
Resensitizing Paclitaxel-Resistant Ovarian Cancer via Targeting Lipid Metabolism Key Enzymes CPT1A, SCD and FASN
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
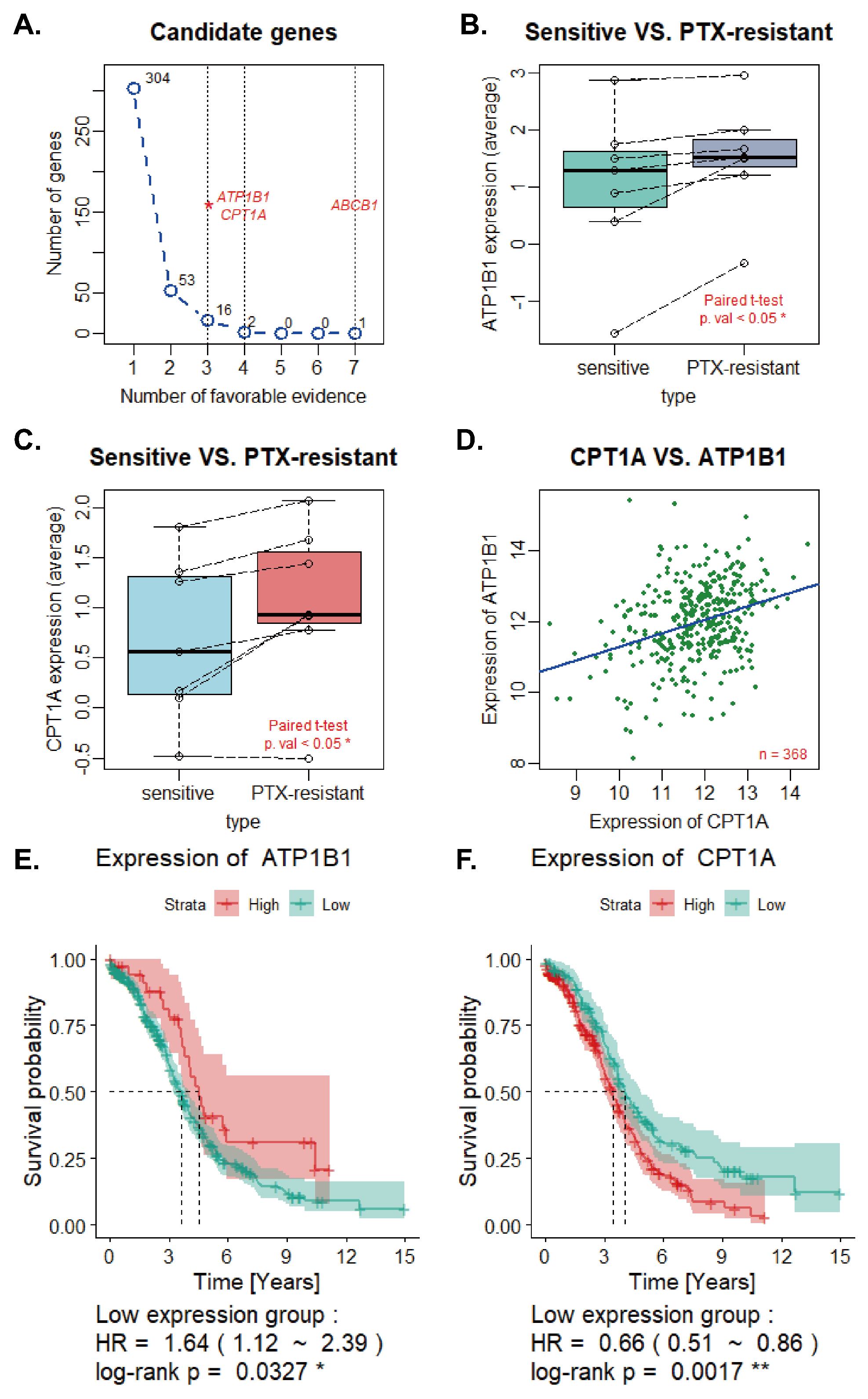
2.1. Screening, Validation of PRGs and Univariate Regression Analysis
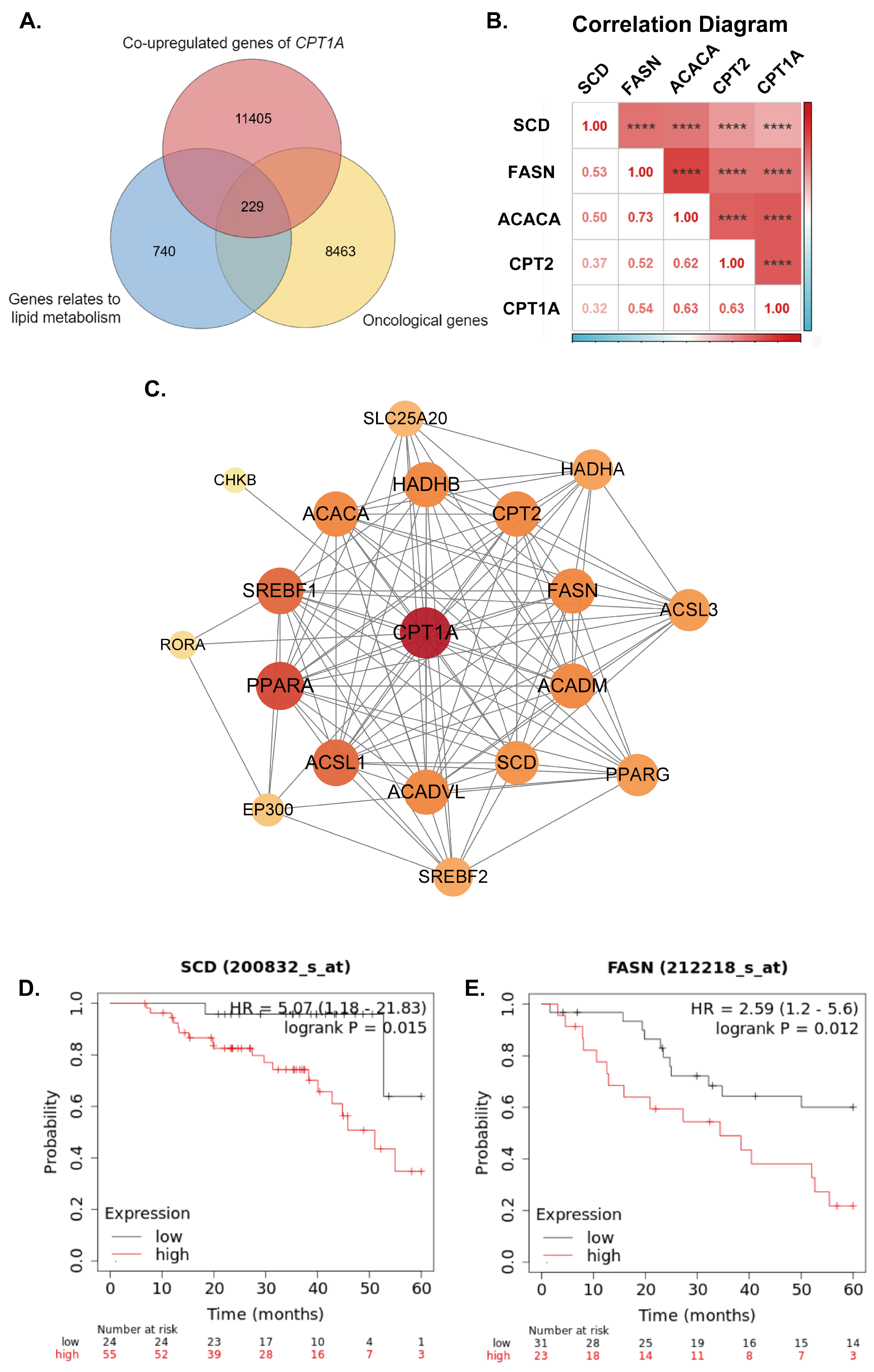
2.2. Overall Survival Analysis and Identification of Synergistic Genes
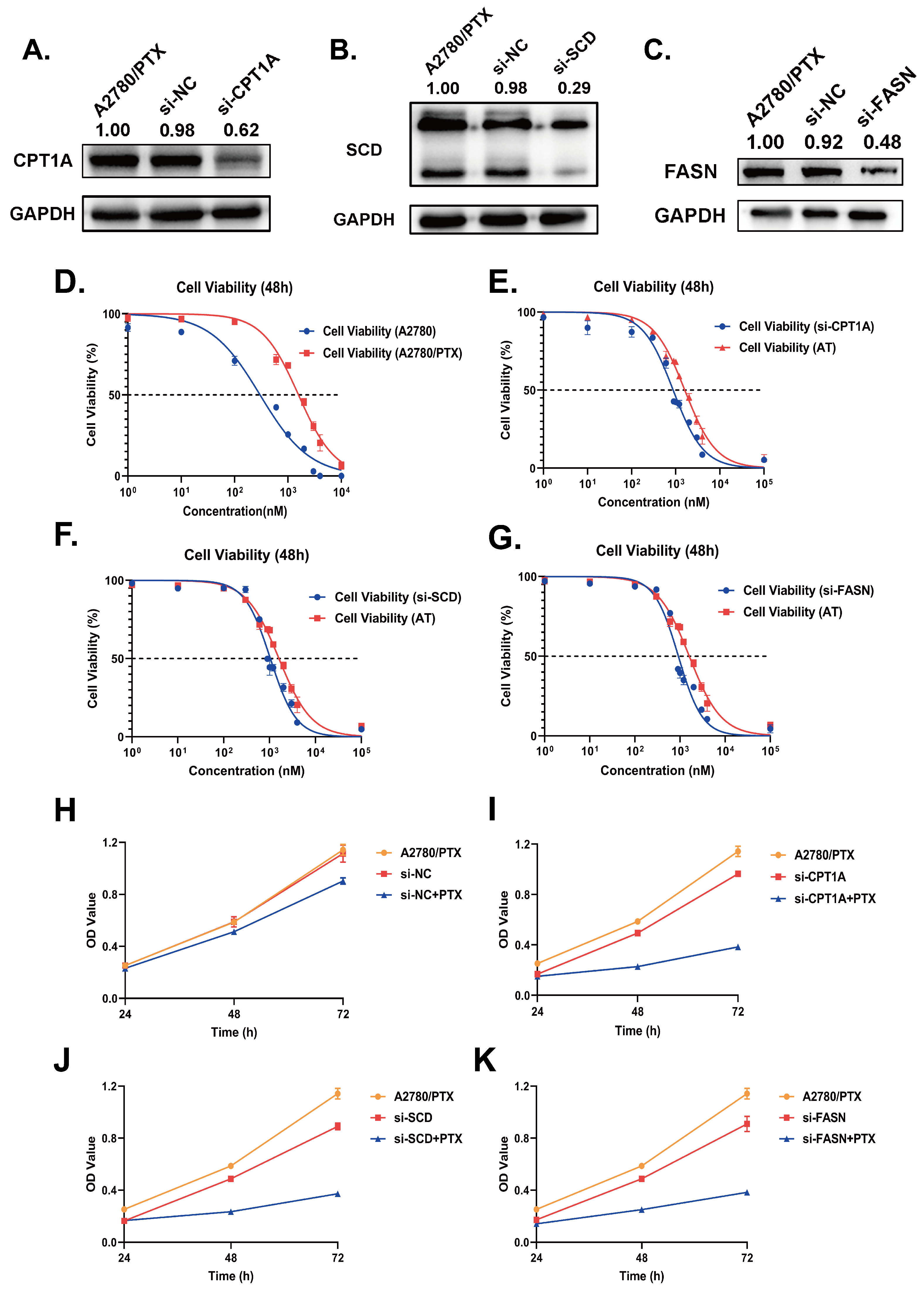
2.3. Validation of Gene Expression Levels of CPT1A, SCD and FASN in Cultured Cell Lines
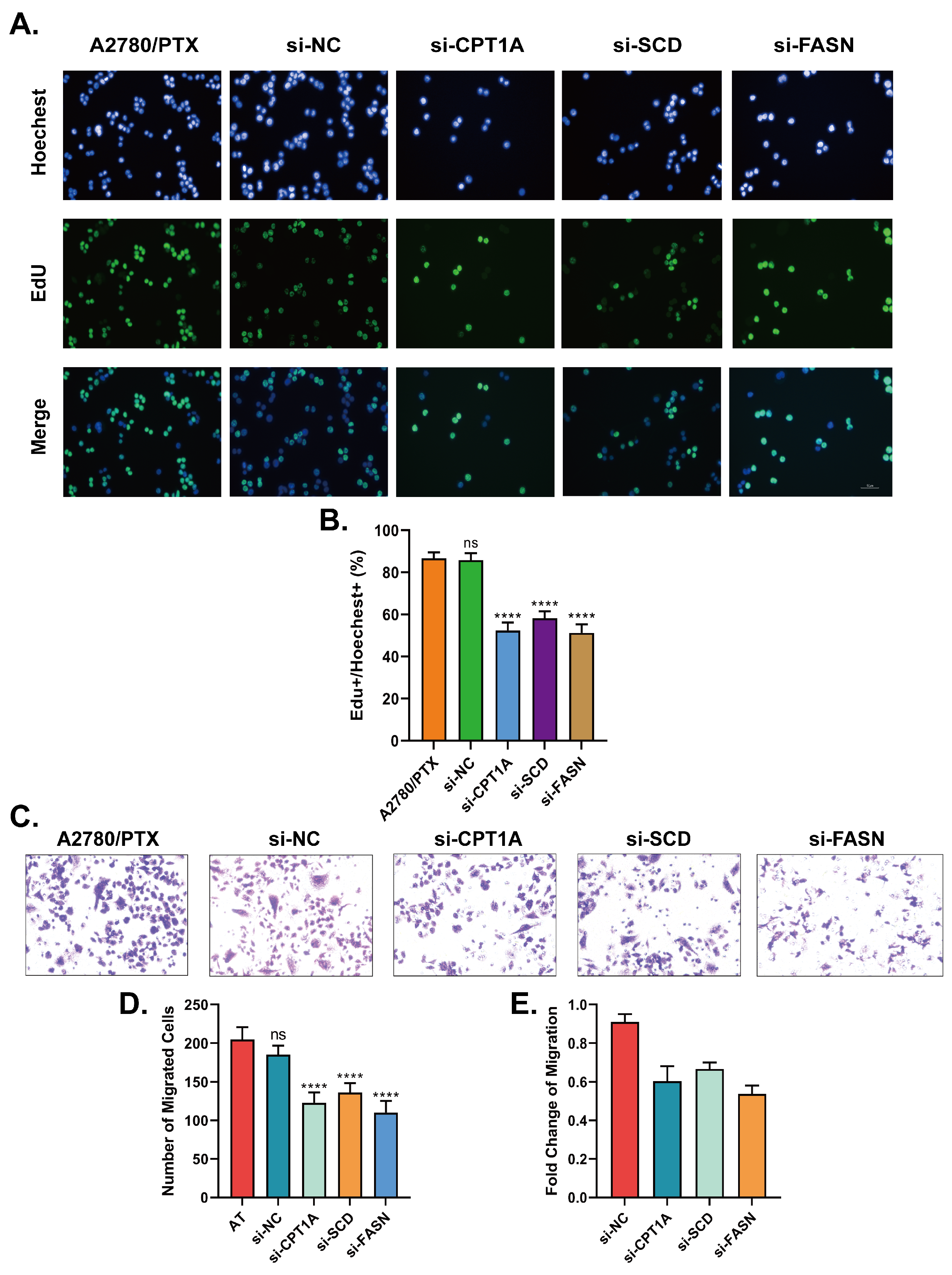
2.4. Down-Regulation of CPT1A, SCD and FASN Resensitizes A2780/PTX to Paclitaxel and Inhibits the Capabilities in Cell Proliferation and Metastasis
2.5. Inhibitors of CPT1A, SCD and FASN Restore Sensitivity towards Paclitaxel and Induce Apoptosis
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Data Process and Candidate Genes Screening
4.3. Overall Survival Analysis and Screening Genes with Synergistic Effect
4.4. Further Identification of CPT1A Synergistic Genes via Protein–Protein Interaction Networks and Survival Analysis
4.5. Cell Culture and Reagents
4.6. Establishment of Paclitaxel-Resistant Cell Lines
4.7. RNA Extraction and Quantitative Real-Time PCR
4.8. Western Blot
4.9. Transfection of Small Interfering RNA
4.10. Obtention and Validation of Inhibitors of CPT1A, FASN and SCD
4.11. Cell Viability Assay
4.12. Cell Colony Formation Assay
4.13. Transwell Cell Migration Assay
4.14. Flow Cytometry
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A. How Taxol/Paclitaxel Kills Cancer Cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of Microtubule Assembly in Vitro by Taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.P.H.; Horwitz, S.B. Taxol®: The First Microtubule Stabilizing Agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Peng, Z.; Gao, L.; Yang, X.; Sun, Z.; Hou, X.; Li, E.; Zhu, L.; Yang, H. ClC-3 Promotes Paclitaxel Resistance via Modulating Tubulins Polymerization in Ovarian Cancer Cells. Biomed. Pharmacother. 2021, 138, 111407. [Google Scholar] [CrossRef] [PubMed]
- Barbuti, A.M.; Chen, Z.S. Paclitaxel Through the Ages of Anticancer Therapy: Exploring Its Role in Chemoresistance and Radiation Therapy. Cancers 2015, 7, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of Taxane Resistance. Cancers 2020, 12, 3323. [Google Scholar] [CrossRef]
- Silverman, J.A. Multidrug-resistance Transporters. Pharm. Biotechnol. 1999, 12, 353–386. [Google Scholar] [CrossRef]
- Fojo, A.T.; Menefee, M. Microtubule Targeting Agents: Basic Mechanisms of Multidrug Resistance (MDR). Semin. Oncol. 2005, 32, S3–S8. [Google Scholar] [CrossRef]
- Tan, Y.; Li, J.; Zhao, G.; Huang, K.C.; Cardenas, H.; Wang, Y.; Matei, D.; Cheng, J.X. Metabolic Reprogramming from Glycolysis to Fatty Acid Uptake and Beta-Oxidation in Platinum-Resistant Cancer Cells. Nat. Commun. 2022, 13, 4554. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.C.; Richiardone, E.; Jorge, J.; Polónia, B.; Xavier, C.P.; Salaroglio, I.C.; Riganti, C.; Vasconcelos, M.H.; Corbet, C.; Sarmento-Ribeiro, A.B. Impact of Cancer Metabolism on Therapy Resistance–Clinical Implications. Drug Resist. Updates 2021, 59, 100797. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Ye, D.M.; Ye, S.C.; Yu, S.Q.; Shu, F.F.; Xu, S.S.; Chen, Q.Q.; Wang, Y.L.; Tang, Z.T.; Pan, C. Drug-resistance Reversal in Colorectal Cancer Cells by Destruction of Flotillins, the Key Lipid Rafts Proteins. Neoplasma 2019, 66, 576–583. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gašparović, A.Č.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and Cholesterol: Inducers of Cancer Multidrug Resistance and Therapeutic Targets. Drug Resist. Updates 2020, 49, 100670. [Google Scholar] [CrossRef]
- Yu, W.; Lei, Q.; Yang, L.; Qin, G.; Liu, S.; Wang, D.; Ping, Y.; Zhang, Y. Contradictory Roles of Lipid Metabolism in Immune Response within the Tumor Microenvironment. J. Hematol. Oncol. 2021, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Yan, S. Long-chain Acyl-CoA Synthetase in Fatty Acid Metabolism Involved in Liver and Other Diseases: An Update. World J. Gastroenterol. 2015, 21, 3492. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the Leukemia Cell Metabolism by the CPT1a Inhibition: Functional Preclinical Effects in Leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Dong, X.; Xiao, L.; Tan, Z.; Luo, X.; Yang, L.; Li, W.; Shi, F.; Li, Y.; Zhao, L.; et al. CPT1A-mediated Fatty Acid Oxidation Promotes Cell Proliferation via Nucleoside Metabolism in Nasopharyngeal Carcinoma. Cell Death Dis. 2022, 13, 331. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of Fatty Acid Oxidation as a Therapy for MYC-overexpressing Triple-Negative Breast Cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Han, S.; Wei, R.; Zhang, X.; Jiang, N.; Fan, M.; Huang, J.H.; Xie, B.; Zhang, L.; Miao, W.; Butler, A.C.P.; et al. CPT1A/2-Mediated FAO Enhancement—A Metabolic Target in Radioresistant Breast Cancer. Front. Oncol. 2019, 9, 1201. [Google Scholar] [CrossRef]
- Chen, M.; Chao, B.; Xu, J.; Liu, Z.; Tao, Y.; He, J.; Wang, J.; Yang, H.; Luo, X.; Qi, H. CPT1A Modulates PI3K/Akt/mTOR Pathway to Promote Preeclampsia. Placenta 2023, 133, 23–31. [Google Scholar] [CrossRef]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; Van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte Lipolysis Links Obesity to Breast Cancer Growth: Adipocyte-Derived Fatty Acids Drive Breast Cancer Cell Proliferation and Migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Z.; Zhao, X.; Ruan, S.; Zhang, X.; Wang, S.; Huang, T. CPT1A Regulates Breast Cancer-Associated Lymphangiogenesis via VEGF Signaling. Biomed. Pharmacother. 2018, 106, 1–7. [Google Scholar] [CrossRef]
- Huang, D.; Chowdhury, S.; Wang, H.; Savage, S.R.; Ivey, R.G.; Kennedy, J.J.; Whiteaker, J.R.; Lin, C.; Hou, X.; Oberg, A.L.; et al. Multiomic Analysis Identifies CPT1A as a Potential Therapeutic Target in Platinum-Refractory, High-Grade Serous Ovarian Cancer. Cell Rep. Med. 2021, 2, 100471. [Google Scholar] [CrossRef]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef]
- Stoykova, G.E.; Schlaepfer, I.R. Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. Int. J. Mol. Sci. 2019, 20, 2626. [Google Scholar] [CrossRef] [PubMed]
- Ezzeddini, R.; Taghikhani, M.; Somi, M.H.; Samadi, N.; Rasaee, M.J. Clinical Importance of FASN in Relation to HIF-1α and SREBP-1c in Gastric Adenocarcinoma. Life Sci. 2019, 224, 169–176. [Google Scholar] [CrossRef]
- Luu, T.H.; Bard, J.M.; Carbonnelle, D.; Chaillou, C.; Huvelin, J.M.; Bobin-Dubigeon, C.; Nazih, H. Lithocholic Bile Acid Inhibits Lipogenesis and Induces Apoptosis in Breast Cancer Cells. Cell. Oncol. 2018, 41, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Becker, K.G.; Wood Iii, W.H.; Zhang, Y.; Morin, P.J. Gene Expression and Pathway Analysis of Ovarian Cancer Cells Selected for Resistance to Cisplatin, Paclitaxel, or Doxorubicin. J. Ovarian Res. 2011, 4, 21. [Google Scholar] [CrossRef]
- Yang, R.; Yi, M.; Xiang, B. Novel Insights on Lipid Metabolism Alterations in Drug Resistance in Cancer. Front. Cell Dev. Biol. 2022, 10, 875318. [Google Scholar] [CrossRef]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid Metabolism in Cancer Progression and Therapeutic Strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid Metabolism in Cancer Cells under Metabolic Stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the Role of ABC Transporters in Multidrug-Resistant Cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Boumahdi, S.; De Sauvage, F.J. The Great Escape: Tumour Cell Plasticity in Resistance to Targeted Therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avancès, C.; Allory, Y.; De La Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of De Novo Lipogenesis Stearoyl-CoA Desaturase 1 Inhibition Interf. Oncogenic Signaling Blocks Prostate Cancer Progress. Mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef] [PubMed]
- Vagin, O.; Tokhtaeva, E.; Sachs, G. The Role of the B1 Subunit of the Na,K-ATPase and Its Glycosylation in Cell-Cell Adhesion. J. Biol. Chem. 2006, 281, 39573–39587. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.A.; Palmer, L.G.; Quan, K.; Harper, J.F.; Ball, W.J.; Bander, N.H.; Soler, A.P.; Rajasekaran, A.K. Na,K-ATPase β-Subunit Is Required for Epithelial Polarization, Suppression of Invasion, and Cell Motility. Mol. Biol. Cell 2001, 12, 279–295. [Google Scholar] [CrossRef]
- Huynh, T.P.; Barwe, S.P.; Lee, S.J.; McSpadden, R.; Franco, O.E.; Hayward, S.W.; Damoiseaux, R.; Grubbs, S.S.; Petrelli, N.J.; Rajasekaran, A.K. Glucocorticoids Suppress Renal Cell Carcinoma Progression by Enhancing Na,K-ATPase Beta-1 Subunit Expression. PLoS ONE 2015, 10, e0122442. [Google Scholar] [CrossRef] [PubMed]
- Rotoli, D.; Cejas, M.M.; Maeso, M.d.C.; Pérez-Rodríguez, N.D.; Morales, M.; Ávila, J.; Mobasheri, A.; Martín-Vasallo, P. The Na, K-ATPase β-Subunit Isoforms Expression in Glioblastoma Multiforme: Moonlighting Roles. Int. J. Mol. Sci. 2017, 18, 2369. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H. Reprogramming of Glucose, Fatty Acid and Amino Acid Metabolism for Cancer Progression. Cell. Mol. Life Sci. CMLS 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic Pathways Promoting Cancer Cell Survival and Growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty Acid Oxidation and Carnitine Palmitoyltransferase I: Emerging Therapeutic Targets in Cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- McGarry, J.D.; Brown, N.F. The Mitochondrial Carnitine Palmitoyltransferase System—From Concept to Molecular Analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Tan, Z.; Shi, F.; Tang, M.; Xie, L.; Zhao, L.; Li, Y.; Hu, J.; Zhou, M.; Bode, A.; et al. PGC1α/CEBPB/CPT1A Axis Promotes Radiation Resistance of Nasopharyngeal Carcinoma through Activating Fatty Acid Oxidation. Cancer Sci. 2019, 110, 2050–2062. [Google Scholar] [CrossRef]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de Novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Hudgins, L.C.; Hellerstein, M.; Seidman, C.; Neese, R.; Diakun, J.; Hirsch, J. Human Fatty Acid Synthesis Is Stimulated by a Eucaloric Low Fat, High Carbohydrate Diet. J. Clin. Investig. 1996, 97, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Gansler, T.S.; Hardman, W.; Hunt, D.A.; Schaffel, S.; Hennigar, R.A. Increased Expression of Fatty Acid Synthase (OA-519) in Ovarian Neoplasms Predicts Shorter Survival. Hum. Pathol. 1997, 28, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Coleman, C.; Sen, T. Stearoyl Coenzyme A Desaturase-1: Multitasker in Cancer, Metabolism, and Ferroptosis. Trends Cancer 2023, 9, 480–489. [Google Scholar] [CrossRef]
- Scaglia, N.; Chisholm, J.W.; Igal, R.A. Inhibition of StearoylCoA Desaturase-1 Inactivates Acetyl-CoA Carboxylase and Impairs Proliferation in Cancer Cells: Role of AMPK. PLoS ONE 2009, 4, e6812. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Tan, Y.; Cardenas, H.; Vayngart, D.; Wang, Y.; Huang, H.; Keathley, R.; Wei, J.J.; Ferreira, C.R.; Orsulic, S.; et al. Ovarian Cancer Cell Fate Regulation by the Dynamics between Saturated and Unsaturated Fatty Acids. Proc. Natl. Acad. Sci. USA 2022, 119, e2203480119. [Google Scholar] [CrossRef]
- Roongta, U.V.; Pabalan, J.G.; Wang, X.; Ryseck, R.P.; Fargnoli, J.; Henley, B.J.; Yang, W.P.; Zhu, J.; Madireddi, M.T.; Lawrence, R.M.; et al. Cancer Cell Dependence on Unsaturated Fatty Acids Implicates Stearoyl-CoA Desaturase as a Target for Cancer Therapy. Mol. Cancer Res. 2011, 9, 1551–1561. [Google Scholar] [CrossRef]
- Nashed, M.; Chisholm, J.W.; Igal, R.A. Stearoyl-CoA Desaturase Activity Modulates the Activation of Epidermal Growth Factor Receptor in Human Lung Cancer Cells. Exp. Biol. Med. 2012, 237, 1007–1017. [Google Scholar] [CrossRef]
- Tan, Z.; Xiao, L.; Tang, M.; Bai, F.; Li, J.; Li, L.; Shi, F.; Li, N.; Li, Y.; Du, Q.; et al. Targeting CPT1A-mediated Fatty Acid Oxidation Sensitizes Nasopharyngeal Carcinoma to Radiation Therapy. Theranostics 2018, 8, 2329–2347. [Google Scholar] [CrossRef]
- Nallanthighal, S.; Rada, M.; Heiserman, J.P.; Cha, J.; Sage, J.; Zhou, B.; Yang, W.; Hu, Y.; Korgaonkar, C.; Hanos, C.T.; et al. Inhibition of Collagen XI Alpha 1-Induced Fatty Acid Oxidation Triggers Apoptotic Cell Death in Cisplatin-Resistant Ovarian Cancer. Cell Death Dis. 2020, 11, 258. [Google Scholar] [CrossRef]
- Rada, M.; Nallanthighal, S.; Cha, J.; Ryan, K.; Sage, J.; Eldred, C.; Ullo, M.; Orsulic, S.; Cheon, D.J. Inhibitor of Apoptosis Proteins (IAPs) Mediate Collagen Type XI Alpha 1-Driven Cisplatin Resistance in Ovarian Cancer. Oncogene 2018, 37, 4809–4820. [Google Scholar] [CrossRef] [PubMed]
- Bauerschlag, D.O.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty Acid Synthase Overexpression: Target for Therapy and Reversal of Chemoresistance in Ovarian Cancer. J. Transl. Med. 2015, 13, 146. [Google Scholar] [CrossRef]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; deSouza, N.M.; Chung, Y.L. The Effect of FASN Inhibition on the Growth and Metabolism of a Cisplatin-resistant Ovarian Carcinoma Model. Int. J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.K.F.; Lau, E.Y.T.; Leung, D.H.W.; Lo, J.; Ho, N.P.Y.; Cheng, L.K.W.; Ma, S.; Lin, C.H.; Copland, J.A.; Ding, J.; et al. Stearoyl-CoA Desaturase Regulates Sorafenib Resistance via Modulation of ER Stress-Induced Differentiation. J. Hepatol. 2017, 67, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes Promote Ovarian Cancer Metastasis and Provide Energy for Rapid Tumor Growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes Activate Mitochondrial Fatty Acid Oxidation and Autophagy to Promote Tumor Growth in Colon Cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; LeGonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S.; et al. Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef]
- Lee, C.k.; Jeong, S.h.; Jang, C.; Bae, H.; Kim, Y.H.; Park, I.; Kim, S.K.; Koh, G.Y. Tumor Metastasis to Lymph Nodes Requires YAP-dependent Metabolic Adaptation. Science 2019, 363, 644–649. [Google Scholar] [CrossRef]
- Wang, X.; Yung, M.M.H.; Sharma, R.; Chen, F.; Poon, Y.T.; Lam, W.Y.; Li, B.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. Epigenetic Silencing of miR-33b Promotes Peritoneal Metastases of Ovarian Cancer by Modulating the TAK1/FASN/CPT1A/NF-κB Axis. Cancers 2021, 13, 4795. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, H.; Li, J.; Fang, X.; Pan, H.; Yuan, X.; Zhang, P. Up-Regulated FASN Expression Promotes Transcoelomic Metastasis of Ovarian Cancer Cell through Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2014, 15, 11539–11554. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on Fat: The Role of Lipid Synthesis in Cancer Metabolism and Tumour Development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef] [PubMed]
- Coley, H.M. Development of Drug-Resistant Models. In Cancer Cell Culture; Humana Press: Clifton, NJ, USA, 2003; Volume 88, pp. 267–274. [Google Scholar] [CrossRef]


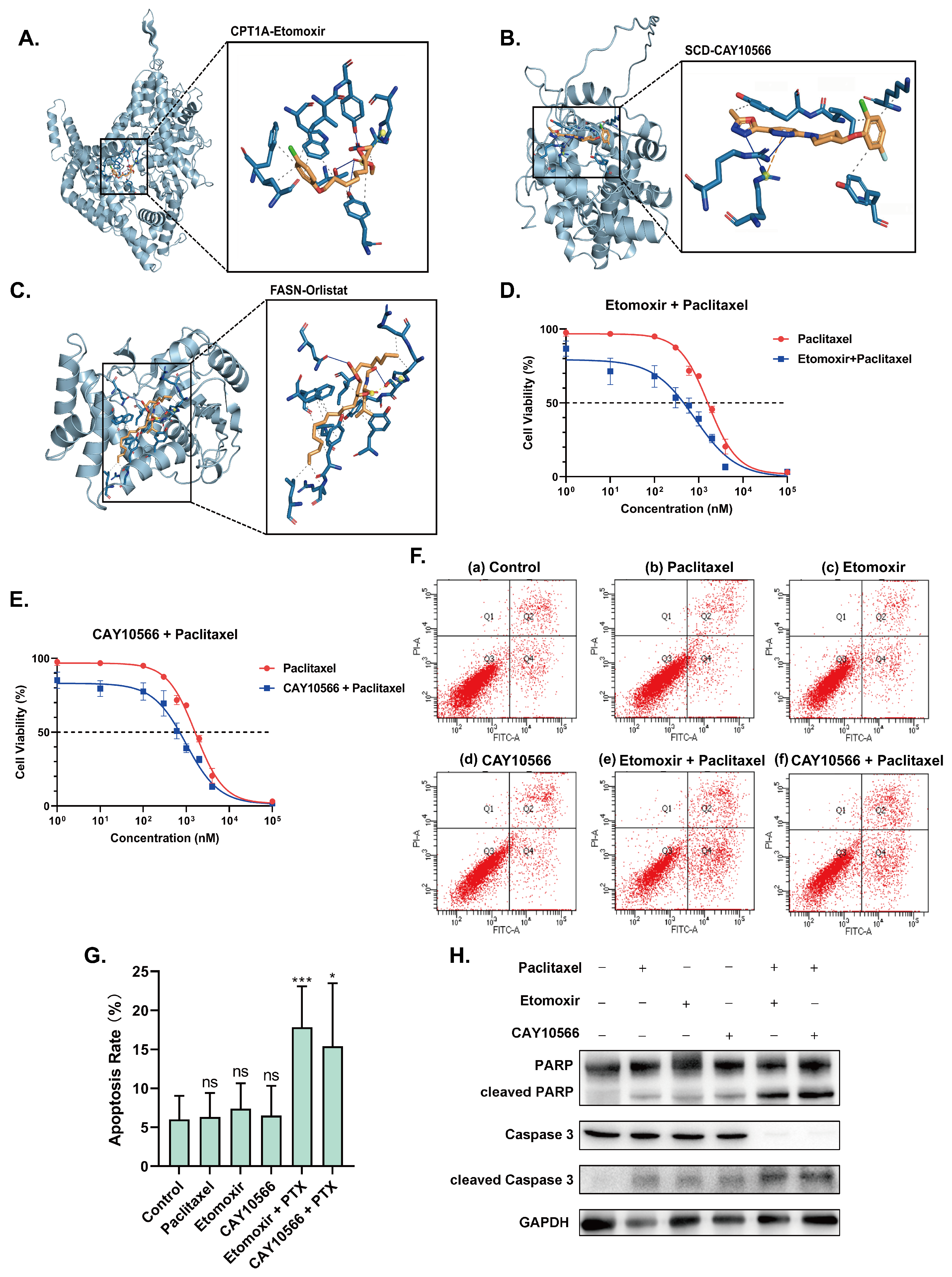
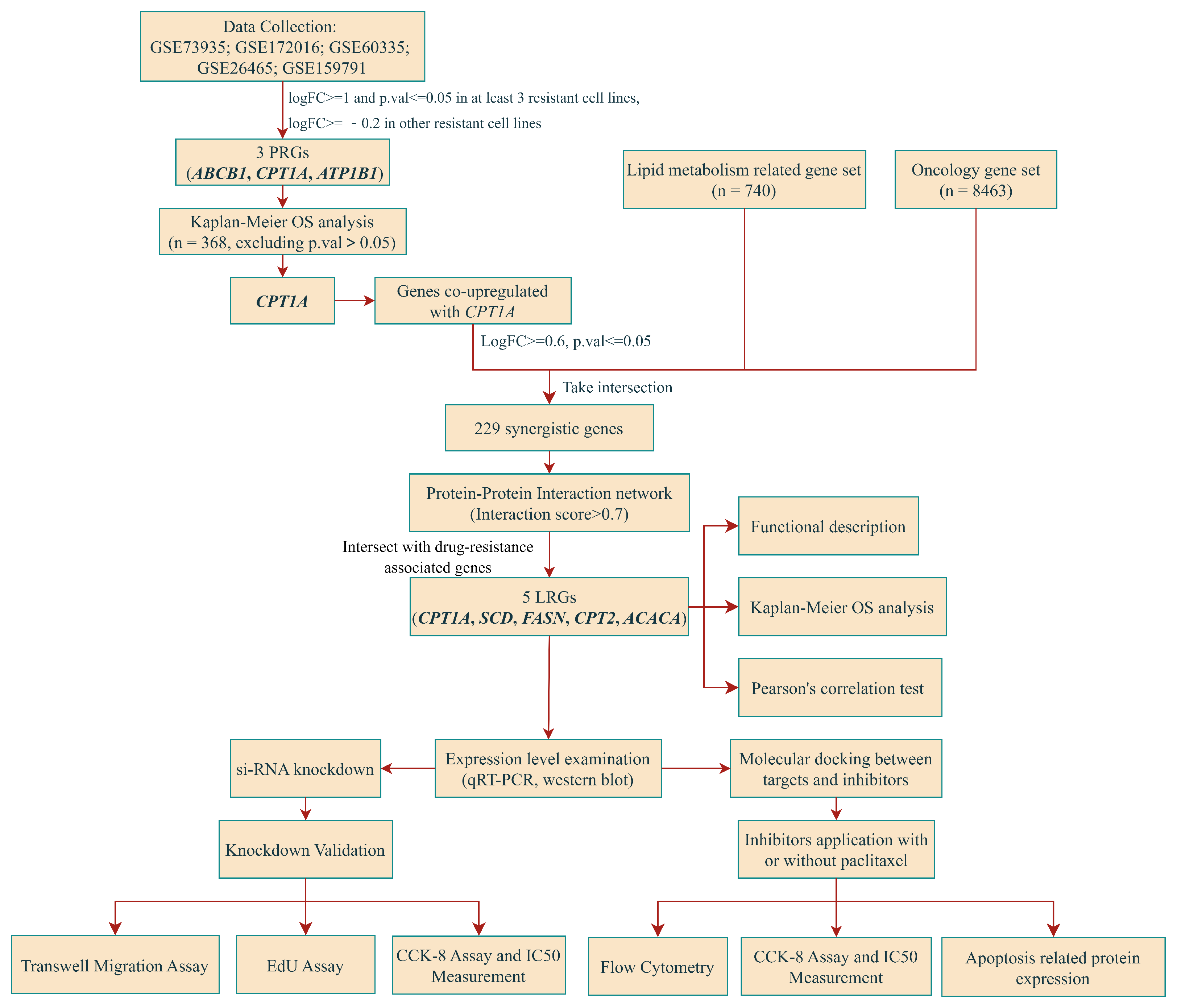

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Characteristics | All (N = 368) |
|---|---|
| Age at diagnosis | |
| Median | 59 |
| Range | 30–87 |
| > 65 | 114 (31%) |
| Grade | |
| G1 | 1 (0.3%) |
| G2 | 39 (10.6%) |
| G3 | 319 (86.7%) |
| G4 | 1 (0.3%) |
| GB | 2 (0.5%) |
| GX | 6 (1.6%) |
| FIGO stage | |
| Stage I | 1 (0.3%) |
| Stage II | 21 (5.7%) |
| Stage III | 289 (78.5%) |
| Stage IV | 57 (15.5%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Q.; Liu, Z.; Wang, T.; Zhao, P.; Liu, M.; Wang, Y.; Zhao, W.; Yuan, Y.; Li, S. Resensitizing Paclitaxel-Resistant Ovarian Cancer via Targeting Lipid Metabolism Key Enzymes CPT1A, SCD and FASN. Int. J. Mol. Sci. 2023, 24, 16503. https://doi.org/10.3390/ijms242216503
Ma Q, Liu Z, Wang T, Zhao P, Liu M, Wang Y, Zhao W, Yuan Y, Li S. Resensitizing Paclitaxel-Resistant Ovarian Cancer via Targeting Lipid Metabolism Key Enzymes CPT1A, SCD and FASN. International Journal of Molecular Sciences. 2023; 24(22):16503. https://doi.org/10.3390/ijms242216503
Chicago/Turabian StyleMa, Qinsiyu, Zhan’ao Liu, Tengyu Wang, Pengfei Zhao, Mingrui Liu, Yifang Wang, Weitong Zhao, Ying Yuan, and Shuo Li. 2023. "Resensitizing Paclitaxel-Resistant Ovarian Cancer via Targeting Lipid Metabolism Key Enzymes CPT1A, SCD and FASN" International Journal of Molecular Sciences 24, no. 22: 16503. https://doi.org/10.3390/ijms242216503
APA StyleMa, Q., Liu, Z., Wang, T., Zhao, P., Liu, M., Wang, Y., Zhao, W., Yuan, Y., & Li, S. (2023). Resensitizing Paclitaxel-Resistant Ovarian Cancer via Targeting Lipid Metabolism Key Enzymes CPT1A, SCD and FASN. International Journal of Molecular Sciences, 24(22), 16503. https://doi.org/10.3390/ijms242216503