
CRISPR-Cas9 Mediated Stable Expression of Exogenous Proteins in the CHO Cell Line through Site-Specific Integration

Abstract
:1. Introduction
2. Results
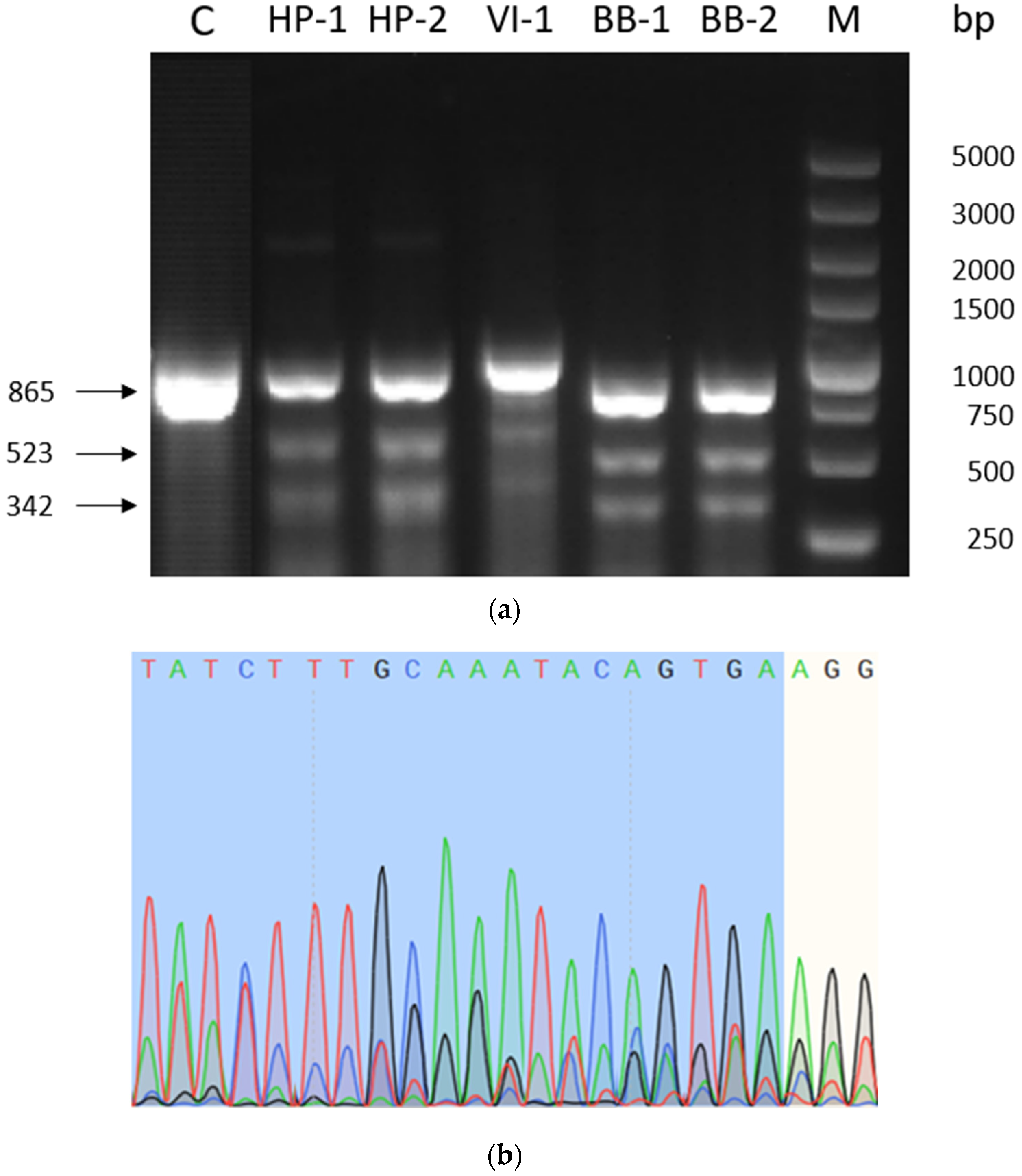
2.1. Validation of sgRNA Modification Efficiency
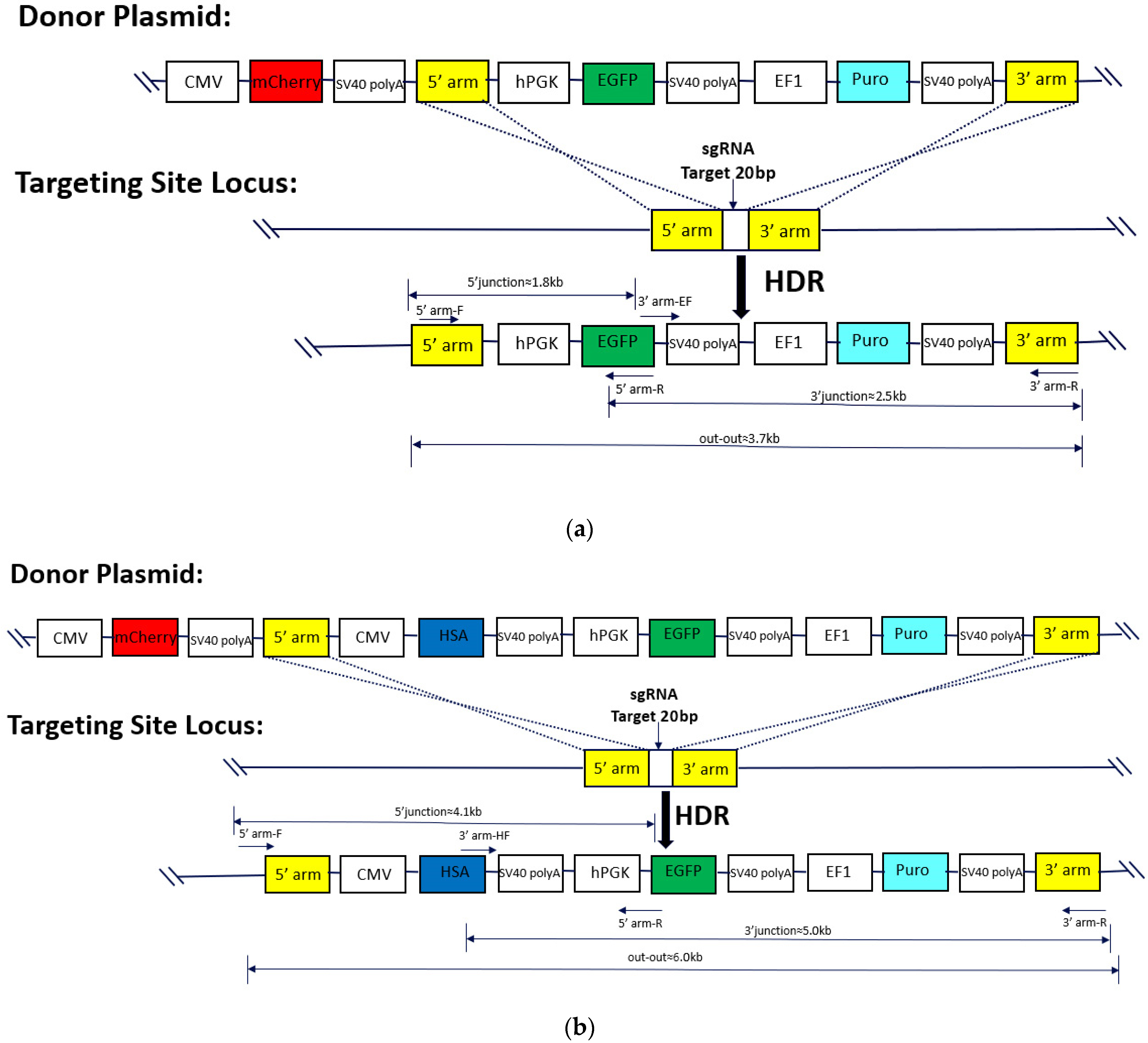
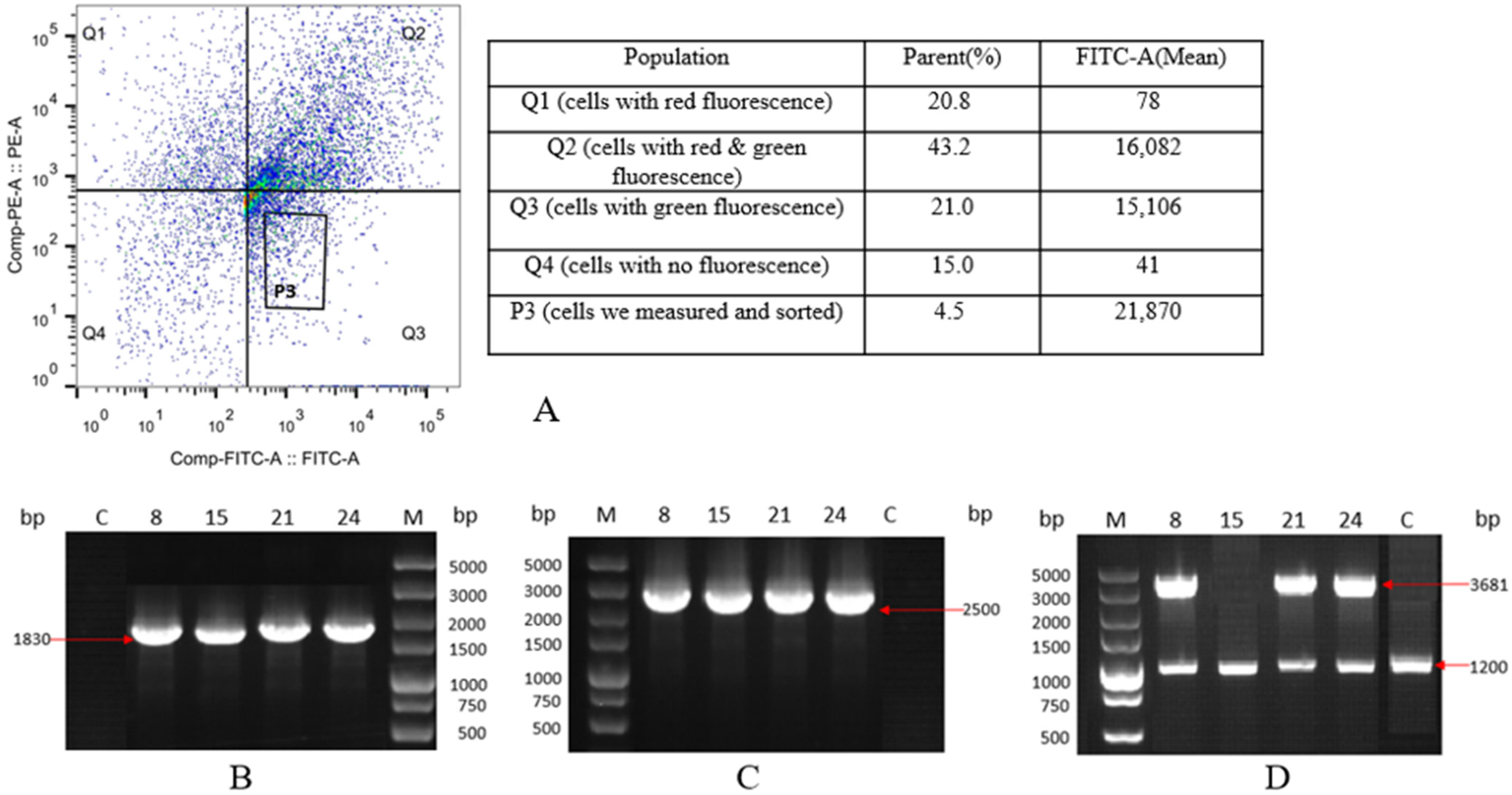
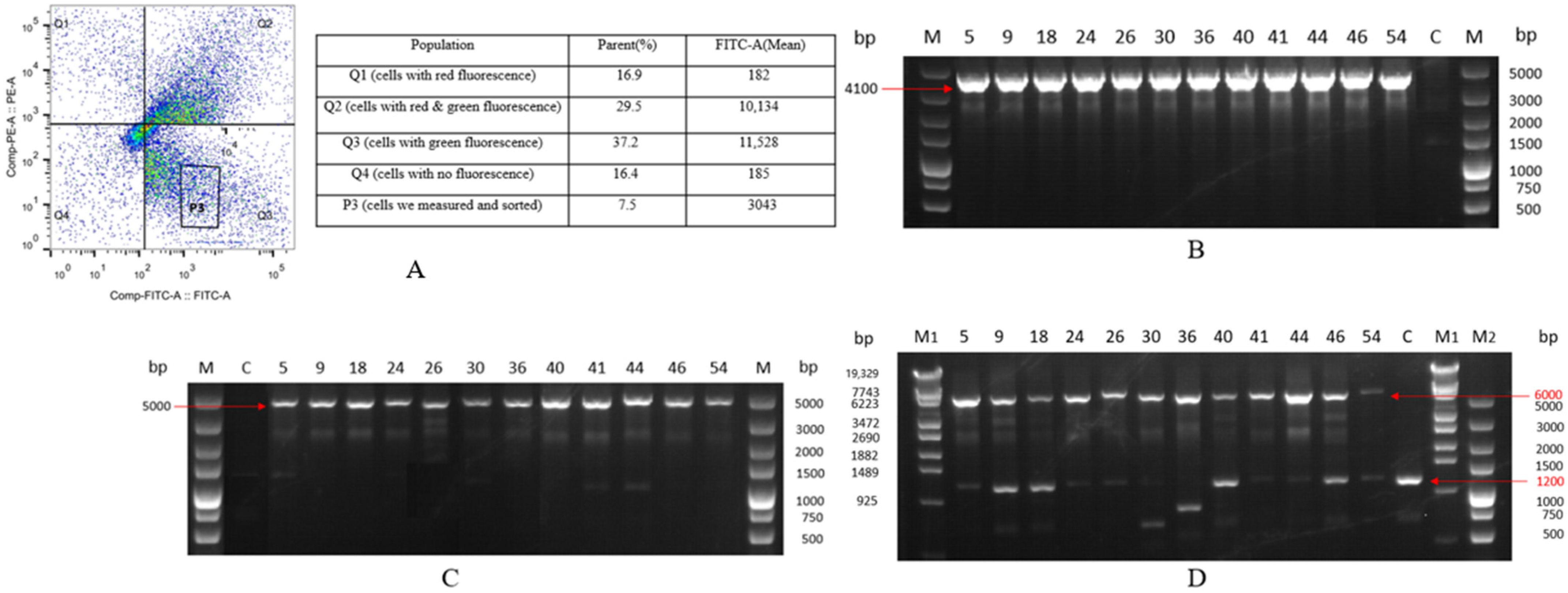
2.2. Selection and Characterization of Monoclonal Cell Lines with Integrated EGFP Gene
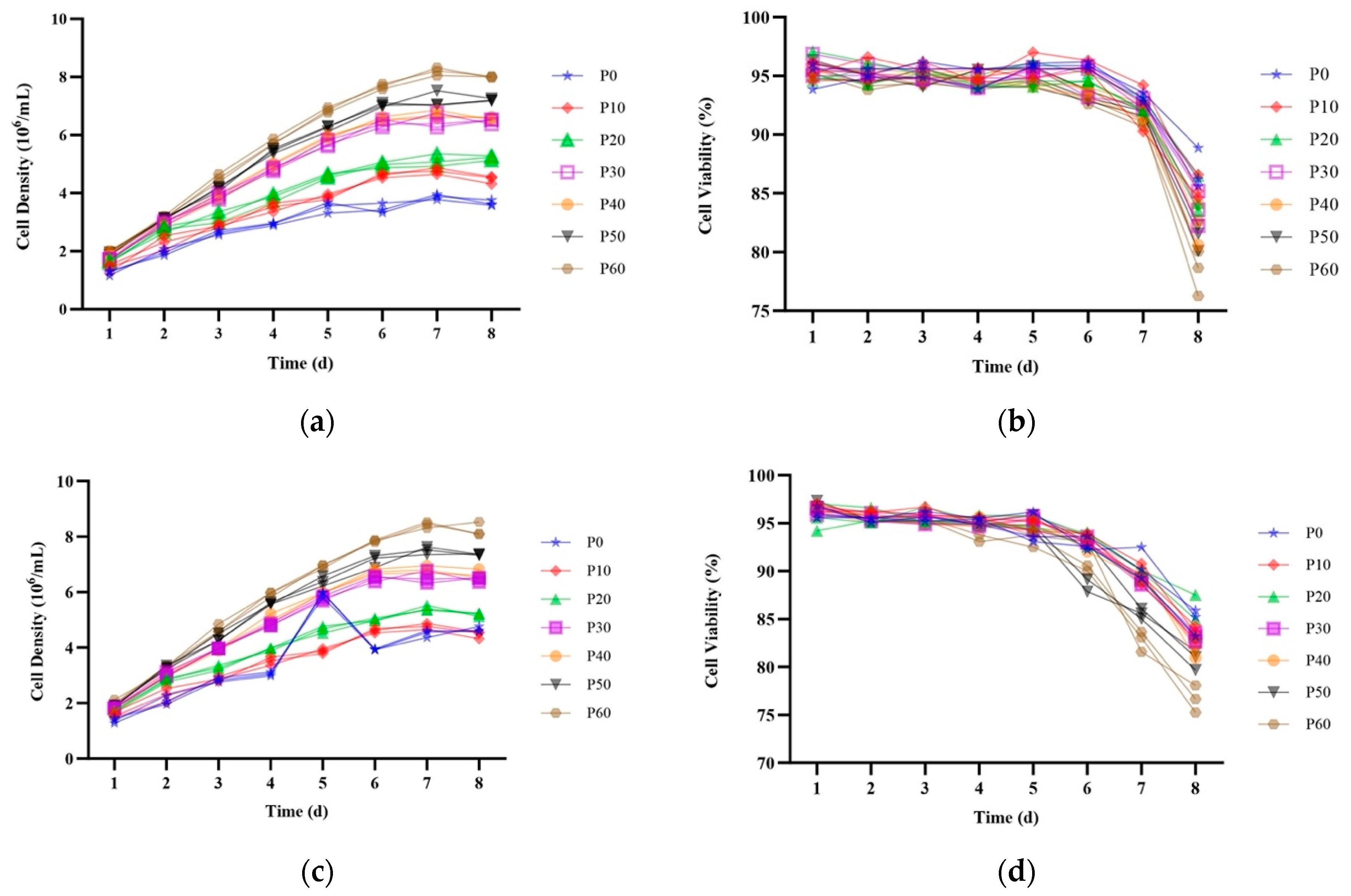
2.3. Stability of EGFP Expression over 60 Passages in Site-Specific Integrant Cell Lines
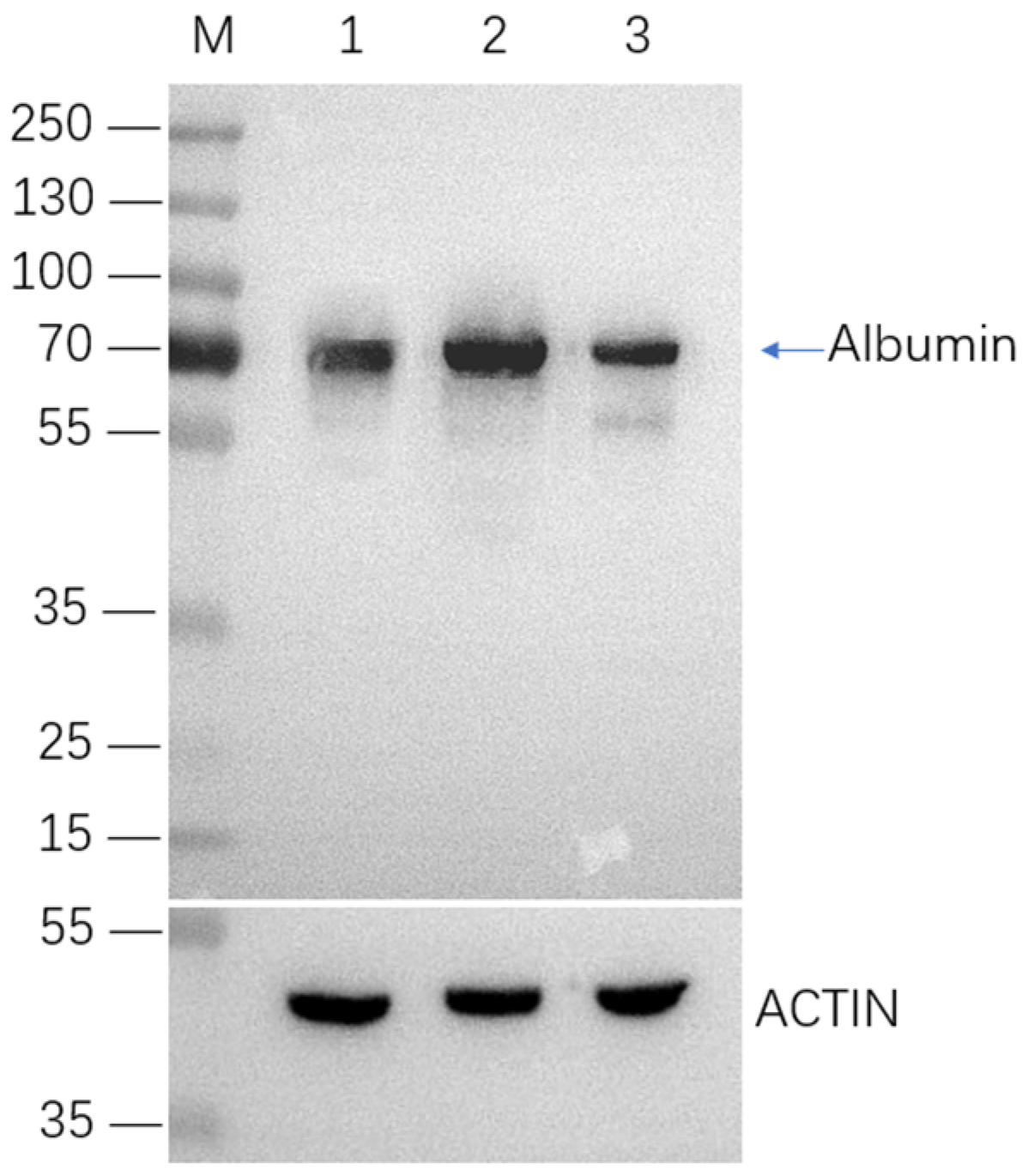
2.4. Selection and Characterization of Monoclonal Cell Lines with Integrated HSA Gene
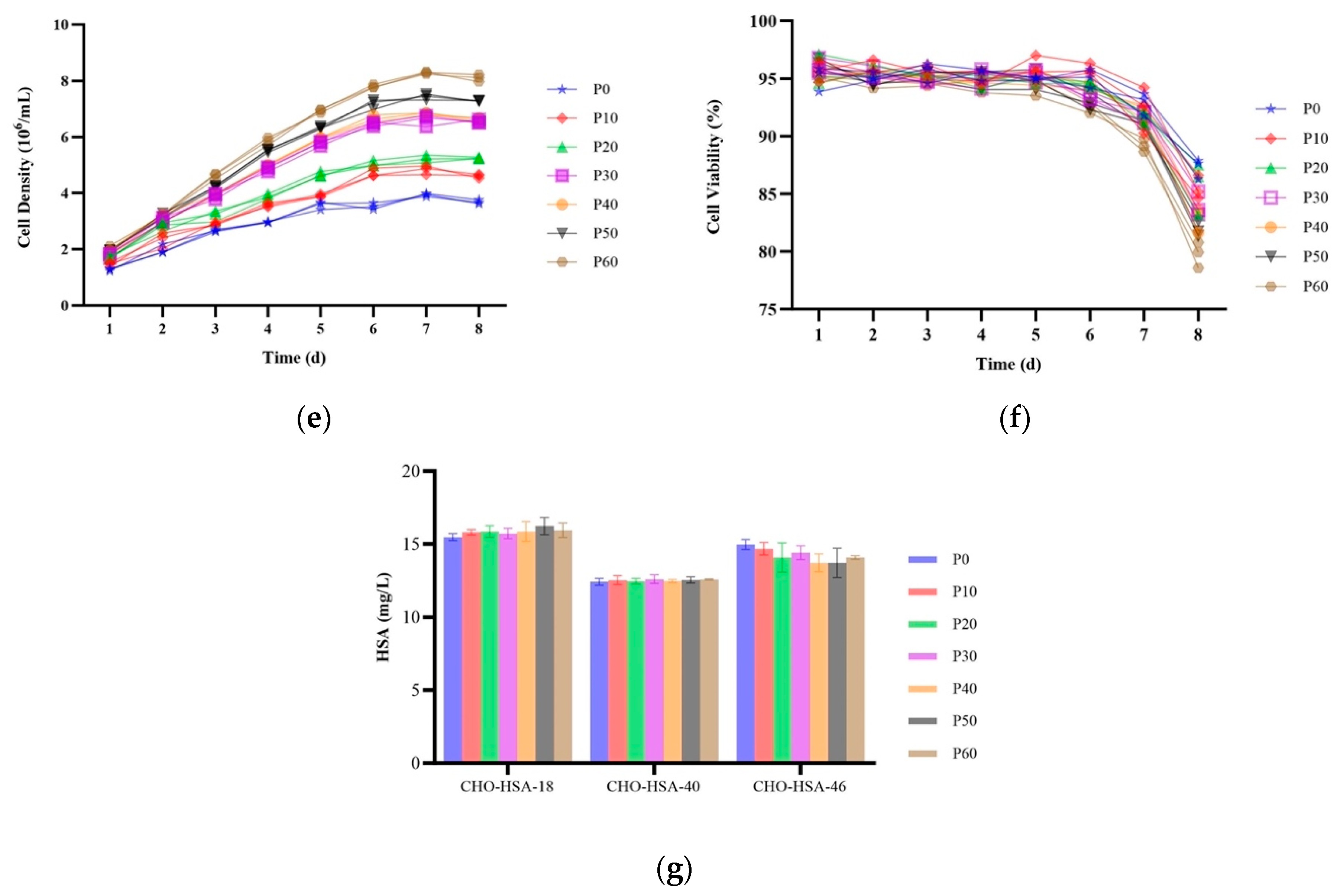
2.5. HSA Expression in Site-Specific Integrant Cell Lines over 60 Passages
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Expression System Construction and Transfection
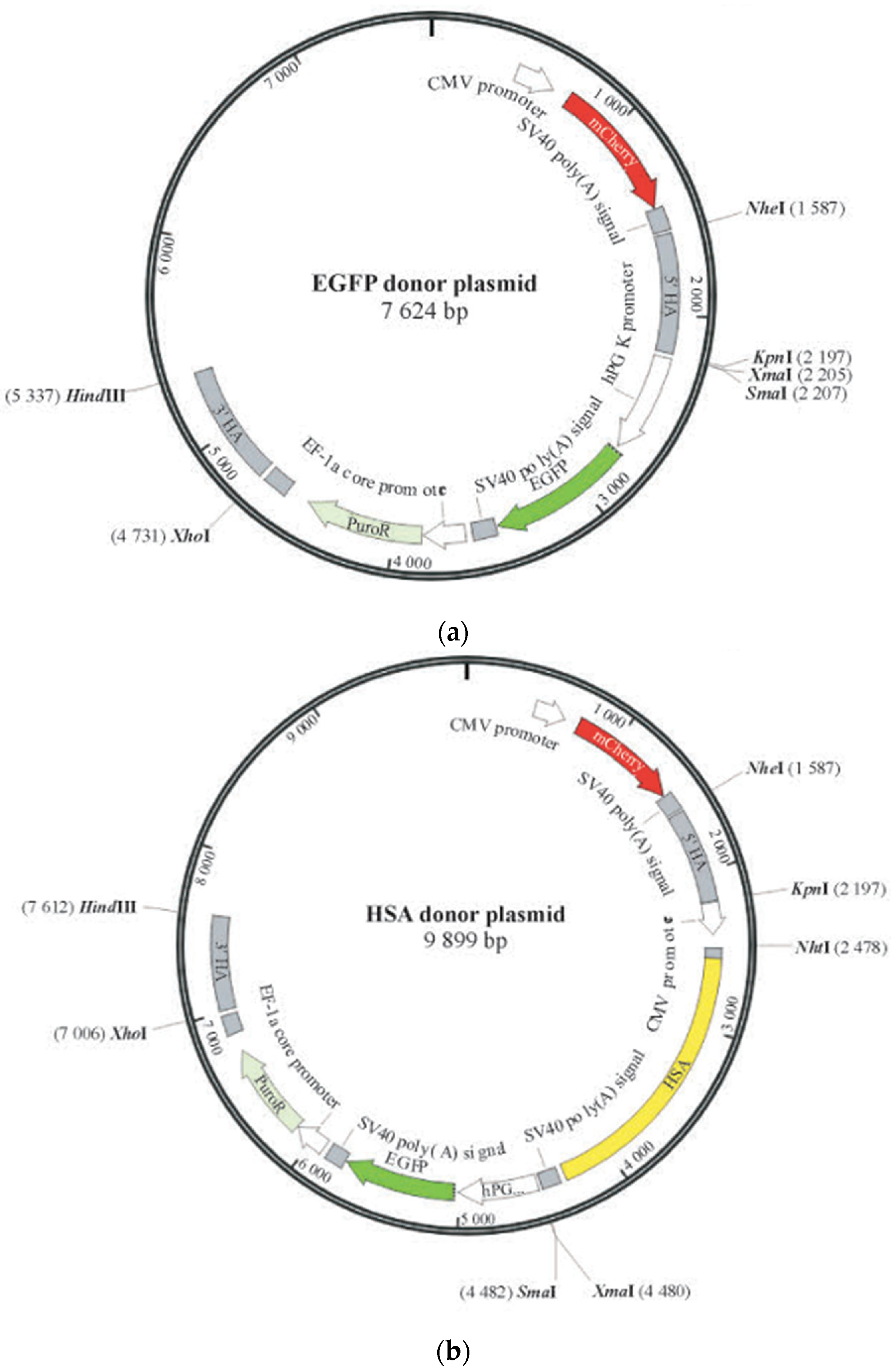
4.3. Donor Plasmid Design and Recombinant Monoclonal Confirmation
4.4. Determination of Targeted Protein Expression
4.5. Identification of Cell Line and Exogenous Protein Stability
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischer, S.; Handrick, R.; Otte, K. The art of CHO cell engineering: A comprehensive retrospect and future perspectives. Biotechnol. Adv. 2015, 33, 1878–1896. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, Y.G.; Lee, G.M. CHO cells in biotechnology for production of recombinant proteins: Current state and further potential. Appl. Microbiol. Biotechnol. 2012, 93, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Dahodwala, H.; Lee, K.H. The fickle CHO: A review of the causes, implications, and potential alleviation of the CHO cell line instability problem. Curr. Opin. Biotechnol. 2019, 60, 128–137. [Google Scholar] [CrossRef]
- Butler, M. Animal cell cultures: Recent achievements and perspectives in the production of biopharmaceuticals. Appl. Microbiol. Biotechnol. 2005, 68, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kallehauge, T.B.; Pedersen, L.E.; Kildegaard, H.F. Site-specific integration in CHO cells mediated by CRISPR/Cas9 and homology-directed DNA repair pathway. Sci. Rep. 2015, 5, 8572. [Google Scholar] [CrossRef] [PubMed]
- Wurm, F. CHO quasispecies-implications for manufacturing processes. Processes 2013, 1, 296–311. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, J.; Luo, M.; Luo, H.; Zhao, M.; Han, L.; Zhang, M.; Yang, H.; Xie, Y.; Jiang, H.; et al. Rapid development of stable transgene CHO cell lines by CRISPR/Cas9-mediated site-specific integration into C12orf35. Appl. Microbiol. Biotechnol. 2018, 102, 6105–6117. [Google Scholar] [CrossRef]
- Krämer, O.; Klausing, S.; Noll, T. Methods in mammalian cell line engineering: From random mutagenesis to sequence-specific approaches. Appl. Microbiol. Biotechnol. 2010, 88, 425–436. [Google Scholar] [CrossRef]
- Kawahara, A.H.; Yu, H.; Ota, S.; Taimatsu, K. Site-specific integration of exogenous genes using genome editing technologies in zebrafish. Int. J. Mol. Sci. 2016, 17, 727. [Google Scholar] [CrossRef]
- Grav, L.M.; la Cour Karottki, K.J.; Lee, J.S.; Kildegaard, H.F. Application of CRISPR/Cas9 genome editing to improve recombinant protein production in CHO cells. Methods. Mol. Biol. 2017, 1603, 101–118. [Google Scholar]
- Xu, X.; Nagarajan, H.; Lewis, N.E.; Pan, S.; Cai, Z.; Liu, X.; Chen, W.; Xie, M.; Wang, W.; Hammond, S. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat. Biotechnol. 2011, 29, 735–741. [Google Scholar] [CrossRef]
- Lewis, N.E.; Liu, X.; Li, Y.; Nagarajan, H.; Yerganian, G.; O’Brien, E.; Bordbar, A.; Roth, A.M.; Rosenbloom, J.; Bian, C. Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nat. Biotechnol. 2013, 31, 759–765. [Google Scholar] [CrossRef]
- Eisenhut, P.; Klanert, G.; Weinguny, M.; Baier, L.; Jadhav, V.; Ivansson, D.; Borth, N.A. CRISPR/Cas9 based engineering strategy for overexpression of multiple genes in Chinese hamster ovary cells. Metabol. Eng. 2018, 1, 72–81. [Google Scholar] [CrossRef]
- Kawabe, Y.; Komatsu, S.; Komatsu, S.; Murakami, M.; Ito, A.; Sakuma, T.; Nakamura, T.; Yamamoto, T.; Kamihira, M. Targeted knock-in of an scFv-Fc antibody gene into the hprt locus of Chinese hamster ovary cells using CRISPR/Cas9 and CRIS-PITCh systems. J. Biosci. Bioeng. 2018, 125, 599–605. [Google Scholar] [CrossRef]
- Chi, X.; Zheng, Q.; Jiang, R.; Chen-Tsai, R.Y.; Kong, L.J. A system for site-specific integration of transgenes in mammalian cells. PLoS ONE 2019, 14, e0219842. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, Y.; Gong, X.; Jin, J.; Li, H. Site-specific integration of light chain and heavy chain genes of antibody into CHO-K1 stable hot spot and detection of antibody and fusion protein expression level. Prep. Biochem. Biotechnol. 2019, 49, 384–390. [Google Scholar] [CrossRef]
- Cheng, J.K.; Lewis, A.M.; Kim, D.S.; Dyess, T.; Alper, H.S. Identifying and retargeting transcriptional hot spots in the human genome. Biotechnol. J. 2016, 11, 1100–1109. [Google Scholar] [CrossRef]
- Zhou, S.; Ding, X.; Yang, L.; Chen, Y.; Gong, X.; Jin, J.; Li, H. Discovery of a stable expression hot spot in the genome of Chinese hamster ovary cells using lentivirus-based random integration. Biotechnol. Biotechnol. Equip. 2019, 33, 605–612. [Google Scholar] [CrossRef]
- Bailey, A.L.; Diane, H.; Ray, F.; Dickson, A.J. Determination of Chinese hamster ovary cell line stability and recombinant antibody expression during long-term culture. Biotechnol. Bioeng. 2012, 109, 2093–2103. [Google Scholar] [CrossRef]
- Hamaker, N.K.; Lee, K.H. A Site-Specific integration reporter system that enables rapid evaluation of CRISPR/Cas9-mediated genome editing strategies in CHO cells. Biotechnol. J. 2020, 15, e2000057. [Google Scholar] [CrossRef]
- Sun, T.; Li, C.D.; Han, L.; Jiang, H.; Xie, Y.Q.; Zhang, B.H.; Qian, X.P.; Lu, H.L.; Zhu, J.W. Functional knockout of FUT8 in Chinese hamster ovary cells using CRISPR/Cas9 to produce a defucosylated antibody. Eng. Life Sci. 2015, 15, 660–666. [Google Scholar] [CrossRef]
- Ding, X.; Chen, Y.; Yang, Z. A comprehensive evaluation of stable expression “hot spot” in the ScltI gene of Chinese hamster ovary cells. Appl. Microbiol. Biotechnol. 2023, 107, 1299–1309. [Google Scholar] [CrossRef]
- Lee, J.S.; Grav, L.M.; Pedersen, L.E.; Lee, G.M.; Kildegaard, H.F. Accelerated homology-directed targeted integration of transgenes in Chinese hamster ovary cells via CRISPR/Cas9 and fluorescent enrichment. Biotechnol. Bioeng. 2016, 113, 2518–2523. [Google Scholar] [CrossRef]
- Lai, T.; Yang, Y.; Ng, S.K. Advances in mammalian cell line develop- ment technologies for recombinant protein production. Pharmaceuticals 2013, 6, 579–603. [Google Scholar] [CrossRef]
- Hiller, G.W.; Ovalle, A.M.; Gagnon, M.P.; Curran, M.L.; Wang, W.G. Cell- controlled hybrid perfusion fed-batch CHO cell process provides significant productivity improvement over conventional fed-batch cultures. Biotechnol. Bioeng. 2017, 114, 1438–1447. [Google Scholar] [CrossRef]
- Josse, L.; Xie, J.; Proud, C.G.; Smales, C.M. mTORC1 signalling and eIF4E/4E-BP1 translation initiation factor stoichiometry influence recombinant protein productivity from GS-CHOK1 cells. Biochem. J. 2016, 473, 4651–4664. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engi- neering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Wang, X.; Kawabe, Y.; Kato, R.; Hada, T.; Ito, A.; Yamana, Y.; Kondo, M.; Kamihira, M. Accumulative scFv-Fc antibody gene integration into the hprt chromosomal locus of Chinese hamster ovary cells. J. Biosci. Bioeng. 2017, 124, 583–590. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Primer Sequence (5′→3′) |
|---|---|
| sgRNA-F | AGCCTAGTGCTCCTGATACG |
| sgRNA-R | AGACCGATACCAGGATCTTG |
| BB-F | AGCCTAGTGCTCCTGATACG |
| BB-R | TTCTGCTGTGGACTCTGAAG |
| VI-F | AACCACCAGGTCAGAAATCC |
| VI-R | CAGAGGCCAATCAGCAGTAG |
| HP-F | GCTGTGCATTGAAACCCATG |
| HP-R | TGGGTATGGAGATGGGGCGG |
| Primer | Primer Sequence (5′→3′) |
|---|---|
| 5′ arm-F/Oo-F | CCTGTCCACGTCTAAGTATC |
| 5′ arm-R | GTCCATGCCGAGAGTGATCC |
| 3′ arm-EGFP-F | AAGGGCGAGGAGCTGTTCAC |
| 3′ arm-HSA-F | GGTACTGCTGCTCTGGGTTC |
| 3′ arm-R/Oo-R | GCTTTGCTCCGAAGTCCATC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Habib, A.; Zhao, G.; Ding, X. CRISPR-Cas9 Mediated Stable Expression of Exogenous Proteins in the CHO Cell Line through Site-Specific Integration. Int. J. Mol. Sci. 2023, 24, 16767. https://doi.org/10.3390/ijms242316767
Huang Z, Habib A, Zhao G, Ding X. CRISPR-Cas9 Mediated Stable Expression of Exogenous Proteins in the CHO Cell Line through Site-Specific Integration. International Journal of Molecular Sciences. 2023; 24(23):16767. https://doi.org/10.3390/ijms242316767
Chicago/Turabian StyleHuang, Zhipeng, Arslan Habib, Guoping Zhao, and Xiaoming Ding. 2023. "CRISPR-Cas9 Mediated Stable Expression of Exogenous Proteins in the CHO Cell Line through Site-Specific Integration" International Journal of Molecular Sciences 24, no. 23: 16767. https://doi.org/10.3390/ijms242316767