
TRPC6 Deletion Enhances eNOS Expression and Reduces LPS—Induced Acute Lung Injury
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
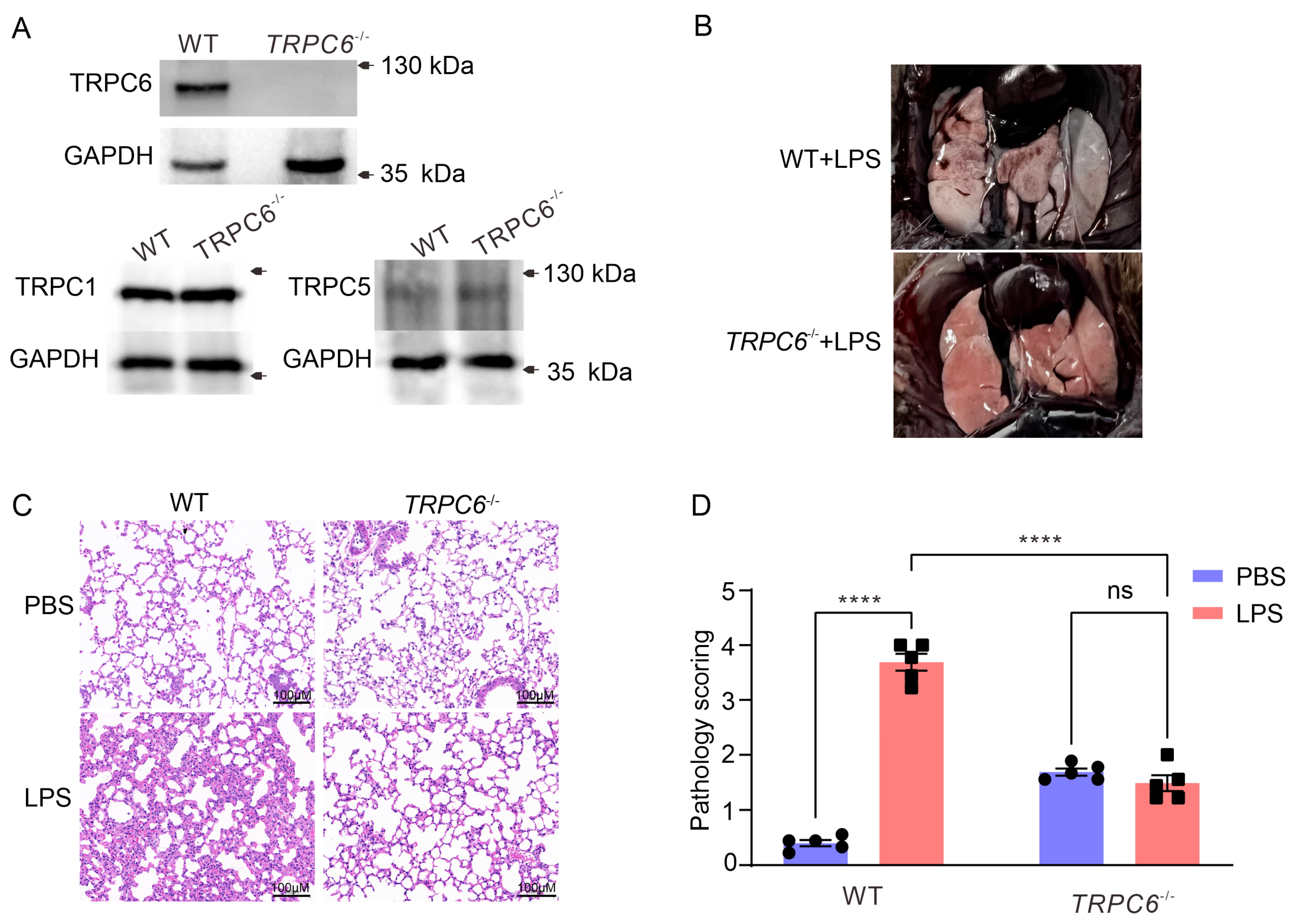
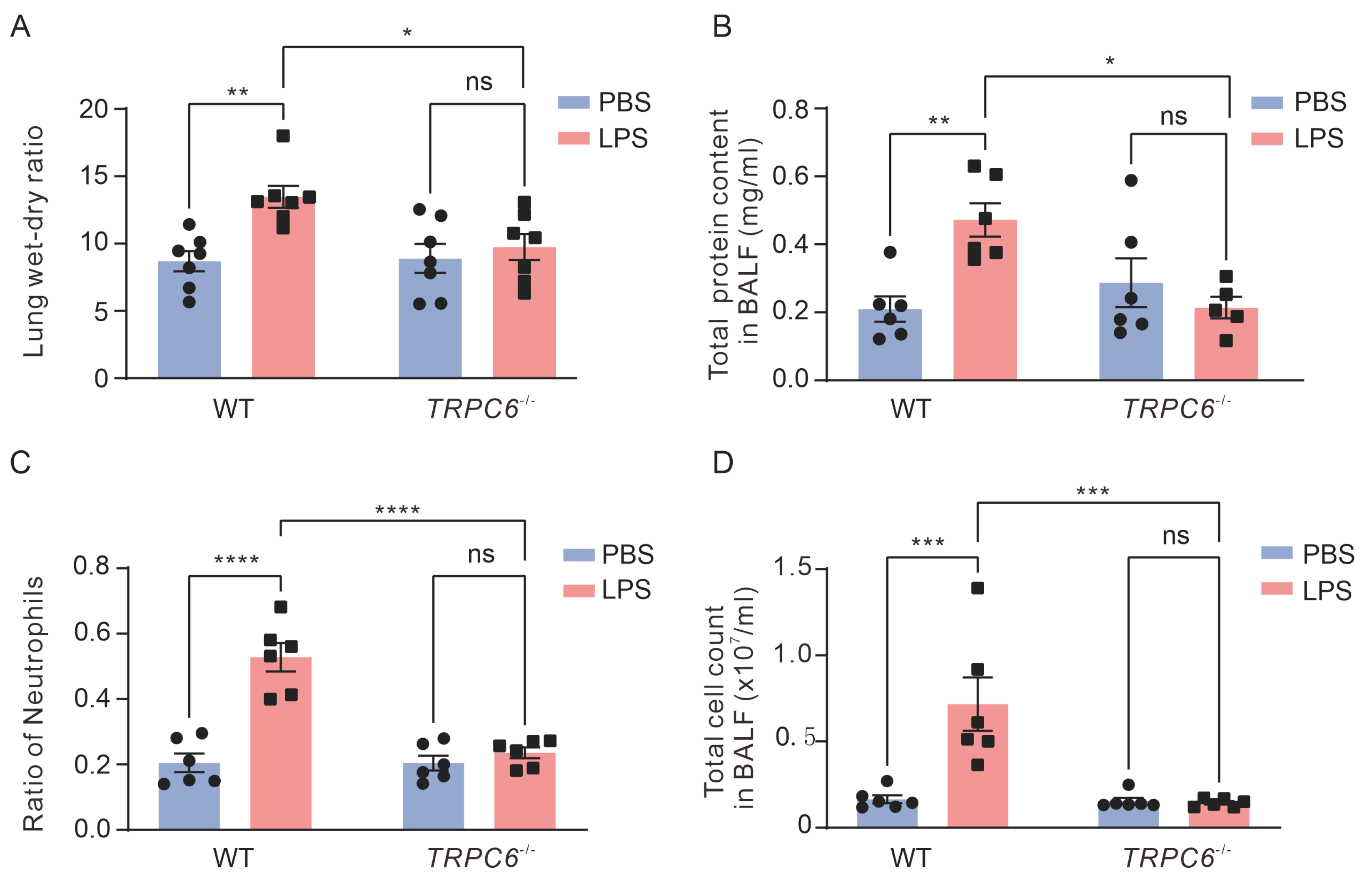
2.1. Knockout of the TRPC6 Alleviates Acute Lung Inflammation in Lps—Treated Mice
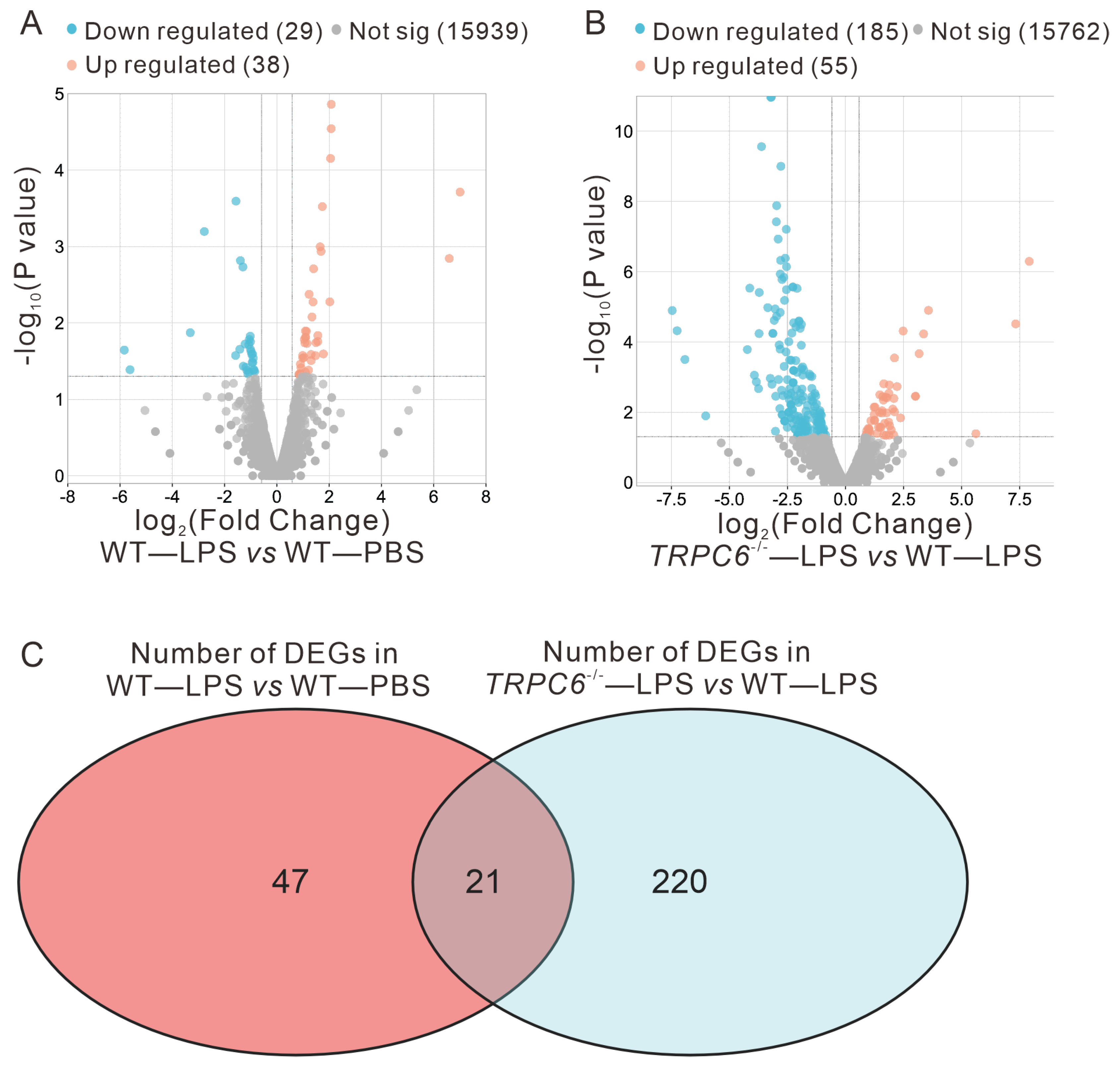
2.2. Effect of TRPC6 Knockout on Altered Gene Expression in the Lungs of LPS—Treated Mice
2.2.1. Differential Expression Analysis Results
2.2.2. GO and KEGG Enrichment Analysis Results
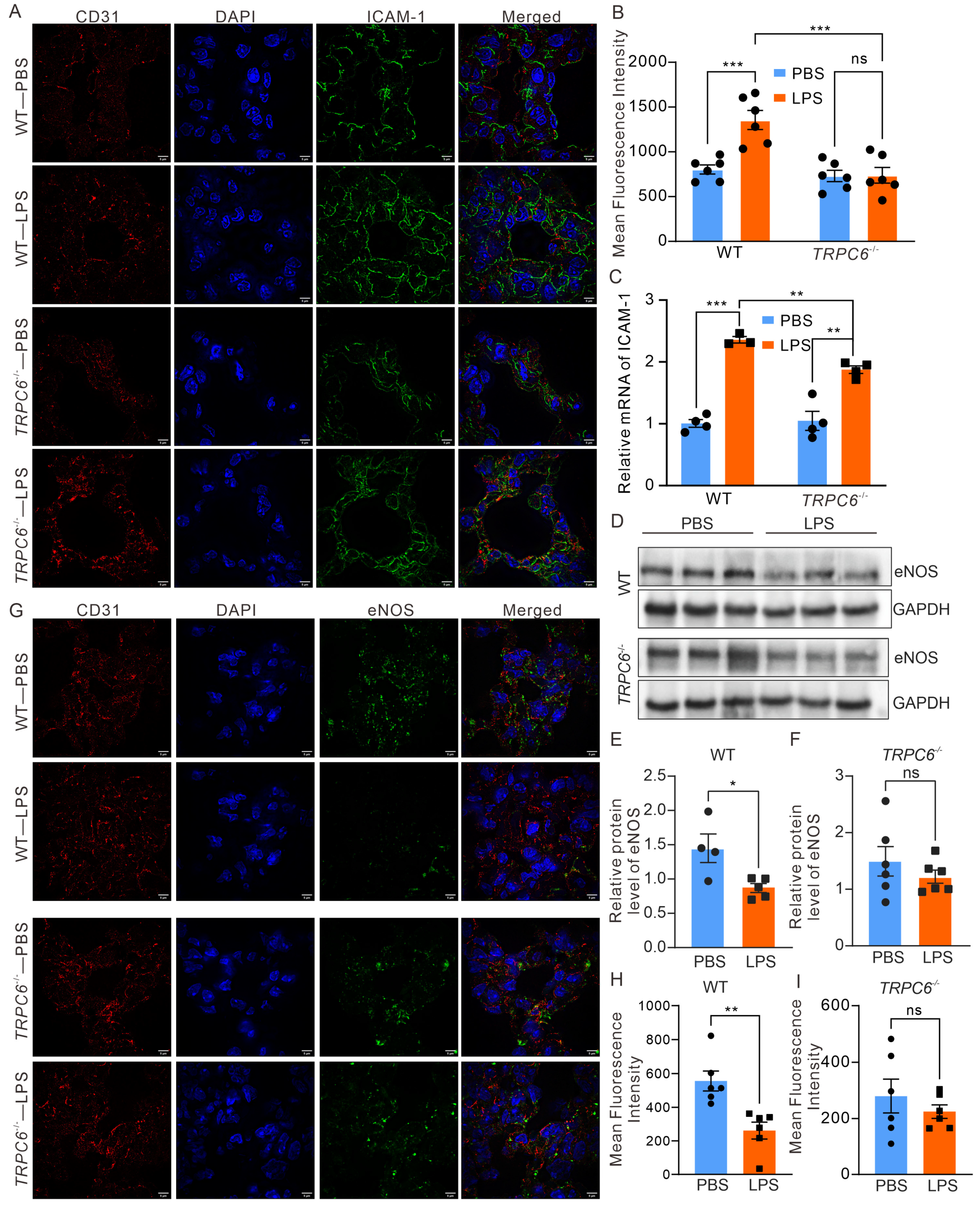
2.3. Deletion of TRPC6 Ameliorated the Downregulation of eNOS Expression in the Lungs of Mice Treated with LPS
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Animals
4.3. Mouse Model of ALI
4.4. Evaluation of Lung Injury
4.5. Cell Culture
4.6. Mitochondrial ROS (mROS) Measurement
4.7. Western Blot
4.8. Reverse Transcription Polymerase Chain Reaction (RT—PCR) Analysis
4.9. Transcriptome Sequencing
4.10. Evans Blue
4.11. Modified Transwell Assay
4.12. Immunofluorescence Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, D.; Zhang, H.; Wu, Q.; Li, F.; Wang, Y.; Liu, S.; Wang, J. Sestrin 2 protects against LPS-induced acute lung injury by inducing mitophagy in alveolar macrophages. Life Sci. 2021, 267, 118941. [Google Scholar] [CrossRef]
- Kulkarni, H.S.; Lee, J.S.; Bastarache, J.A.; Kuebler, W.M.; Downey, G.P.; Albaiceta, G.M.; Altemeier, W.A.; Artigas, A.; Bates, J.H.T.; Calfee, C.S.; et al. Update on the Features and Measurements of Experimental Acute Lung Injury in Animals: An Official American Thoracic Society Workshop Report. Am. J. Respir. Cell Mol. Biol. 2022, 66, e1–e14. [Google Scholar] [CrossRef] [PubMed]
- Jayasimhan, D.; Foster, S.; Chang, C.L.; Hancox, R.J. Cardiac biomarkers in acute respiratory distress syndrome: A systematic review and meta-analysis. J. Intensive Care 2021, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, L.; Wang, M.M.; Cao, L.P.; Sun, Z.R.; Yang, Z.Z.; Zhang, W.; Zhang, P.; Nie, S.N. Pravastatin attenuates sepsis-induced acute lung injury through decreasing pulmonary microvascular permeability via inhibition of Cav-1/eNOS pathway. Int. Immunopharmacol. 2021, 100, 108077. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Bichsel, C.; Norris, A.L.; Thorpe, J.; Pevsner, J.; Alexandrescu, S.; Pinto, A.; Zurakowski, D.; Kleiman, R.J.; Sahin, M.; et al. Endothelial GNAQ p.R183Q Increases ANGPT2 (Angiopoietin-2) and Drives Formation of Enlarged Blood Vessels. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e27–e43. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef]
- Filippini, A.; D’Amore, A.; D’Alessio, A. Calcium Mobilization in Endothelial Cell Functions. Int. J. Mol. Sci. 2019, 20, 4525. [Google Scholar] [CrossRef]
- Harraz, O.F.; Jensen, L.J. Vascular calcium signalling and ageing. J. Physiol. 2021, 599, 5361–5377. [Google Scholar] [CrossRef]
- Hao, Y.; Wang, Z.; Frimpong, F.; Chen, X. Calcium-Permeable Channels and Endothelial Dysfunction in Acute Lung Injury. Curr. Issues Mol. Biol. 2022, 44, 2217–2229. [Google Scholar] [CrossRef]
- Wang, H.; Sun, X.; Lu, Q.; Zemskov, E.A.; Yegambaram, M.; Wu, X.; Wang, T.; Tang, H.; Black, S.M. The mitochondrial redistribution of eNOS is involved in lipopolysaccharide induced inflammasome activation during acute lung injury. Redox Biol. 2021, 41, 101878. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca2+ Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Mederos y Schnitzler, M.; Brandes, R.P.; Grimminger, F.; Meissner, M.; Freichel, M.; et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 2012, 3, 649. [Google Scholar] [CrossRef] [PubMed]
- Tauseef, M.; Knezevic, N.; Chava, K.R.; Smith, M.; Sukriti, S.; Gianaris, N.; Obukhov, A.G.; Vogel, S.M.; Schraufnagel, D.E.; Dietrich, A.; et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 2012, 209, 1953–1968. [Google Scholar] [CrossRef] [PubMed]
- Akahori, D.; Inui, N.; Inoue, Y.; Yasui, H.; Hozumi, H.; Suzuki, Y.; Karayama, M.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; et al. Effect of Hypoxia on Pulmonary Endothelial Cells from Bleomycin-Induced Pulmonary Fibrosis Model Mice. Int. J. Mol. Sci. 2022, 23, 8996. [Google Scholar] [CrossRef] [PubMed]
- Khodadadi, S.; Zabihi, N.A.; Niazmand, S.; Abbasnezhad, A.; Mahmoudabady, M.; Rezaee, S.A. Teucrium polium improves endothelial dysfunction by regulating eNOS and VCAM-1 genes expression and vasoreactivity in diabetic rat aorta. Biomed. Pharmacother. 2018, 103, 1526–1530. [Google Scholar] [CrossRef]
- Urban, N.; Wang, L.; Kwiek, S.; Rademann, J.; Kuebler, W.M.; Schaefer, M. Identification and Validation of Larixyl Acetate as a Potent TRPC6 Inhibitor. Mol. Pharmacol. 2016, 89, 197–213. [Google Scholar] [CrossRef]
- Sharma, A.; Ahmad, S.; Ahmad, T.; Ali, S.; Syed, M.A. Mitochondrial dynamics and mitophagy in lung disorders. Life Sci. 2021, 284, 119876. [Google Scholar] [CrossRef]
- Chen, X.; Taylor-Nguyen, N.N.; Riley, A.M.; Herring, B.P.; White, F.A.; Obukhov, A.G. The TRPC6 inhibitor, larixyl acetate, is effective in protecting against traumatic brain injury-induced systemic endothelial dysfunction. J. Neuroinflamm. 2019, 16, 21. [Google Scholar] [CrossRef]
- Solopov, P.; Colunga Biancatelli, R.M.L.; Dimitropoulou, C.; Catravas, J.D. Sex-Related Differences in Murine Models of Chemically Induced Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 5909. [Google Scholar] [CrossRef]
- Weber, E.W.; Han, F.; Tauseef, M.; Birnbaumer, L.; Mehta, D.; Muller, W.A. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J. Exp. Med. 2015, 212, 1883–1899. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhu, Y.; Gao, G.; Zhang, Y. Long noncoding RNA SNHG16 targets miR-146a-5p/CCL5 to regulate LPS-induced WI-38 cell apoptosis and inflammation in acute pneumonia. Life Sci. 2019, 228, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Xu, M.M.; Fan, C.; Feng, C.L.; Lu, Q.K.; Lu, H.M.; Xiang, C.G.; Bai, F.; Wang, H.Y.; Wu, Y.W.; et al. STING inhibitor ameliorates LPS-induced ALI by preventing vascular endothelial cells-mediated immune cells chemotaxis and adhesion. Acta Pharmacol. Sin. 2022, 43, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Hirota, J.A.; Hiebert, P.R.; Gold, M.; Wu, D.; Graydon, C.; Smith, J.A.; Ask, K.; McNagny, K.; Granville, D.J.; Knight, D.A. Granzyme B deficiency exacerbates lung inflammation in mice after acute lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 453–462. [Google Scholar] [CrossRef]
- Hezam, K.; Wang, C.; Fu, E.; Zhou, M.; Liu, Y.; Wang, H.; Zhu, L.; Han, Z.; Han, Z.C.; Chang, Y.; et al. Superior protective effects of PGE2 priming mesenchymal stem cells against LPS-induced acute lung injury (ALI) through macrophage immunomodulation. Stem Cell Res. Ther. 2023, 14, 48. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Zhang, Y.; Hu, Q.P.; Li, J.Q.; Liu, Y.T.; Wu, Q.G.; Wu, J.G.; Lai, X.P.; Zhang, Z.D.; Li, X.; et al. Anti-inflammatory effects of rosmarinic acid-4-O-β-D-glucoside in reducing acute lung injury in mice infected with influenza virus. Antivir. Res. 2017, 144, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Lou, S.; Zheng, F.; Gao, H.; Wang, N.; Tian, S.; Huang, G.; Zhao, H. Hydnocarpin D attenuates lipopolysaccharide-induced acute lung injury via MAPK/NF-κB and Keap1/Nrf2/HO-1 pathway. Phytomedicine 2022, 101, 154143. [Google Scholar] [CrossRef]
- Yang, H.; Lv, H.; Li, H.; Ci, X.; Peng, L. Oridonin protects LPS-induced acute lung injury by modulating Nrf2-mediated oxidative stress and Nrf2-independent NLRP3 and NF-κB pathways. Cell. Commun. Signal. CCS 2019, 17, 62. [Google Scholar] [CrossRef]
- Ju, M.; Liu, B.; He, H.; Gu, Z.; Liu, Y.; Su, Y.; Zhu, D.; Cang, J.; Luo, Z. MicroRNA-27a alleviates LPS-induced acute lung injury in mice via inhibiting inflammation and apoptosis through modulating TLR4/MyD88/NF-κB pathway. Cell. Cycle (Georget. Tex.) 2018, 17, 2001–2018. [Google Scholar] [CrossRef]
- Nakuluri, K.; Nishad, R.; Mukhi, D.; Kumar, S.; Nakka, V.P.; Kolligundla, L.P.; Narne, P.; Natuva, S.S.K.; Phanithi, P.B.; Pasupulati, A.K. Cerebral ischemia induces TRPC6 via HIF1α/ZEB2 axis in the glomerular podocytes and contributes to proteinuria. Sci. Rep. 2019, 9, 17897. [Google Scholar] [CrossRef]
- Alfieri, A.; Ong, A.C.; Kammerer, R.A.; Solanky, T.; Bate, S.; Tasab, M.; Brown, N.J.; Brookes, Z.L. Angiopoietin-1 regulates microvascular reactivity and protects the microcirculation during acute endothelial dysfunction: Role of eNOS and VE-cadherin. Pharmacol. Res. 2014, 80, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Nakahara, T.; Araho, D.; Murakami, J.; Nishimura, M. Glycolipids from spinach suppress LPS-induced vascular inflammation through eNOS and NK-κB signaling. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 91, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.C.; Mao, A.R.; Fang, Y.Q.; Fan, Y.H.; Wang, F.F.; Ma, J.; You, P. Simvastatin decreases hyperbaric oxygen-induced acute lung injury by upregulating eNOS. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L287–L297. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Jeffers, L.A.; Koval, M. Measurement of Lung Vessel and Epithelial Permeability In Vivo with Evans Blue. In Permeability Barrier: Methods in Molecular Biology; Springer: Berlin, Germany; Humana: New York, NY, USA, 2021; Volume 2367, pp. 137–148. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, S.; Li, L.; Huang, X.; Xing, S.; Zhang, Y.; Qiu, G.; Han, Z.; Shang, Y.; Sun, D.E.; et al. Sparse deconvolution improves the resolution of live-cell super-resolution fluorescence microscopy. Nat. Biotechnol. 2022, 40, 606–617. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zhang, X.; Guo, J.; Yang, S.; Yang, F.; Chen, X. TRPC6 Deletion Enhances eNOS Expression and Reduces LPS—Induced Acute Lung Injury. Int. J. Mol. Sci. 2023, 24, 16756. https://doi.org/10.3390/ijms242316756
Wang M, Zhang X, Guo J, Yang S, Yang F, Chen X. TRPC6 Deletion Enhances eNOS Expression and Reduces LPS—Induced Acute Lung Injury. International Journal of Molecular Sciences. 2023; 24(23):16756. https://doi.org/10.3390/ijms242316756
Chicago/Turabian StyleWang, Mengyuan, Xingfang Zhang, Juan Guo, Shangze Yang, Fang Yang, and Xingjuan Chen. 2023. "TRPC6 Deletion Enhances eNOS Expression and Reduces LPS—Induced Acute Lung Injury" International Journal of Molecular Sciences 24, no. 23: 16756. https://doi.org/10.3390/ijms242316756