, Spin-Isolated Tyrosine and Phenylalanine for Protein NMR Applications. Org. Lett. 2021, 23, 6288–6292. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Fatima, K.; Dudi, R.K.; Tabassum, M.; Iqbal, H.; Kumar, Y.; Luqman, S.; Mondhe, D.M.; Chanda, D.; Khan, F.; et al. Antiproliferative Activity of Diarylnaphthylpyrrolidine Derivative via Dual Target Inhibition. Eur. J. Med. Chem. 2020, 188, 111986. [Google Scholar] [CrossRef]
- Portolani, C.; Centonze, G.; Luciani, S.; Pellegrini, A.; Righi, P.; Mazzanti, A.; Ciogli, A.; Sorato, A.; Bencivenni, G. Synthesis of Atropisomeric Hydrazides by One-Pot Sequential Enantio- and Diastereoselective Catalysis. Angew. Chem. Int. Ed. 2022, 61, e202209895. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuzovlev, A.S.; Zybalov, M.D.; Golovin, A.V.; Gureev, M.A.; Kasatkina, M.A.; Biryukov, M.V.; Belik, A.R.; Silonov, S.A.; Yunin, M.A.; Zigangirova, N.A.; et al. Naphthyl-Substituted Indole and Pyrrole Carboxylic Acids as Effective Antibiotic Potentiators—Inhibitors of Bacterial Cystathionine γ-Lyase. Int. J. Mol. Sci. 2023, 24, 16331. https://doi.org/10.3390/ijms242216331
Kuzovlev AS, Zybalov MD, Golovin AV, Gureev MA, Kasatkina MA, Biryukov MV, Belik AR, Silonov SA, Yunin MA, Zigangirova NA, et al. Naphthyl-Substituted Indole and Pyrrole Carboxylic Acids as Effective Antibiotic Potentiators—Inhibitors of Bacterial Cystathionine γ-Lyase. International Journal of Molecular Sciences. 2023; 24(22):16331. https://doi.org/10.3390/ijms242216331
Chicago/Turabian StyleKuzovlev, Andrey S., Mikhail D. Zybalov, Andrey V. Golovin, Maxim A. Gureev, Mariia A. Kasatkina, Mikhail V. Biryukov, Albina R. Belik, Sergey A. Silonov, Maxim A. Yunin, Nailya A. Zigangirova, and et al. 2023. "Naphthyl-Substituted Indole and Pyrrole Carboxylic Acids as Effective Antibiotic Potentiators—Inhibitors of Bacterial Cystathionine γ-Lyase" International Journal of Molecular Sciences 24, no. 22: 16331. https://doi.org/10.3390/ijms242216331
APA StyleKuzovlev, A. S., Zybalov, M. D., Golovin, A. V., Gureev, M. A., Kasatkina, M. A., Biryukov, M. V., Belik, A. R., Silonov, S. A., Yunin, M. A., Zigangirova, N. A., Reshetnikov, V. V., Isakova, Y. E., Porozov, Y. B., & Ivanov, R. A. (2023). Naphthyl-Substituted Indole and Pyrrole Carboxylic Acids as Effective Antibiotic Potentiators—Inhibitors of Bacterial Cystathionine γ-Lyase. International Journal of Molecular Sciences, 24(22), 16331. https://doi.org/10.3390/ijms242216331