

From Molecular Insights to Clinical Perspectives in Drug-Associated Bullous Pemphigoid
 , , ,
, , , 
Abstract
:1. Introduction
2. Pathogenesis of Bullous Pemphigoid (BP)
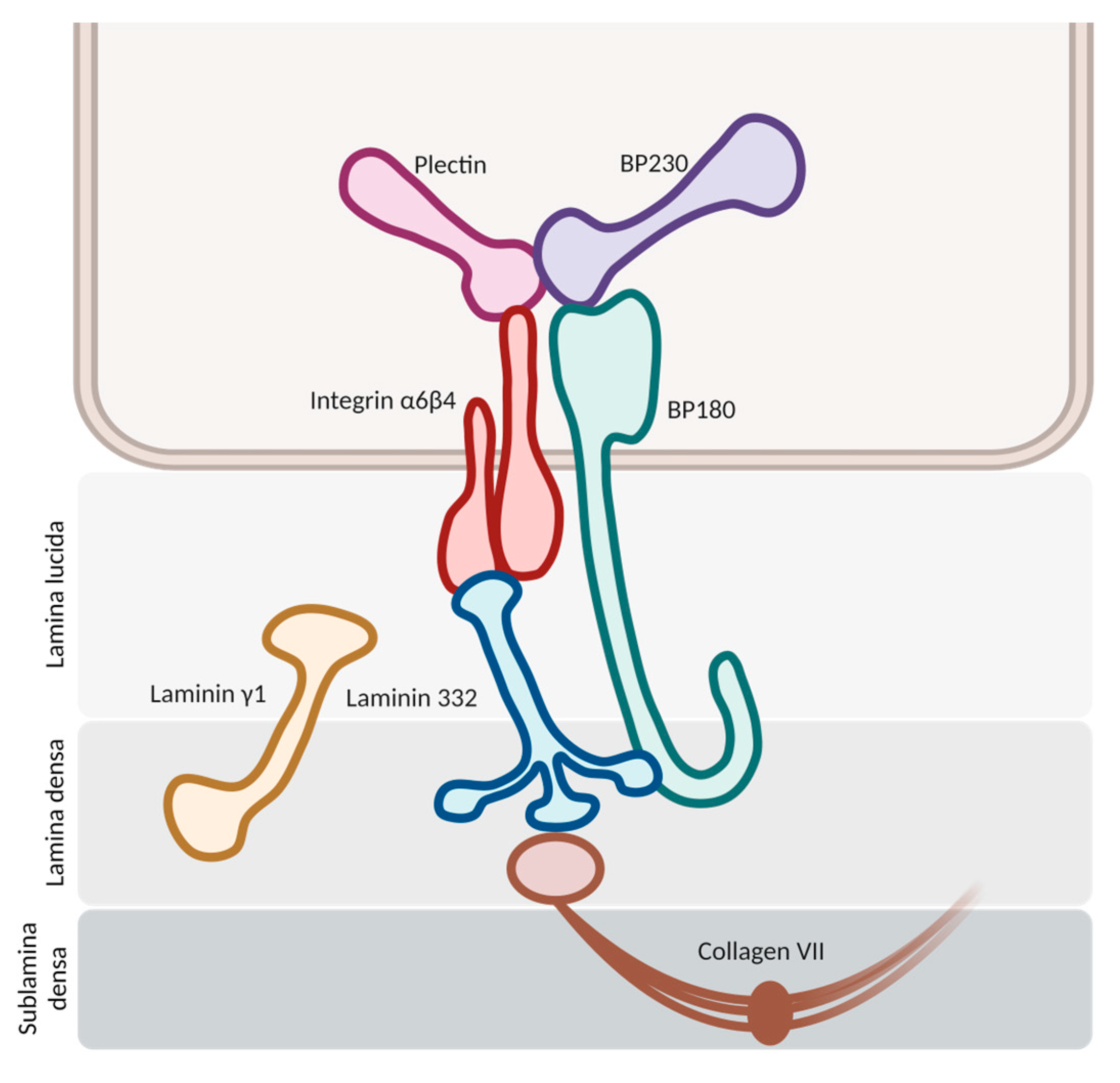
2.1. Antigenic Targets
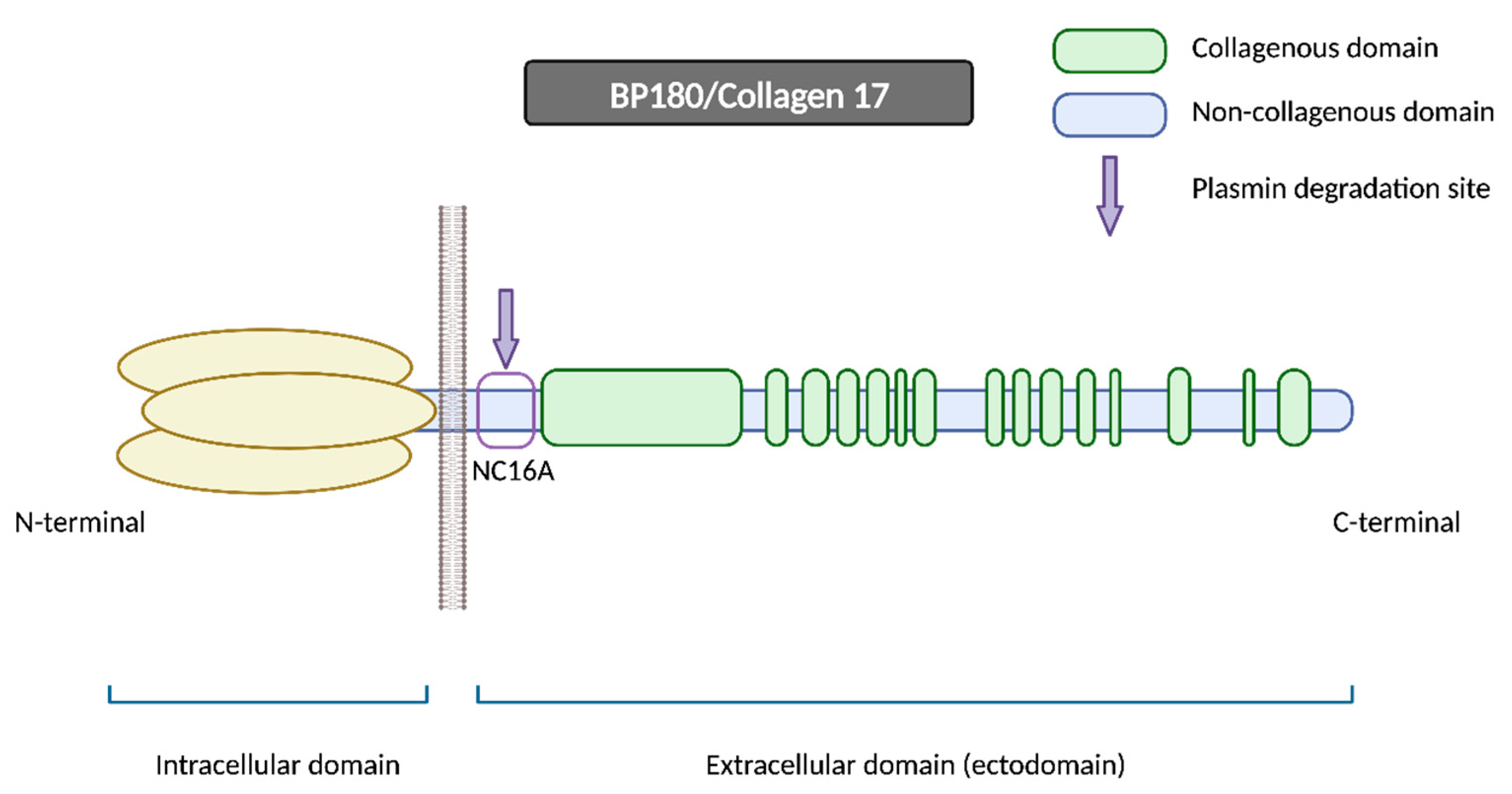
2.1.1. BP180
2.1.2. BP230
2.2. Hypothesis of Blister Formation
2.2.1. Complement-Dependent Immune Response
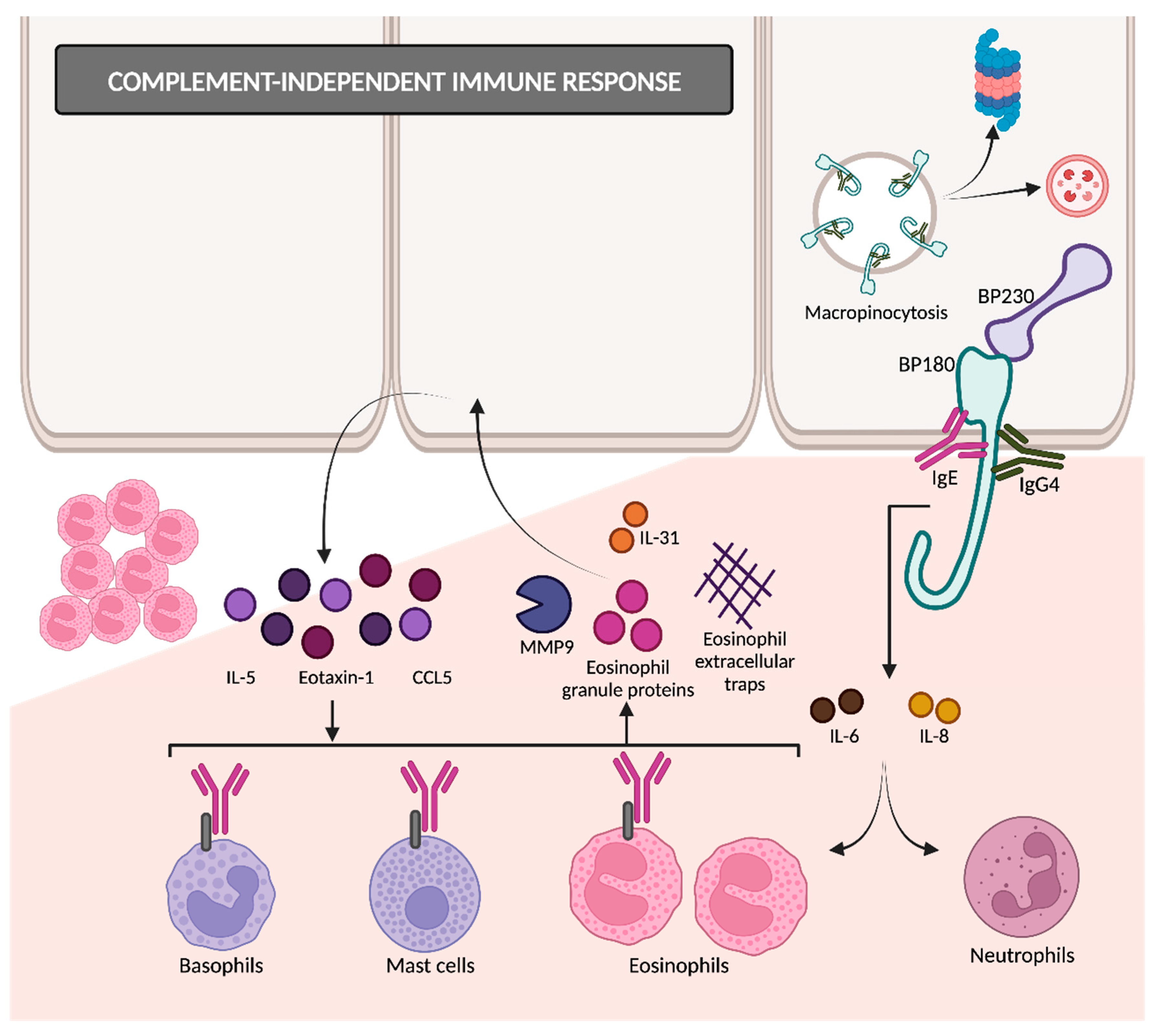
2.2.2. Complement-Independent Immune Response
2.3. Breakdown of Self-Tolerance
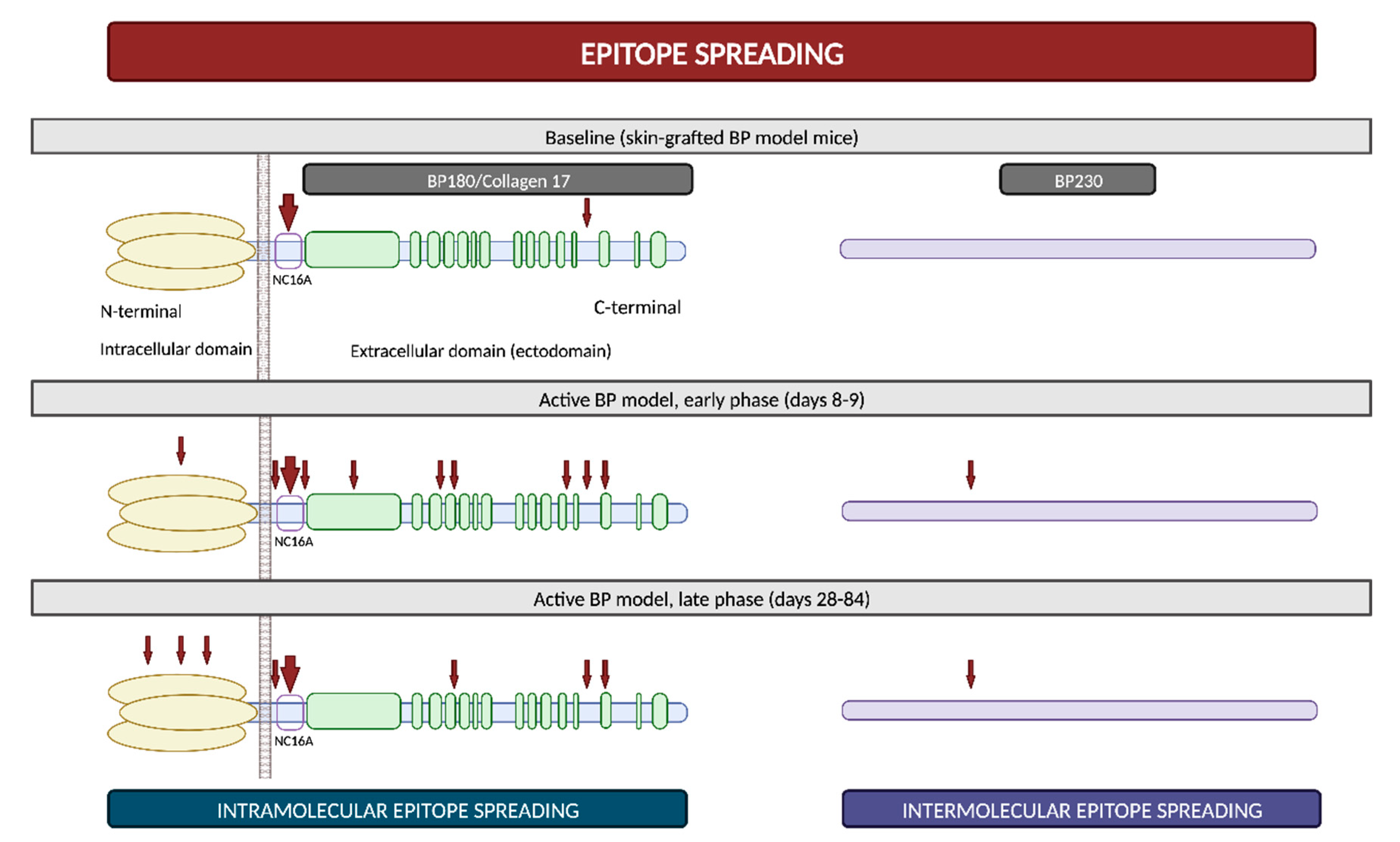
2.4. Epitope Spreading
3. General Aspects of Drug-Associated Bullous Pemphigoid (DABP)
3.1. Drugs Related to DABP
3.2. Pathogenic Mechanisms
- Molecular mimicry: many drugs bind to RNA and proteins in a way that closely resembles the interaction pattern observed with viruses. This similarity raises the possibility that these drugs might be erroneously recognized as microbial antigens. The immune system’s misidentification of drugs in predisposed individuals could result in the activation of CD4+ T cells and the subsequent initiation of the autoimmune cascade [7,63].
- Antigenic haptens: some drugs may have the ability to function as antigenic haptens that can bind to and modify protein molecules within the lamina lucida of the BMZ. Such interactions might induce the modification of their antigenic properties, thereby acting as neoantigens. Alternatively, this phenomenon could lead to the exposure of a previously hidden antigenic site, supporting the drug-triggering epitope spreading theory [4,7,63,66].
- Direct immune dysregulation: drugs may cause immune reorganization, disrupting the endogenous regulatory processes that prevent the development of several diseases. Alterations in T-regulatory cell functions may suppress “forbidden” B cell clones and then result in the release of autoantibodies against the BMZ [63,66].
- Non-immunological mechanisms: thiol-containing drugs may directly interact with the sulfhydryl groups present in the BMZ proteins and subsequently disrupt the dermoepidermal junction without the involvement of immunological mechanisms. However, this dermoepidermal cleavage may also expose new, hidden antigenic sites [7,63].
- Thiol-based drugs: they might induce BP acting as haptens or directly disrupting the dermoepidermal junction, as previously described. Moreover, penicillamine, a specific thiol-based drug, could decrease the activity of T-regulatory cells [62]. Many drugs, such as furosemide, hydrochlorothiazide, spironolactone, penicillins or sulfasalazine, contain sulfur atoms within their molecules, yet not as part of a thiol group. However, it is hypothesized that they may be able to form thiol groups during their metabolism, thereby inducing BP through a similar mechanism to thiol-based drugs [67].
- Phenol-based drugs: these medications incorporate a phenyl group in their molecular structure and are thought to interfere with the integrity of the BMZ, consequently revealing hidden epitopes. Examples of these phenol drugs are non-steroidal anti-inflammatory drugs (NSAID), cephalosporins, angiotensin II receptor blockers (ARB) and selective serotonin reuptake inhibitors (SSRI).
- Non-thiol non-phenol-based drugs: the number of these drugs is continuously growing, although the precise underlying mechanisms remain largely undefined.
3.3. General Differences between DABP and Idiopathic BP
3.3.1. Clinical Differences
3.3.2. Histological and Laboratory Differences
4. Dipeptidyl Peptidase 4 Inhibitor-Associated Bullous Pemphigoid (DPP4i-BP)
4.1. Epidemiology of DPP4i-BP
4.1.1. General Risk of BP Development
4.1.2. Risk of BP Development in Patients with Diabetes Mellitus in Absence of DPP4i
4.1.3. Risk of BP Development in Patients with Diabetes Mellitus in Absence of DPP4i
4.1.4. Latency Period
4.2. Pathogenesis of DPP4i-BP
4.2.1. Genetic Predisposition
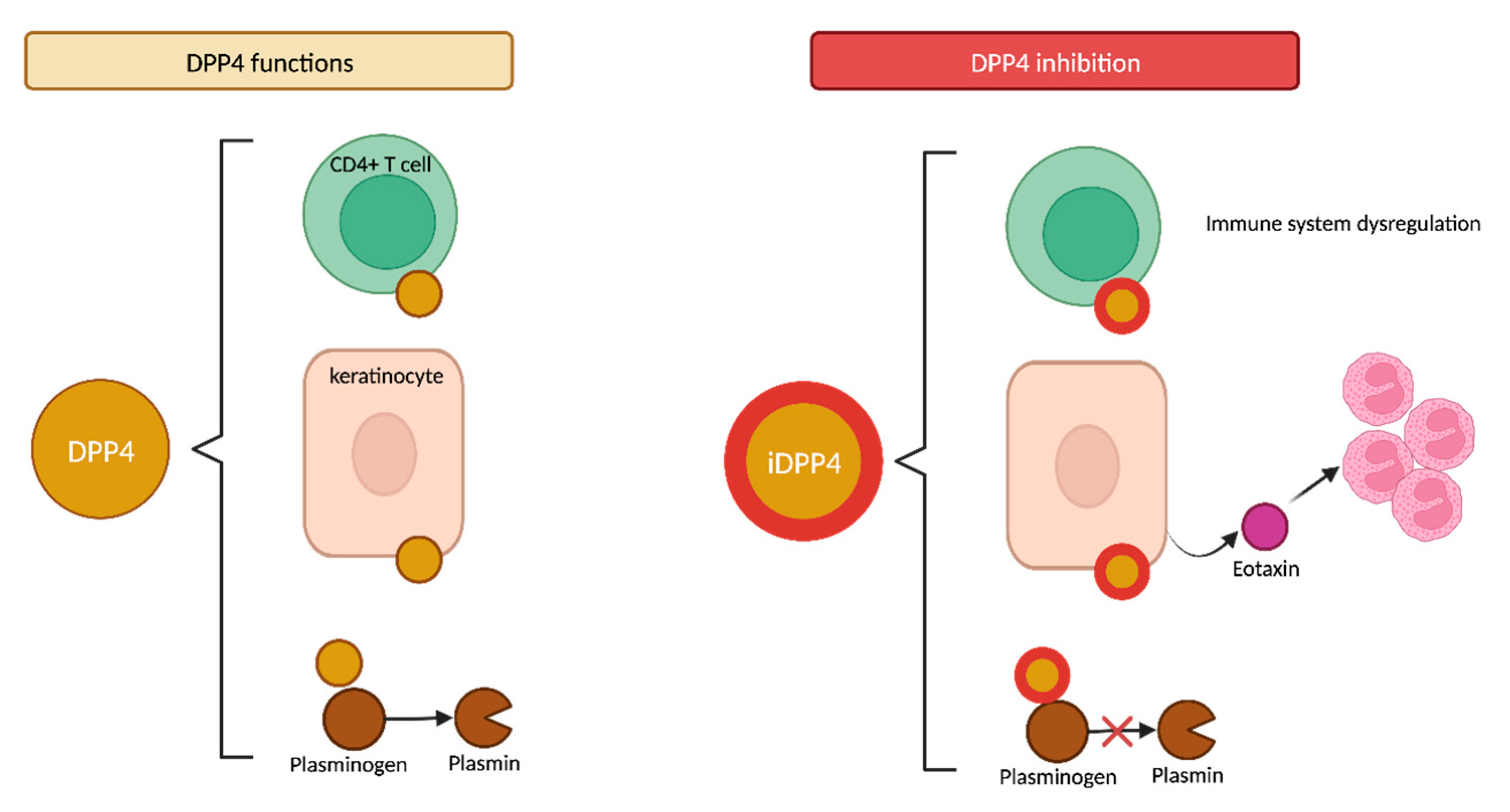
4.2.2. DPP4 Functions
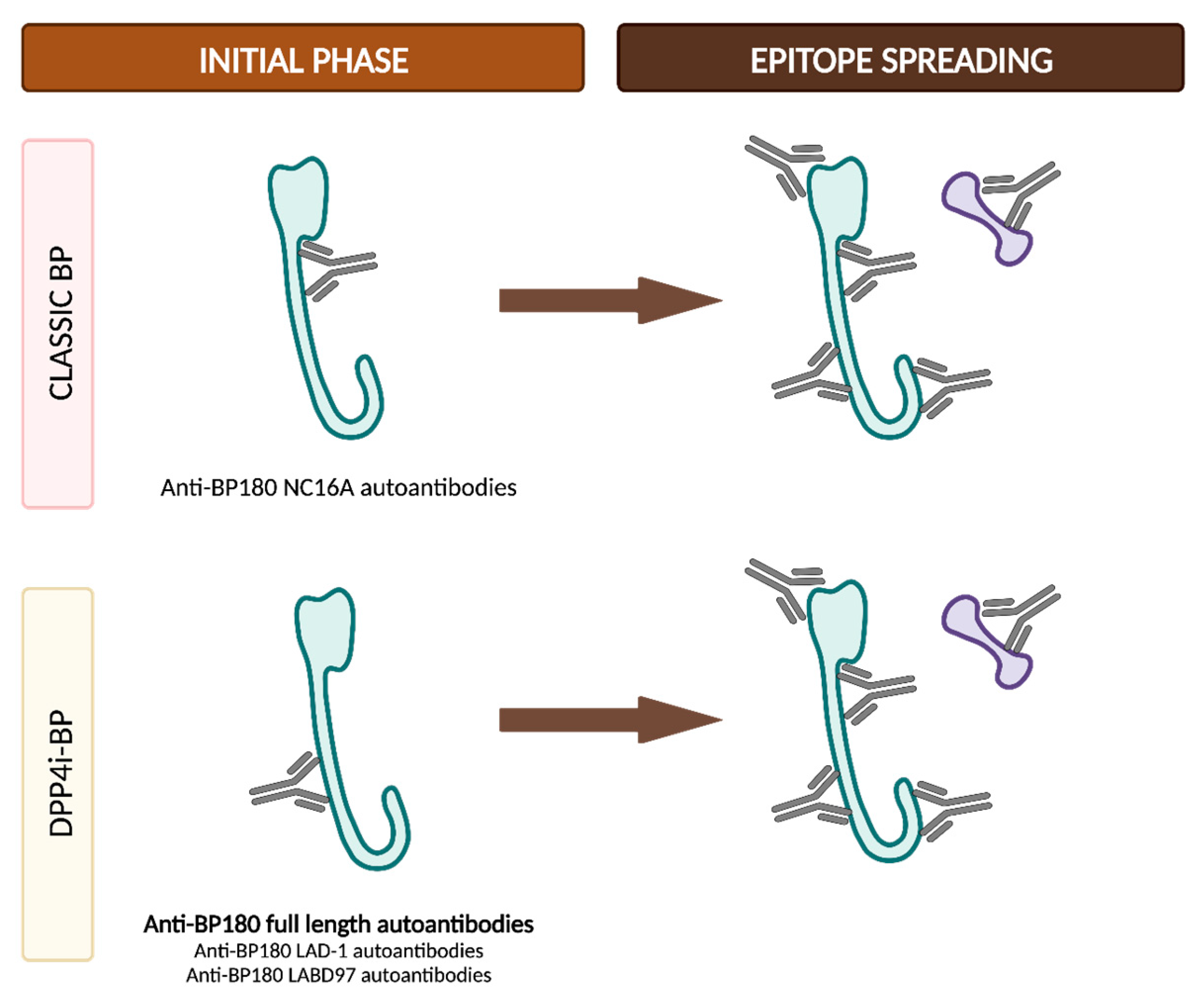
4.2.3. DPP4i-BP Autoantibodies and Epitope Spreading
4.2.4. Currently Described Immune and Pathogenic Mechanisms
4.3. Clinical and Immunological Distinct Features in Classic and DPP4i-Associated BP
4.3.1. Effect of DPP4i Withdrawal
4.3.2. DPP4i-BP Clinical Subtypes
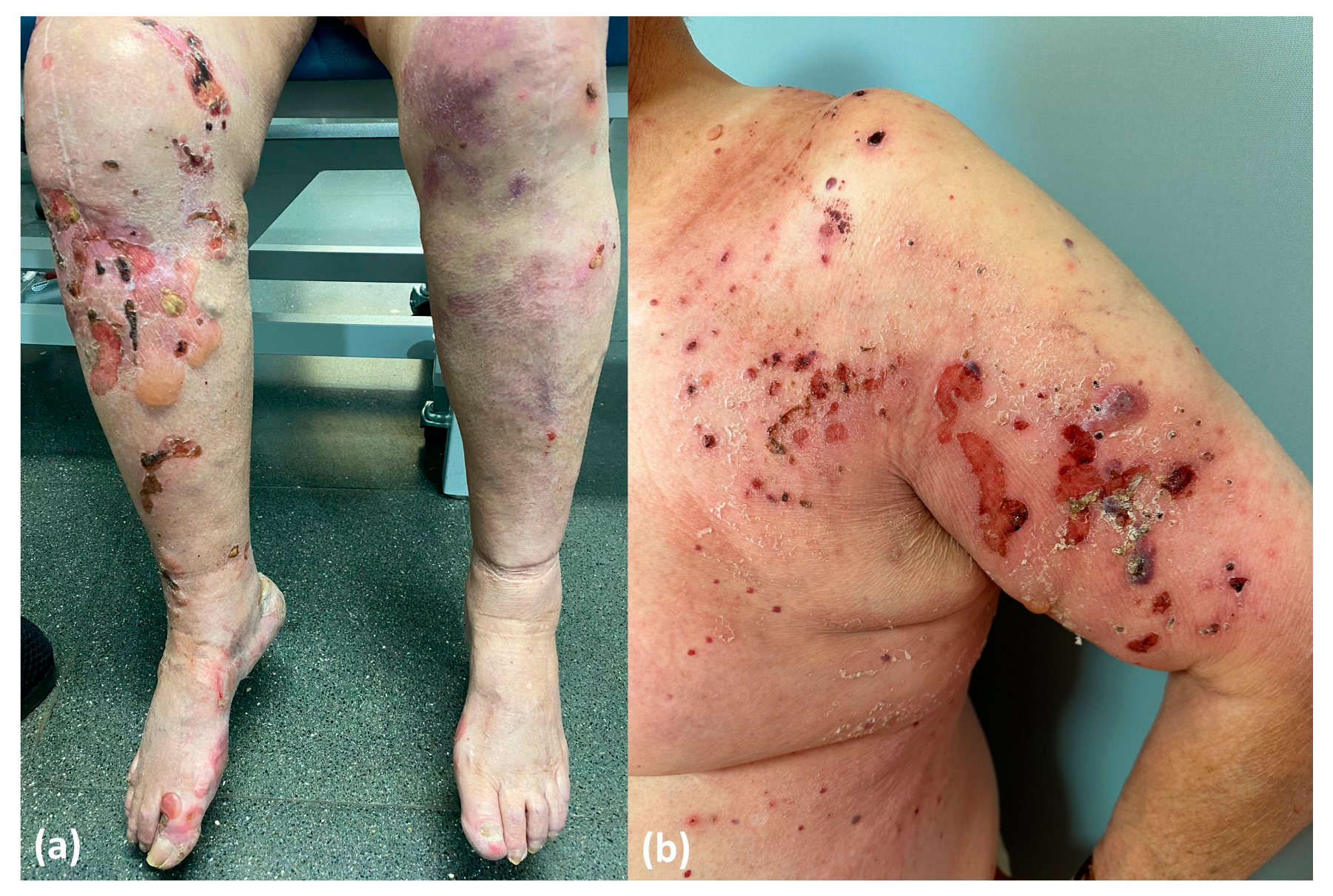
- Drug-induced BP. This subtype would represent the true drug-related BP and would appear de novo in patients with no prior genetic predisposition. Patients in this category exhibit distinctive features, including a non-inflammatory phenotype (Figure 10a), negative results for anti-BP180 NC16A autoantibodies and positivity for other epitopes within BP180, such as full-length autoantibodies, LAD-1 and LABD97. Additionally, they often display lower levels of tissue and peripheral eosinophilia. In these patients, discontinuation of DPP4i is considered mandatory to restrain the stimulus for the immune system and to ultimately achieve disease control.
- Drug-triggered BP. This subtype would occur in patients already predisposed to developing BP, and the initiation of DPP4i treatment would merely precipitate the onset of the bullous disease. These patients typically display the characteristic features observed in classic BP, including an inflammatory phenotype (Figure 10b) and the positivity and high titers of anti-BP180 NC16A autoantibodies, along with higher levels of peripheral and tissue eosinophilia. In this subgroup of patients, discontinuing DPP4i is also advisable to eliminate at least one of the contributing factors to BP etiopathogenesis. However, it is important to note that BP may persist even after discontinuing gliptin treatment.
5. Bullous Pemphigoid Associated with Antineoplastic Drugs
5.1. Immune Checkpoint Inhibitor-Associated Bullous Pemphigoid (ICI-BP)
5.1.1. Epidemiology of ICI-BP
5.1.2. Pathogenesis of ICI-BP
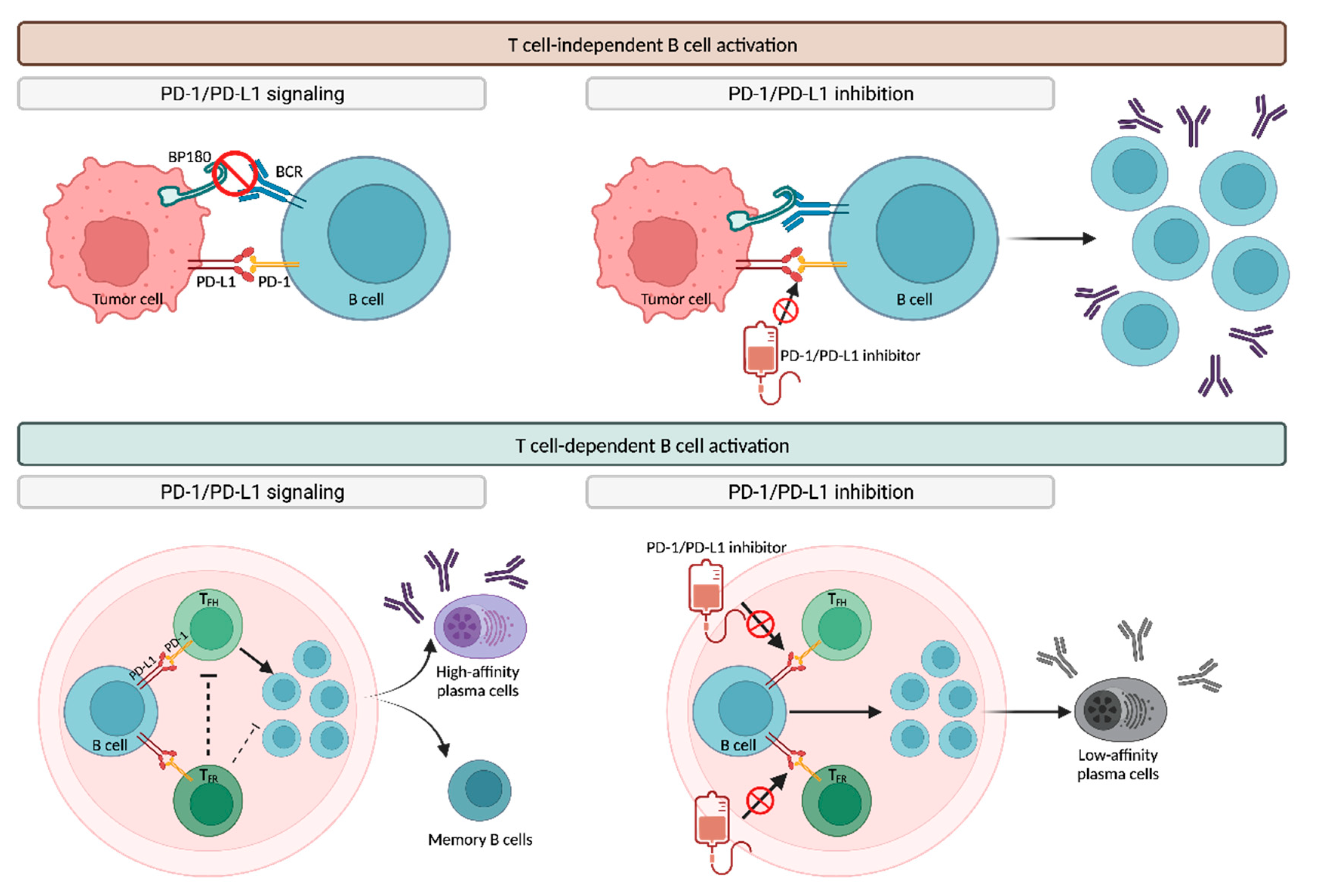
T-Cell-Independent B Cell Activation
T-Cell-Dependent B Cell Activation
Autoantigens and Epitope Spreading
5.1.3. Clinical Course and Management of ICI-BP
5.2. Other Antineoplastic Medications
- Recombinant interleukin-2 (IL-2). Aldesleukin’s potential association with DABP is plausible due to the overexpression of IL-2 and its receptor in BP [140].
- Epidermal growth factor receptor (EGFR) inhibitors. Erlotinib has been associated with BP development in a patient with lung adenocarcinoma, possibly linked to the expression of EGFR in basal keratinocytes [141].
- Mammalian target of rapamycin (mTOR) inhibitors. Three cases of sirolimus- and everolimus-related BP in kidney transplant recipients have been reported in the literature. The pathogenic mechanisms could be attributed to the role of mTOR in the cell cycle or to the imbalance between cell-mediated and humoral immunity induced by mTOR inhibitors. Additionally, it might also be associated with factors related to the renal graft itself [142,143].
- BRAF inhibitors. Dabrafenib might induce a pemphigoid-like reaction with typical clinical and histological features, despite negative DIF [144].
- Cyclin-dependent kinase 4/6 (CDK4/6) inhibitors. BP has recently been linked with novel targeted antineoplastic therapies such as palbociclib, but the underlying pathogenic mechanisms remain unknown [145].
6. Bullous Pemphigoid Associated with Biologic Agents (BIBP)
6.1. Pathogenesis of BIBP
6.1.1. TNF-α Pathway
6.1.2. IL-17/23 Pathway
6.2. Clinical Features of BIBP
7. Bullous Pemphigoid Associated with Other Drugs
7.1. Bullous Pemphigoid Associated with Diuretics
- Loop diuretics (furosemide, bumetanide, torsemide). Some case–control and database studies suggest a link between furosemide and DABP development (OR 3.3–3.8) [72,158,159], while other studies do not find this association with loop diuretics [3,64]. Interestingly, furosemide-induced BP may primarily affects sun-exposed areas due to the drug’s well-known photosensitivity [160]. In one case report, switching from furosemide to bumetanide resulted in the complete clinical remission of BP [161], although bumetanide itself has also been associated with BP development in some patients [162,163]. Finally, torsemide has been linked to DABP in one case report due to its temporal relationship and structural similarity to furosemide [164].
- Aldosterone antagonists (spironolactone). Similar to furosemide, studies on the association between spironolactone and BP exhibit conflicting results [3,61,78,159]. However, a meta-analysis conducted by Liu et al., which included all previous case–control studies, reported a significant association between the use of aldosterone antagonists and BP, with a pooled OR of 1.75 (95% CI, 1.28–2.40; I2 = 4%) [64].
- Acetazolamide. A single case report highlights a patient who experienced a relapse of well-controlled BP lesions one month after initiating acetazolamide. The authors suggest that this diuretic, commonly used in ophthalmology, may have triggered the BP’s recurrence due to its structural similarity to other diuretics like furosemide [166].
7.2. Bullous Pemphigoid Associated with Neurological Drugs
7.3. Bullous Pemphigoid Associated with Cardiovascular Drugs
7.4. Bullous Pemphigoid Associated with Antimicrobial Agents
8. Conclusions
9. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Montagnon, C.M.; Tolkachjov, S.N.; Murrell, D.F.; Camilleri, M.J.; Lehman, J.S. Subepithelial Autoimmune Blistering Dermatoses: Clinical Features and Diagnosis. J. Am. Acad. Dermatol. 2021, 85, 1–14. [Google Scholar] [CrossRef]
- Aoki, V.; Miyamoto, D. Unfolding the Worldwide Incidence of Bullous Pemphigoid: What Are We Missing? Br. J. Dermatol. 2022, 186, 386–387. [Google Scholar] [CrossRef]
- Bastuji-Garin, S.; Joly, P.; Lemordant, P.; Sparsa, A.; Bedane, C.; Delaporte, E.; Roujeau, J.-C.; Bernard, P.; Guillaume, J.-C.; Ingen-Housz-Oro, S.; et al. Risk Factors for Bullous Pemphigoid in the Elderly: A Prospective Case–Control Study. J. Investig. Dermatol. 2011, 131, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Verheyden, M.; Bilgic, A.; Murrell, D. A Systematic Review of Drug-Induced Pemphigoid. Acta Derm. Venereol. 2020, 100, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.; Santi, C.G.; Aoki, V.; Maruta, C.W. Bullous Pemphigoid. An. Bras. Dermatol. 2019, 94, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Langan, S.M.; Smeeth, L.; Hubbard, R.; Fleming, K.M.; Smith, C.J.P.; West, J. Bullous Pemphigoid and Pemphigus Vulgaris—Incidence and Mortality in the UK: Population Based Cohort Study. BMJ 2008, 337, a180. [Google Scholar] [CrossRef] [PubMed]
- Moro, F.; Fania, L.; Sinagra, J.L.M.; Salemme, A.; Di Zenzo, G. Bullous Pemphigoid: Trigger and Predisposing Factors. Biomolecules 2020, 10, 1432. [Google Scholar] [CrossRef] [PubMed]
- Borradori, L.; Van Beek, N.; Feliciani, C.; Tedbirt, B.; Antiga, E.; Bergman, R.; Böckle, B.C.; Caproni, M.; Caux, F.; Chandran, N.S.; et al. Updated S2 K Guidelines for the Management of Bullous Pemphigoid Initiated by the European Academy of Dermatology and Venereology (EADV). J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1689–1704. [Google Scholar] [CrossRef] [PubMed]
- Clapé, A.; Muller, C.; Gatouillat, G.; Le Jan, S.; Barbe, C.; Pham, B.-N.; Antonicelli, F.; Bernard, P. Mucosal Involvement in Bullous Pemphigoid Is Mostly Associated with Disease Severity and to Absence of Anti-BP230 Autoantibody. Front. Immunol. 2018, 9, 479. [Google Scholar] [CrossRef]
- Sárdy, M.; Kostaki, D.; Varga, R.; Peris, K.; Ruzicka, T. Comparative Study of Direct and Indirect Immunofluorescence and of Bullous Pemphigoid 180 and 230 Enzyme-Linked Immunosorbent Assays for Diagnosis of Bullous Pemphigoid. J. Am. Acad. Dermatol. 2013, 69, 748–753. [Google Scholar] [CrossRef]
- Yang, A.; Xuan, R.; Murrell, D.F. A New Indirect Immunofluorescence BIOCHIP Method for the Serological Diagnosis of Bullous Pemphigoid: A Review of Literature. Australas. J. Dermatol. 2019, 60, e173–e177. [Google Scholar] [CrossRef]
- Yang, M.; Wu, H.; Zhao, M.; Chang, C.; Lu, Q. The Pathogenesis of Bullous Skin Diseases. J. Transl. Autoimmun. 2019, 2, 100014. [Google Scholar] [CrossRef] [PubMed]
- Hirako, Y.; Usukura, J.; Nishizawa, Y.; Owaribe, K. Demonstration of the Molecular Shape of BP180, a 180-kDa Bullous Pemphigoid Antigen and Its Potential for Trimer Formation. J. Biol. Chem. 1996, 271, 13739–13745. [Google Scholar] [CrossRef] [PubMed]
- Tuusa, J.; Kokkonen, N.; Tasanen, K. BP180/Collagen XVII: A Molecular View. Int. J. Mol. Sci. 2021, 22, 12233. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, L.; Xia, Y. BP180 Is Critical in the Autoimmunity of Bullous Pemphigoid. Front. Immunol. 2017, 8, 1752. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, H. What’s New in the Pathogeneses and Triggering Factors of Bullous Pemphigoid. J. Dermatol. 2023, 50, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Amagai, M.; Kuroda-Kinoshita, K.; Hashimoto, T.; Shirakata, Y.; Hashimoto, K.; Nishikawa, T. BP180 ELISA Using Bacterial Recombinant NC16a Protein as a Diagnostic and Monitoring Tool for Bullous Pemphigoid. J. Dermatol. Sci. 2002, 30, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.; Nishie, W.; Mai, Y.; Wada, M.; Natsuga, K.; Ujiie, H.; Iwata, H.; Yamagami, J.; Shimizu, H. Autoantibody Profile Differentiates between Inflammatory and Noninflammatory Bullous Pemphigoid. J. Investig. Dermatol. 2016, 136, 2201–2210. [Google Scholar] [CrossRef]
- Nishie, W.; Lamer, S.; Schlosser, A.; Licarete, E.; Franzke, C.-W.; Hofmann, S.C.; Jackow, J.; Sitaru, C.; Bruckner-Tuderman, L. Ectodomain Shedding Generates Neoepitopes on Collagen XVII, the Major Autoantigen for Bullous Pemphigoid. J. Immunol. 2010, 185, 4938–4947. [Google Scholar] [CrossRef]
- Hiroyasu, S.; Turner, C.T.; Richardson, K.C.; Granville, D.J. Proteases in Pemphigoid Diseases. Front. Immunol. 2019, 10, 1454. [Google Scholar] [CrossRef]
- Seppänen, A.; Suuronen, T.; Hofmann, S.C.; Majamaa, K.; Alafuzoff, I. Distribution of Collagen XVII in the Human Brain. Brain Res. 2007, 1158, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Hurskainen, T.; Moilanen, J.; Sormunen, R.; Franzke, C.-W.; Soininen, R.; Loeffek, S.; Huilaja, L.; Nuutinen, M.; Bruckner-Tuderman, L.; Autio-Harmainen, H.; et al. Transmembrane Collagen XVII Is a Novel Component of the Glomerular Filtration Barrier. Cell Tissue Res. 2012, 348, 579–588. [Google Scholar] [CrossRef]
- Barrick, B.J.; Ida, C.M.; Laniosz, V.; Jentoft, M.E.; Sominidi-Damodaran, S.; Wieland, C.N.; Meves, A.; Lehman, J.S. Bullous Pemphigoid, Neurodegenerative Disease, and Hippocampal BP180 Expression: A Retrospective Postmortem Neuropathologic Study. J. Investig. Dermatol. 2016, 136, 2090–2092. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, M.; Zillikens, D.; Schmidt, E. Pemphigoid Diseases: Pathogenesis, Diagnosis, and Treatment. Autoimmunity 2012, 45, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Meijer, J.M.; Diercks, G.F.H.; De Lang, E.W.G.; Pas, H.H.; Jonkman, M.F. Assessment of Diagnostic Strategy for Early Recognition of Bullous and Nonbullous Variants of Pemphigoid. JAMA Dermatol. 2019, 155, 158. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.; Vinay, K.; Borradori, L.; Amber, K.T. Insights into the Pathogenesis of Bullous Pemphigoid: The Role of Complement-Independent Mechanisms. Front. Immunol. 2022, 13, 912876. [Google Scholar] [CrossRef]
- Romeijn, T.R.; Jonkman, M.F.; Knoppers, C.; Pas, H.H.; Diercks, G.F.H. Complement in Bullous Pemphigoid: Results from a Large Observational Study. Br. J. Dermatol. 2017, 176, 517–519. [Google Scholar] [CrossRef]
- Ständer, S.; Holtsche, M.M.; Schmidt, E.; Hammers, C.M.; Zillikens, D.; Ludwig, R.J.; Kridin, K. Presence of Cutaneous Complement Deposition Distinguishes between Immunological and Histological Features of Bullous Pemphigoid—Insights from a Retrospective Cohort Study. J. Clin. Med. 2020, 9, 3928. [Google Scholar] [CrossRef]
- Solimani, F.; Didona, D.; Li, J.; Bao, L.; Patel, P.M.; Gasparini, G.; Kridin, K.; Cozzani, E.; Hertl, M.; Amber, K.T. Characterizing the Proteome of Bullous Pemphigoid Blister Fluid Utilizing Tandem Mass Tag Labeling Coupled with LC–MS/MS. Arch. Dermatol. Res. 2021, 314, 921–928. [Google Scholar] [CrossRef]
- Chiorean, R.M.; Baican, A.; Mustafa, M.B.; Lischka, A.; Leucuta, D.-C.; Feldrihan, V.; Hertl, M.; Sitaru, C. Complement-Activating Capacity of Autoantibodies Correlates with Disease Activity in Bullous Pemphigoid Patients. Front. Immunol. 2018, 9, 2687. [Google Scholar] [CrossRef]
- Ujiie, H.; Sasaoka, T.; Izumi, K.; Nishie, W.; Shinkuma, S.; Natsuga, K.; Nakamura, H.; Shibaki, A.; Shimizu, H. Bullous Pemphigoid Autoantibodies Directly Induce Blister Formation without Complement Activation. J. Immunol. 2014, 193, 4415–4428. [Google Scholar] [CrossRef] [PubMed]
- Dainichi, T.; Nishie, W.; Yamagami, Y.; Sonobe, H.; Ujiie, H.; Kaku, Y.; Kabashima, K. Bullous Pemphigoid Suggestive of Complement-independent Blister Formation with Anti- BP 180 IgG4 Autoantibodies. Br. J. Dermatol. 2016, 175, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Zhang, Y.; Li, N.; Wang, G.; Liu, Z. The Autoimmune Skin Disease Bullous Pemphigoid: The Role of Mast Cells in Autoantibody-Induced Tissue Injury. Front. Immunol. 2018, 9, 407. [Google Scholar] [CrossRef]
- Schmidt, E.; Wehr, B.; Tabengwa, E.M.; Reimer, S.; Bröcker, E.-B.; Zillikens, D. Elevated Expression and Release of Tissue-Type, but Not Urokinase-Type, Plasminogen Activator after Binding of Autoantibodies to Bullous Pemphigoid Antigen 180 in Cultured Human Keratinocytes. Clin. Exp. Immunol. 2004, 135, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Kamio, N.; Aoyama, Y.; Yamamoto, Y.; Hirako, Y.; Owaribe, K.; Kitajima, Y. IgG from Patients with Bullous Pemphigoid Depletes Cultured Keratinocytes of the 180-kDa Bullous Pemphigoid Antigen (Type XVII Collagen) and Weakens Cell Attachment. J. Investig. Dermatol. 2009, 129, 919–926. [Google Scholar] [CrossRef]
- Hiroyasu, S.; Ozawa, T.; Kobayashi, H.; Ishii, M.; Aoyama, Y.; Kitajima, Y.; Hashimoto, T.; Jones, J.C.R.; Tsuruta, D. Bullous Pemphigoid IgG Induces BP180 Internalization via a Macropinocytic Pathway. Am. J. Pathol. 2013, 182, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.; Reimer, S.; Jainta, S.; Bröcker, E.-B.; Zillikens, D.; Kruse, N.; Marinkovich, M.P.; Marinkovich, M.P.; Giudice, G.J. Autoantibodies to BP180 Associated with Bullous Pemphigoid Release Interleukin-6 and Interleukin-8 from Cultured Human Keratinocytes. J. Investig. Dermatol. 2000, 115, 842–848. [Google Scholar] [CrossRef]
- Tukaj, S.; Grüner, D.; Tukaj, C.; Zillikens, D.; Kasperkiewicz, M. Calcitriol Exerts Anti-Inflammatory Effects in Keratinocytes Treated with Autoantibodies from a Patient with Bullous Pemphigoid. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 288–292. [Google Scholar] [CrossRef]
- Dainichi, T.; Chow, Z.; Kabashima, K. IgG4, Complement, and the Mechanisms of Blister Formation in Pemphigus and Bullous Pemphigoid. J. Dermatol. Sci. 2017, 88, 265–270. [Google Scholar] [CrossRef]
- Sitaru, C.; Mihai, S.; Zillikens, D. The Relevance of the IgG Subclass of Autoantibodies for Blister Induction in Autoimmune Bullous Skin Diseases. Arch. Dermatol. Res. 2007, 299, 1–8. [Google Scholar] [CrossRef]
- Lamb, P.M.; Patton, T.; Deng, J.-S. The Predominance of IgG4 in Prodromal Bullous Pemphigoid. Int. J. Dermatol. 2008, 47, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Ohzono, A.; Teye, K.; Numata, S.; Hiroyasu, S.; Tsuruta, D.; Hachiya, T.; Kuroda, K.; Hashiguchi, M.; Kawakami, T.; et al. Detection of IgE Autoantibodies to BP180 and BP230 and Their Relationship to Clinical Features in Bullous Pemphigoid. Br. J. Dermatol. 2017, 177, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, N.; Lüttmann, N.; Huebner, F.; Recke, A.; Karl, I.; Schulze, F.S.; Zillikens, D.; Schmidt, E. Correlation of Serum Levels of IgE Autoantibodies Against BP180 with Bullous Pemphigoid Disease Activity. JAMA Dermatol. 2017, 153, 30. [Google Scholar] [CrossRef] [PubMed]
- Messingham, K.N.; Crowe, T.P.; Fairley, J.A. The Intersection of IgE Autoantibodies and Eosinophilia in the Pathogenesis of Bullous Pemphigoid. Front. Immunol. 2019, 10, 2331. [Google Scholar] [CrossRef]
- Maglie, R.; Solimani, F.; Didona, D.; Pipitò, C.; Antiga, E.; Di Zenzo, G. The Cytokine Milieu of Bullous Pemphigoid: Current and Novel Therapeutic Targets. Front. Med. 2023, 10, 1128154. [Google Scholar] [CrossRef]
- Quaglino, P.; Antiga, E.; Comessatti, A.; Caproni, M.; Nardò, T.; Ponti, R.; Novelli, M.; Osella-Abate, S.; Fabbri, P.; Bernengo, M.G. Circulating CD4+ CD25brightFOXP3+ Regulatory T-Cells Are Significantly Reduced in Bullous Pemphigoid Patients. Arch. Dermatol. Res. 2012, 304, 639–645. [Google Scholar] [CrossRef]
- Antiga, E.; Quaglino, P.; Volpi, W.; Pierini, I.; Del Bianco, E.; Bianchi, B.; Novelli, M.; Savoia, P.; Bernengo, M.G.; Fabbri, P.; et al. Regulatory T Cells in Skin Lesions and Blood of Patients with Bullous Pemphigoid. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 222–230. [Google Scholar] [CrossRef]
- Gambichler, T.; Tsitlakidon, A.; Skrygan, M.; Höxtermann, S.; Susok, L.; Hessam, S. T Regulatory Cells and Other Lymphocyte Subsets in Patients with Bullous Pemphigoid. Clin. Exp. Dermatol. 2017, 42, 632–637. [Google Scholar] [CrossRef]
- Muramatsu, K.; Zheng, M.; Yoshimoto, N.; Ito, T.; Ujiie, I.; Iwata, H.; Shimizu, H.; Ujiie, H. Regulatory T Cell Subsets in Bullous Pemphigoid and Dipeptidyl Peptidase-4 Inhibitor-Associated Bullous Pemphigoid. J. Dermatol. Sci. 2020, 100, 23–30. [Google Scholar] [CrossRef]
- Cao, T.; Shao, S.; Fang, H.; Li, B.; Wang, G. Role of Regulatory Immune Cells and Molecules in Autoimmune Bullous Dermatoses. Front. Immunol. 2019, 10, 1746. [Google Scholar] [CrossRef]
- Didona, D.; Di Zenzo, G. Humoral Epitope Spreading in Autoimmune Bullous Diseases. Front. Immunol. 2018, 9, 779. [Google Scholar] [CrossRef] [PubMed]
- Di Zenzo, G.; Calabresi, V.; Olasz, E.B.; Zambruno, G.; Yancey, K.B. Sequential Intramolecular Epitope Spreading of Humoral Responses to Human BPAG2 in a Transgenic Model. J. Investig. Dermatol. 2010, 130, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, H.; Yoshimoto, N.; Natsuga, K.; Muramatsu, K.; Iwata, H.; Nishie, W.; Shimizu, H. Immune Reaction to Type XVII Collagen Induces Intramolecular and Intermolecular Epitope Spreading in Experimental Bullous Pemphigoid Models. Front. Immunol. 2019, 10, 1410. [Google Scholar] [CrossRef] [PubMed]
- Di Zenzo, G.; Thoma-Uszynski, S.; Calabresi, V.; Fontao, L.; Hofmann, S.C.; Lacour, J.-P.; Sera, F.; Bruckner-Tuderman, L.; Zambruno, G.; Borradori, L.; et al. Demonstration of Epitope-Spreading Phenomena in Bullous Pemphigoid: Results of a Prospective Multicenter Study. J. Investig. Dermatol. 2011, 131, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Mignogna, M.D.; Fortuna, G.; Leuci, S.; Stasio, L.; Mezza, E.; Ruoppo, E. Lichen Planus Pemphigoides, a Possible Example of Epitope Spreading. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontology 2010, 109, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Kwan, J.M.; Ahmed, A.R. Relationship between Radiation Therapy and Bullous Pemphigoid. Dermatology 2014, 229, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Amber, K.T.; Zikry, J.; Hertl, M. A Multi-Hit Hypothesis of Bullous Pemphigoid and Associated Neurological Disease: Is HLA-DQB1*03:01, a Potential Link between Immune Privileged Antigen Exposure and Epitope Spreading? HLA 2017, 89, 127–134. [Google Scholar] [CrossRef]
- Bean, S.F.; Good, R.A.; Windhorst, D.B. Bullous Pemphigoid in an 11-Year-Old Boy. Arch. Dermatol. 1970, 102, 205–208. [Google Scholar] [CrossRef]
- Naranjo, C.A.; Busto, U.; Sellers, E.M.; Sandor, P.; Ruiz, I.; Roberts, E.A.; Janecek, E.; Domecq, C.; Greenblatt, D.J. A Method for Estimating the Probability of Adverse Drug Reactions. Clin. Pharmacol. Ther. 1981, 30, 239–245. [Google Scholar] [CrossRef]
- Karch, F.E.; Lasagna, L. Toward the Operational Identification of Adverse Drug Reactions. Clin. Pharmacol. Ther. 1977, 21, 247–254. [Google Scholar] [CrossRef]
- Tan, C.W.X.; Pang, Y.; Sim, B.; Thirumoorthy, T.; Pang, S.M.; Lee, H.Y. The Association between Drugs and Bullous Pemphigoid. Br. J. Dermatol. 2017, 176, 549–551. [Google Scholar] [CrossRef]
- Vassileva, S. Drug-Induced Pemphigoid: Bullous and Cicatricial. Clin. Dermatol. 1998, 16, 379–387. [Google Scholar] [CrossRef]
- Stavropoulos, P.G.; Soura, E.; Antoniou, C. Drug-Induced Pemphigoid: A Review of the Literature. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-D.; Chen, W.-T.; Chi, C.-C. Association Between Medication Use and Bullous Pemphigoid: A Systematic Review and Meta-Analysis. JAMA Dermatol. 2020, 156, 891. [Google Scholar] [CrossRef]
- Molina, G.E.; Yanovsky, R.L.; Wei, E.X.; Chen, S.T. Missed Drug-Induced Bullous Pemphigoid Leads to Longer Immunosuppression than Recognized Cases: A 9-Year Retrospective Review. J. Am. Acad. Dermatol. 2020, 82, 1255–1258. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, V.; Sacerdoti, G. Pemphigus and Bullous Pemphigoid Due to Drugs. Int. J. Dermatol. 1991, 30, 307–312. [Google Scholar] [CrossRef]
- Borch, J.E.; Andersen, K.E.; Clemmensen, O.; Bindslev-Jensen, C. Drug-Induced Bullous Pemphigoid with Positive Patch Test and in Vitro IgE Sensitization. Acta Derm. Venereol. 2005, 85, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, J.; Hazaz, B.; Sandbank, M. Bullous Pemphigoid Mimicking Bullous Erythema Multiforme: An Untoward Side Effect of Penicillins. J. Am. Acad. Dermatol. 1988, 18, 345–349. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The Incretin System: Glucagon-like Peptide-1 Receptor Agonists and Dipeptidyl Peptidase-4 Inhibitors in Type 2 Diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Bellary, S.; Kyrou, I.; Brown, J.E.; Bailey, C.J. Type 2 Diabetes Mellitus in Older Adults: Clinical Considerations and Management. Nat. Rev. Endocrinol. 2021, 17, 534–548. [Google Scholar] [CrossRef]
- García, M.; Aranburu, M.A.; Palacios-Zabalza, I.; Lertxundi, U.; Aguirre, C. Dipeptidyl Peptidase-IV Inhibitors Induced Bullous Pemphigoid: A Case Report and Analysis of Cases Reported in the European Pharmacovigilance Database. J. Clin. Pharm. Ther. 2016, 41, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Béné, J.; Moulis, G.; Bennani, I.; Auffret, M.; Coupe, P.; Babai, S.; Hillaire-Buys, D.; Micallef, J.; Gautier, S.; the French Association of Regional Pharmaco Vigilance Centres. Bullous Pemphigoid and Dipeptidyl Peptidase IV Inhibitors: A Case-Noncase Study in the French Pharmacovigilance Database. Br. J. Dermatol. 2016, 175, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Varpuluoma, O.; Försti, A.-K.; Jokelainen, J.; Turpeinen, M.; Timonen, M.; Huilaja, L.; Tasanen, K. Vildagliptin Significantly Increases the Risk of Bullous Pemphigoid: A Finnish Nationwide Registry Study. J. Investig. Dermatol. 2018, 138, 1659–1661. [Google Scholar] [CrossRef]
- Benzaquen, M.; Borradori, L.; Berbis, P.; Cazzaniga, S.; Valero, R.; Richard, M.-A.; Feldmeyer, L. Dipeptidyl Peptidase IV Inhibitors, a Risk Factor for Bullous Pemphigoid: Retrospective Multicenter Case-Control Study from France and Switzerland. J. Am. Acad. Dermatol. 2018, 78, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Shimauchi, R.; Nishibori, N.; Kawashima, K.; Oshitani, S.; Fujiya, A.; Shibata, T.; Ohashi, N.; Izumi, K.; Nishie, W.; et al. Dipeptidyl Peptidase-4 Inhibitors-Associated Bullous Pemphigoid: A Retrospective Study of 168 Pemphigoid and 9,304 Diabetes Mellitus Patients. J. Diabetes Investig. 2019, 10, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Kridin, K.; Cohen, A.D. Dipeptidyl-Peptidase IV Inhibitor–Associated Bullous Pemphigoid: A Systematic Review and Meta-Analysis. J. Am. Acad. Dermatol. 2021, 85, 501–503. [Google Scholar] [CrossRef]
- Kuwata, H.; Nishioka, Y.; Noda, T.; Kubo, S.; Myojin, T.; Higashino, T.; Takahashi, Y.; Ishii, H.; Imamura, T. Association between Dipeptidyl Peptidase-4 Inhibitors and Increased Risk for Bullous Pemphigoid within 3 Months from First Use: A 5-year Population-based Cohort Study Using the Japanese National Database. J. Diabetes Investig. 2022, 13, 460–467. [Google Scholar] [CrossRef]
- Lee, S.G.; Lee, H.J.; Yoon, M.S.; Kim, D.H. Association of Dipeptidyl Peptidase 4 Inhibitor Use with Risk of Bullous Pemphigoid in Patients with Diabetes. JAMA Dermatol. 2019, 155, 172. [Google Scholar] [CrossRef]
- Kridin, K.; Avni, O.; Damiani, G.; Tzur Bitan, D.; Onn, E.; Weinstein, O.; Cohen, A.D. Dipeptidyl-Peptidase IV Inhibitor (DPP4i) Confers Increased Odds of Bullous Pemphigoid Even Years after Drug Initiation. Arch. Dermatol. Res. 2022, 315, 33–39. [Google Scholar] [CrossRef]
- Guo, W.; Rathi, S.; Marquez, J.; Smith, H.; Kuruvilla, A.; Tonnesen, M.G.; Salvemini, J.N. Prevalence of Diabetes Mellitus in Bullous Pemphigoid Patients in the Absence of Dipeptidyl Peptidase-4 Inhibitors: A Systematic Review and Meta-Analysis. Arch. Dermatol. Res. 2023, 315, 2207–2213. [Google Scholar] [CrossRef]
- Varpuluoma, O.; Försti, A.-K.; Jokelainen, J.; Turpeinen, M.; Timonen, M.; Tasanen, K.; Huilaja, L. Oral Diabetes Medications Other than Dipeptidyl Peptidase 4 Inhibitors Are Not Associated with Bullous Pemphigoid: A Finnish Nationwide Case-Control Study. J. Am. Acad. Dermatol. 2018, 79, 1034–1038.e5. [Google Scholar] [CrossRef]
- Kridin, K. Comment on “Oral Diabetes Medications Other than Dipeptidyl Peptidase-4 Inhibitors Are Not Associated with Bullous Pemphigoid: A Finnish Nationwide Case Control Study”. J. Am. Acad. Dermatol. 2018, 79, e111–e112. [Google Scholar] [CrossRef] [PubMed]
- Schwager, Z.; Mikailov, A.; Lipworth, A.D. Comment on “Oral Diabetes Medications Other than Dipeptidyl Peptidase-4 Inhibitors Are Not Associated with Bullous Pemphigoid: A Finnish Nationwide Case-Control Study” and a Case Report of Glucagon-like Peptide-1 Receptor Agonist–Induced Bullous Pemphigoid. J. Am. Acad. Dermatol. 2019, 80, e189–e190. [Google Scholar] [CrossRef] [PubMed]
- Lambadiari, V.; Kountouri, A.; Kousathana, F.; Korakas, E.; Kokkalis, G.; Theotokoglou, S.; Palaiodimou, L.; Katsimbri, P.; Ikonomidis, I.; Theodoropoulos, K.; et al. The Association of Bullous Pemphigoid with Dipeptidyl-Peptidase 4 Inhibitors: A Ten-Year Prospective Observational Study. BMC Endocr. Disord. 2021, 21, 23. [Google Scholar] [CrossRef]
- Baetta, R.; Corsini, A. Pharmacology of Dipeptidyl Peptidase-4 Inhibitors: Similarities and Differences. Drugs 2011, 71, 1441–1467. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, H.; Muramatsu, K.; Mushiroda, T.; Ozeki, T.; Miyoshi, H.; Iwata, H.; Nakamura, A.; Nomoto, H.; Cho, K.Y.; Sato, N.; et al. HLA-DQB1*03:01 as a Biomarker for Genetic Susceptibility to Bullous Pemphigoid Induced by DPP-4 Inhibitors. J. Investig. Dermatol. 2018, 138, 1201–1204. [Google Scholar] [CrossRef]
- Oyama, N.; Setterfield, J.F.; Powell, A.M.; Sakuma-Oyama, Y.; Albert, S.; Bhogal, B.S.; Vaughan, R.W.; Kaneko, F.; Challacombe, S.J.; Black, M.M. Bullous Pemphigoid Antigen II (BP180) and Its Soluble Extracellular Domains Are Major Autoantigens in Mucous Membrane Pemphigoid: The Pathogenic Relevance to HLA Class II Alleles and Disease Severity: Circulating Autoantibody Profile of MMP. Br. J. Dermatol. 2006, 154, 90–98. [Google Scholar] [CrossRef]
- Lindgren, O.; Varpuluoma, O.; Tuusa, J.; Ilonen, J.; Huilaja, L.; Kokkonen, N.; Tasanen, K. Gliptin-Associated Bullous Pemphigoid and the Expression of Dipeptidyl Peptidase-4/CD26 in Bullous Pemphigoid. Acta Derm. Venereol. 2019, 99, 602–609. [Google Scholar] [CrossRef]
- Klemann, C.; Wagner, L.; Stephan, M.; Von Hörsten, S. Cut to the Chase: A Review of CD26/Dipeptidyl Peptidase-4′s (DPP4) Entanglement in the Immune System. Clin. Exp. Immunol. 2016, 185, 1–21. [Google Scholar] [CrossRef]
- Forssmann, U.; Stoetzer, C.; Stephan, M.; Kruschinski, C.; Skripuletz, T.; Schade, J.; Schmiedl, A.; Pabst, R.; Wagner, L.; Hoffmann, T.; et al. Inhibition of CD26/Dipeptidyl Peptidase IV Enhances CCL11/Eotaxin-Mediated Recruitment of Eosinophils In Vivo. J. Immunol. 2008, 181, 1120–1127. [Google Scholar] [CrossRef]
- Gonzalez-Gronow, M. Dipeptidyl Peptidase IV (DPP IV/CD26) Is a Cell-Surface Plasminogen Receptor. Front. Biosci. 2008, 13, 1610. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Nishie, W.; Izumi, K.; Shimizu, H. Preferential Reactivity of Dipeptidyl Peptidase-IV Inhibitor-Associated Bullous Pemphigoid Autoantibodies to the Processed Extracellular Domains of BP180. Front. Immunol. 2019, 10, 1224. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.C.; Voith, U.; Schönau, V.; Sorokin, L.; Bruckner-Tuderman, L.; Franzke, C.-W. Plasmin Plays a Role in the In Vitro Generation of the Linear IgA Dermatosis Antigen LADB97. J. Investig. Dermatol. 2009, 129, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, H.; Kurihara, Y.; Funakoshi, T.; Umegaki-Arao, N.; Takahashi, H.; Kubo, A.; Tanikawa, A.; Kodani, N.; Minami, Y.; Meguro, S.; et al. Unique Clinical and Serological Features of Bullous Pemphigoid Associated with Dipeptidyl Peptidase-4 Inhibitors. Br. J. Dermatol. 2018, 178, 1462–1463. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Tsunoda, T.; Sato, F.; Yaguchi, Y.; Igarashi, M.; Izumi, K.; Nishie, W.; Ishii, N.; Okamura, K.; Suzuki, T.; et al. Clinical and Immunological Characterization of 14 Cases of Dipeptidyl Peptidase-4 Inhibitor-associated Bullous Pemphigoid: A Single-centre Study. Br. J. Dermatol. 2020, 182, 806–807. [Google Scholar] [CrossRef]
- García-Díez, I.; Ivars-Lleó, M.; López-Aventín, D.; Ishii, N.; Hashimoto, T.; Iranzo, P.; Pujol, R.M.; España, A.; Herrero-Gonzalez, J.E. Bullous Pemphigoid Induced by Dipeptidyl Peptidase-4 Inhibitors. Eight Cases with Clinical and Immunological Characterization. Int. J. Dermatol. 2018, 57, 810–816. [Google Scholar] [CrossRef]
- Sawada, K.; Sawada, T.; Kobayashi, T.; Fujiki, A.; Matsushita, T.; Kawara, S.; Izumi, K.; Nishie, W.; Shimizu, H.; Takehara, K.; et al. A Case of Anti-BP230 Antibody-Positive Bullous Pemphigoid Receiving DPP-4 Inhibitor. Immunol. Med. 2021, 44, 53–55. [Google Scholar] [CrossRef]
- Fania, L.; Salemme, A.; Provini, A.; Pagnanelli, G.; Collina, M.C.; Abeni, D.; Didona, B.; Di Zenzo, G.; Mazzanti, C. Detection and Characterization of IgG, IgE, and IgA Autoantibodies in Patients with Bullous Pemphigoid Associated with Dipeptidyl Peptidase-4 Inhibitors. J. Am. Acad. Dermatol. 2018, 78, 592–595. [Google Scholar] [CrossRef]
- Yamashita, C.; Arase, N.; Higuchi, S.; Arase, H.; Takagi, J.; Nojima, S.; Tanemura, A.; Fujimoto, M. Serum Autoantibodies against the Extracellular Region of A6β4 Integrin in a Patient with Dipeptidyl Peptidase-4 Inhibitor–Induced Bullous Pemphigoid. JAAD Case Rep. 2022, 20, 65–68. [Google Scholar] [CrossRef]
- Mai, Y.; Nishie, W.; Izumi, K.; Yoshimoto, N.; Morita, Y.; Watanabe, M.; Toyonaga, E.; Ujiie, H.; Iwata, H.; Fujita, Y.; et al. Detection of Anti-BP180 NC16A Autoantibodies after the Onset of Dipeptidyl Peptidase-IV Inhibitor-Associated Bullous Pemphigoid: A Report of Three Patients. Br. J. Dermatol. 2018, 179, 790–791. [Google Scholar] [CrossRef]
- Takama, H.; Yoshida, M.; Izumi, K.; Yanagishita, T.; Muto, J.; Ohshima, Y.; Nishie, W.; Shimizu, H.; Akiyama, M.; Watanabe, D. Dipeptidyl Peptidase-4 Inhibitor-Associated Bullous Pemphigoid: Recurrence with Epitope Spreading. Acta Derm. Venereol. 2018, 98, 983–984. [Google Scholar] [CrossRef]
- García-Díez, I.; España, A.; Iranzo, P. Epitope-spreading Phenomena in Dipeptidyl Peptidase-4 Inhibitor-associated Bullous Pemphigoid. Br. J. Dermatol. 2019, 180, 1267–1268. [Google Scholar] [CrossRef] [PubMed]
- Tasanen, K. Dipeptidyl Peptidase-4 Inhibitor-Associated Bullous Pemphigoid. Front. Immunol. 2019, 10, 1238. [Google Scholar] [CrossRef] [PubMed]
- Giusti, D.; Le Jan, S.; Gatouillat, G.; Bernard, P.; Pham, B.N.; Antonicelli, F. Biomarkers Related to Bullous Pemphigoid Activity and Outcome. Exp. Dermatol. 2017, 26, 1240–1247. [Google Scholar] [CrossRef]
- Hung, C.-T.; Chang, Y.-L.; Wang, W.-M. Dipeptidyl Peptidase-4 Inhibitor-Related Bullous Pemphigoid: Clinical, Laboratory, and Histological Features, and Possible Pathogenesis. Int. J. Mol. Sci. 2022, 23, 14101. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Cai, L.; Li, W.; Zhang, L.; Guo, S.; Zhang, R.; Zheng, Y.; Liu, X.; Wang, M.; Zhou, X.; et al. DPP-4 Inhibitors Improve Diabetic Wound Healing via Direct and Indirect Promotion of Epithelial-Mesenchymal Transition and Reduction of Scarring. Diabetes 2018, 67, 518–531. [Google Scholar] [CrossRef]
- Tasanen, K.; Tunggal, L.; Chometon, G.; Bruckner-Tuderman, L.; Aumailley, M. Keratinocytes from Patients Lacking Collagen XVII Display a Migratory Phenotype. Am. J. Pathol. 2004, 164, 2027–2038. [Google Scholar] [CrossRef]
- Nishie, W. Dipeptidyl Peptidase IV Inhibitor-Associated Bullous Pemphigoid: A Recently Recognized Autoimmune Blistering Disease with Unique Clinical, Immunological and Genetic Characteristics. Immunol. Med. 2019, 42, 22–28. [Google Scholar] [CrossRef]
- Nozawa, K.; Suzuki, T.; Kayanuma, G.; Yamamoto, H.; Nagayasu, K.; Shirakawa, H.; Kaneko, S. Lisinopril Prevents Bullous Pemphigoid Induced by Dipeptidyl Peptidase 4 Inhibitors via the Mas Receptor Pathway. Front. Immunol. 2023, 13, 1084960. [Google Scholar] [CrossRef]
- Ständer, S.; Schmidt, E.; Zillikens, D.; Ludwig, R.J.; Kridin, K. More Severe Erosive Phenotype Despite Lower Circulating Autoantibody Levels in Dipeptidyl Peptidase-4 Inhibitor (DPP4i)-Associated Bullous Pemphigoid: A Retrospective Cohort Study. Am. J. Clin. Dermatol. 2021, 22, 117–127. [Google Scholar] [CrossRef]
- He, Y.-L.; Sabo, R.; Campestrini, J.; Wang, Y.; Riviere, G.-J.; Nielsen, J.C.; Rosenberg, M.; Ligueros-Saylan, M.; Howard, D.; Dole, W.P. The Effect of Age, Gender, and Body Mass Index on the Pharmacokinetics and Pharmacodynamics of Vildagliptin in Healthy Volunteers. Br. J. Clin. Pharmacol. 2008, 65, 338–346. [Google Scholar] [CrossRef]
- Kinyó, Á.; Hanyecz, A.; Lengyel, Z.; Várszegi, D.; Oláh, P.; Gyömörei, C.; Kálmán, E.; Berki, T.; Gyulai, R. Clinical, Laboratory and Histological Features of Dipeptidyl Peptidase-4 Inhibitor Related Noninflammatory Bullous Pemphigoid. J. Clin. Med. 2021, 10, 1916. [Google Scholar] [CrossRef] [PubMed]
- Chijiwa, C.; Takeoka, S.; Kamata, M.; Tateishi, M.; Fukaya, S.; Hayashi, K.; Fukuyasu, A.; Tanaka, T.; Ishikawa, T.; Ohnishi, T.; et al. Decrease in Eosinophils Infiltrating into the Skin of Patients with Dipeptidyl Peptidase-4 Inhibitor-Related Bullous Pemphigoid. J. Dermatol. 2018, 45, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Patsatsi, A.; Kyriakou, A.; Meltzanidou, P.; Trigoni, A.; Lamprou, F.; Kokolios, M.; Giannakou, A. Βullous Pemphigoid in Patients with DPP-4 Inhibitors at the Onset of Disease: Does This Differ from Common Bullous Pemphigoid? Eur. J. Dermatol. EJD 2018, 28, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Kridin, K. Dipeptidyl-peptidase IV Inhibitors (DPP4i)-associated Bullous Pemphigoid: Estimating the Clinical Profile and Exploring Intraclass Differences. Dermatol. Ther. 2020, 33, e13790. [Google Scholar] [CrossRef] [PubMed]
- Kridin, K.; Bergman, R. Association of Bullous Pemphigoid with Dipeptidyl-Peptidase 4 Inhibitors in Patients with Diabetes: Estimating the Risk of the New Agents and Characterizing the Patients. JAMA Dermatol. 2018, 154, 1152–1158. [Google Scholar] [CrossRef]
- Patsatsi, A.; Vyzantiadis, T.-A.; Chrysomallis, F.; Devliotou-Panagiotidou, D.; Sotiriadis, D. Medication History of a Series of Patients with Bullous Pemphigoid from Northern Greece—Observations and Discussion. Int. J. Dermatol. 2009, 48, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Plaquevent, M.; Tétart, F.; Fardet, L.; Ingen-Housz-Oro, S.; Valeyrie-Allanore, L.; Bernard, P.; Hebert, V.; Roussel, A.; Avenel-Audran, M.; Chaby, G.; et al. Higher Frequency of Dipeptidyl Peptidase-4 Inhibitor Intake in Bullous Pemphigoid Patients than in the French General Population. J. Investig. Dermatol. 2019, 139, 835–841. [Google Scholar] [CrossRef]
- Armanious, M.; AbuHilal, M. Gliptin-Induced Bullous Pemphigoid: Canadian Case Series of 10 Patients. J. Cutan. Med. Surg. 2021, 25, 163–168. [Google Scholar] [CrossRef]
- Quach, H.T.; Johnson, D.B.; LeBoeuf, N.R.; Zwerner, J.P.; Dewan, A.K. Cutaneous Adverse Events Caused by Immune Checkpoint Inhibitors. J. Am. Acad. Dermatol. 2021, 85, 956–966. [Google Scholar] [CrossRef]
- Merli, M.; Accorinti, M.; Romagnuolo, M.; Marzano, A.; Di Zenzo, G.; Moro, F.; Antiga, E.; Maglie, R.; Cozzani, E.; Parodi, A.; et al. Autoimmune Bullous Dermatoses in Cancer Patients Treated by Immunotherapy: A Literature Review and Italian Multicentric Experience. Front. Med. 2023, 10, 1208418. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Chiu, M.N.; Pilkhwal Sah, S. Adverse Cutaneous Toxicities by PD-1/PD-L1 Immune Checkpoint Inhibitors: Pathogenesis, Treatment, and Surveillance. Cutan. Ocul. Toxicol. 2022, 41, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Kawsar, A.; Edwards, C.; Patel, P.; Heywood, R.M.; Gupta, A.; Mann, J.; Harland, C.; Heelan, K.; Larkin, J.; Lorigan, P.; et al. Checkpoint Inhibitor-Associated Bullous Cutaneous Immune-Related Adverse Events: A Multicentre Observational Study. Br. J. Dermatol. 2022, 187, 981–987. [Google Scholar] [CrossRef]
- Said, J.T.; Liu, M.; Talia, J.; Singer, S.B.; Semenov, Y.R.; Wei, E.X.; Mostaghimi, A.; Nelson, C.A.; Giobbie-Hurder, A.; LeBoeuf, N.R. Risk Factors for the Development of Bullous Pemphigoid in US Patients Receiving Immune Checkpoint Inhibitors. JAMA Dermatol. 2022, 158, 552. [Google Scholar] [CrossRef]
- Siegel, J.; Totonchy, M.; Damsky, W.; Berk-Krauss, J.; Castiglione, F.; Sznol, M.; Petrylak, D.P.; Fischbach, N.; Goldberg, S.B.; Decker, R.H.; et al. Bullous Disorders Associated with Anti–PD-1 and Anti–PD-L1 Therapy: A Retrospective Analysis Evaluating the Clinical and Histopathologic Features, Frequency, and Impact on Cancer Therapy. J. Am. Acad. Dermatol. 2018, 79, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Molle, M.F.; Capurro, N.; Herzum, A.; Micalizzi, C.; Cozzani, E.; Parodi, A. Self-resolving Bullous Pemphigoid Induced by Cemiplimab. Dermatol. Ther. 2022, 35, e15466. [Google Scholar] [CrossRef]
- Mazumder, A.; Darji, K.; Smith, K.; Guo, M. Two Rare Cases of Bullous Pemphigoid Associated with Immune Checkpoint Inhibitors. BMJ Case Rep. 2022, 15, e253059. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Asdourian, M.S.; Shah, N.; Jacoby, T.V.; Reynolds, K.L.; Chen, S.T. Association of Bullous Pemphigoid with Immune Checkpoint Inhibitor Therapy in Patients with Cancer: A Systematic Review. JAMA Dermatol. 2022, 158, 933. [Google Scholar] [CrossRef]
- Ye, Y.; Jing, Y.; Li, L.; Mills, G.B.; Diao, L.; Liu, H.; Han, L. Sex-Associated Molecular Differences for Cancer Immunotherapy. Nat. Commun. 2020, 11, 1779. [Google Scholar] [CrossRef]
- Jang, S.R.; Nikita, N.; Banks, J.; Keith, S.W.; Johnson, J.M.; Wilson, M.; Lu-Yao, G. Association Between Sex and Immune Checkpoint Inhibitor Outcomes for Patients with Melanoma. JAMA Netw. Open 2021, 4, e2136823. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Chung, J.H.; Jo, S.J. Pruritic Bullous Skin Eruption in a Male Patient Receiving Immunotherapy for Oropharyngeal Cancer. Int. J. Dermatol. 2020, 59, 685–686. [Google Scholar] [CrossRef] [PubMed]
- Tsiogka, A.; Bauer, J.; Patsatsi, A. Bullous Pemphigoid Associated with Anti-Programmed Cell Death Protein 1 and Anti-Programmed Cell Death Ligand 1 Therapy: A Review of the Literature. Acta Derm. Venereol. 2021, 101, adv00377. [Google Scholar] [CrossRef]
- Gresham, L.M.; Kirchhof, M.G. A Case of Drug-Induced Bullous Pemphigoid Secondary to Immunotherapy Treated with Upadacitinib: A Case Report. SAGE Open Med. Case Rep. 2023, 11, 2050313X2311609. [Google Scholar] [CrossRef] [PubMed]
- Muntyanu, A.; Netchiporouk, E.; Gerstein, W.; Gniadecki, R.; Litvinov, I.V. Cutaneous Immune-Related Adverse Events (irAEs) to Immune Checkpoint Inhibitors: A Dermatology Perspective on Management. J. Cutan. Med. Surg. 2021, 25, 59–76. [Google Scholar] [CrossRef]
- Morris, L.M.; Lewis, H.A.; Cornelius, L.A.; Chen, D.Y.; Rosman, I.S. Neutrophil-predominant Bullous Pemphigoid Induced by Checkpoint Inhibitors: A Case Series. J. Cutan. Pathol. 2020, 47, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.T.; Geskin, L. A Case of Nivolumab-Induced Bullous Pemphigoid: Review of Dermatologic Toxicity Associated with Programmed Cell Death Protein-1/Programmed Death Ligand-1 Inhibitors and Recommendations for Diagnosis and Management. Oncologist 2018, 23, 1119–1126. [Google Scholar] [CrossRef]
- Nelson, C.A.; Singer, S.; Chen, T.; Puleo, A.E.; Lian, C.G.; Wei, E.X.; Giobbie-Hurder, A.; Mostaghimi, A.; LeBoeuf, N.R. Bullous Pemphigoid after Anti–Programmed Death-1 Therapy: A Retrospective Case-Control Study Evaluating Impact on Tumor Response and Survival Outcomes. J. Am. Acad. Dermatol. 2022, 87, 1400–1402. [Google Scholar] [CrossRef]
- Tang, K.; Seo, J.; Tiu, B.C.; Le, T.K.; Pahalyants, V.; Raval, N.S.; Ugwu-Dike, P.O.; Zubiri, L.; Naranbhai, V.; Carrington, M.; et al. Association of Cutaneous Immune-Related Adverse Events with Increased Survival in Patients Treated with Anti–Programmed Cell Death 1 and Anti–Programmed Cell Death Ligand 1 Therapy. JAMA Dermatol. 2022, 158, 189. [Google Scholar] [CrossRef]
- Hofmann, M.; Audring, H.; Sterry, W.; Trefzer, U. Interleukin-2-Associated Bullous Drug Dermatosis. Dermatology 2005, 210, 74–75. [Google Scholar] [CrossRef]
- Stingeni, L.; Bianchi, L.; Minotti, V.; Lisi, P. Erlotinib-Induced Bullous Pemphigoid. J. Am. Acad. Dermatol. 2012, 67, e199–e201. [Google Scholar] [CrossRef]
- Atzori, L.; Conti, B.; Zucca, M.; Pau, M. Bullous Pemphigoid Induced by M-TOR Inhibitors in Renal Transplant Recipients. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1626–1630. [Google Scholar] [CrossRef] [PubMed]
- De Simone, C.; Caldarola, G.; Castriota, M.; Salerno, M.P.; Citterio, F. Bullous Pemphigoid in a Transplant Recipient: Is This a Sign of Allograft Rejection? Eur. J. Dermatol. 2012, 22, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Satta, R.; Onnis, G.; Gunnella, S.; Montesu, M.A.; Agnoletti, A.F.; Cozzani, E. Dabrafenib-Induced Pemphigoid-like Reaction. Clin. Exp. Dermatol. 2018, 43, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.; Hill, A.; Fearfield, L.; Johnston, S.; Parton, M.; Heelan, K. Cutaneous Toxicities Occurring during Palbociclib (CDK4/6 Inhibitor) and Endocrine Therapy in Patients with Advanced Breast Cancer: A Single-Centre Experience. Breast Cancer Res. Treat. 2021, 188, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Husein-ElAhmed, H.; Steinhoff, M. Bullous Pemphigoid Induced by Biologic Drugs in Psoriasis: A Systematic Review. J. Dermatol. Treat. 2022, 33, 2886–2893. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.-H.; Zuo, Y.-G. Paradoxical Phenomena of Bullous Pemphigoid Induced and Treated by Identical Biologics. Front. Immunol. 2023, 13, 1050373. [Google Scholar] [CrossRef]
- Genovese, G.; Di Zenzo, G.; Cozzani, E.; Berti, E.; Cugno, M.; Marzano, A.V. New Insights into the Pathogenesis of Bullous Pemphigoid: 2019 Update. Front. Immunol. 2019, 10, 1506. [Google Scholar] [CrossRef]
- Rhodes, L.E.; Hashim, A.; McLaughlin, P.J.; Friedmann, P.S. Blister Fluid Cytokines in Cutaneous Inflammatory Bullous Disorders. Acta Derm Venereol. 1999, 79, 288–290. [Google Scholar] [CrossRef]
- Ameglio, F.; D’Auria, L.; Cordiali-Fei, P.; Mussi, A.; Valenzano, L.; D’Agosto, G.; Ferraro, C.; Bonifati, C.; Giacalone, B. Bullous Pemphigoid and Pemphigus Vulgaris: Correlated Behaviour of Serum VEGF, sE-Selectin and TNF-Alpha Levels. J. Biol. Regul. Homeost. Agents 1997, 11, 148–153. [Google Scholar]
- Liu, L.Y.; Bates, M.E.; Jarjour, N.N.; Busse, W.W.; Bertics, P.J.; Kelly, E.A.B. Generation of Th1 and Th2 Chemokines by Human Eosinophils: Evidence for a Critical Role of TNF-α. J. Immunol. 2007, 179, 4840–4848. [Google Scholar] [CrossRef] [PubMed]
- Romagnuolo, M.; Moltrasio, C.; Iannone, C.; Gattinara, M.; Cambiaghi, S.; Marzano, A.V. Pyoderma Gangrenosum Following Anti-TNF Therapy in Chronic Recurrent Multifocal Osteomyelitis: Drug Reaction or Cutaneous Manifestation of the Disease? A Critical Review on the Topic with an Emblematic Case Report. Front. Med. 2023, 10, 1197273. [Google Scholar] [CrossRef] [PubMed]
- Gounni Abdelilah, S.; Wellemans, V.; Agouli, M.; Guenounou, M.; Hamid, Q.; Beck, L.A.; Lamkhioued, B. Increased Expression of Th2-Associated Chemokines in Bullous Pemphigoid Disease. Role of Eosinophils in the Production and Release of These Chemokines. Clin. Immunol. 2006, 120, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.-H.; Tsai, T.-F. Development of Bullous Pemphigoid during Secukinumab Treatment for Psoriasis. J. Dermatol. 2017, 44, e220–e221. [Google Scholar] [CrossRef]
- Giusti, D.; Bini, E.; Terryn, C.; Didier, K.; Le Jan, S.; Gatouillat, G.; Durlach, A.; Nesmond, S.; Muller, C.; Bernard, P.; et al. NET Formation in Bullous Pemphigoid Patients with Relapse Is Modulated by IL-17 and IL-23 Interplay. Front. Immunol. 2019, 10, 701. [Google Scholar] [CrossRef]
- Maronese, C.A.; Cassano, N.; Genovese, G.; Foti, C.; Vena, G.A.; Marzano, A.V. The Intriguing Links between Psoriasis and Bullous Pemphigoid. J. Clin. Med. 2022, 12, 328. [Google Scholar] [CrossRef] [PubMed]
- Warner, C.; Kwak, Y.; Glover, M.H.B.; Davis, L.S. Bullous Pemphigoid Induced by Hydrochlorothiazide Therapy. J. Drugs Dermatol. JDD 2014, 13, 360–362. [Google Scholar]
- Tanaka, H.; Ishii, T. Analysis of Patients with Drug-induced Pemphigoid Using the Japanese Adverse Drug Event Report Database. J. Dermatol. 2019, 46, 240–244. [Google Scholar] [CrossRef]
- Lloyd-Lavery, A.; Chi, C.-C.; Wojnarowska, F.; Taghipour, K. The Associations Between Bullous Pemphigoid and Drug Use: A UK Case-Control Study. JAMA Dermatol. 2013, 149, 58. [Google Scholar] [CrossRef]
- Takeichi, S.; Kubo, Y.; Arase, S.; Hashimoto, T.; Ansai, S. Brunsting-Perry Type Localized Bullous Pemphigoid, Possibly Induced by Furosemide Administration and Sun Exposure. Eur. J. Dermatol. 2009, 19, 500–503. [Google Scholar] [CrossRef]
- Lee, J.J.; Downham, T.F. Furosemide-Induced Bullous Pemphigoid: Case Report and Review of Literature. J. Drugs Dermatol. JDD 2006, 5, 562–564. [Google Scholar] [PubMed]
- Ambur, A.B.; Nathoo, R.; Saeed, S. A Case of Drug-Induced Bullous Pemphigoid with an Isomorphic Response and Updated Review of Koebnerization in Bullous Diseases. Cureus 2021, 13, e20647. [Google Scholar] [CrossRef] [PubMed]
- Boulinguez, B.; Bernard, B.; Bedane, B.; Le Brun, L.B.; Bonnetblanc, B. Bonnetblanc Bullous Pemphigoid Induced by Bumetanide. Br. J. Dermatol. 1998, 138, 548–549. [Google Scholar] [CrossRef]
- Wurtz, P.; Borucki, R.; Georgesen, C. A Case of Bullous Pemphigoid Induced by Torsemide. JAAD Case Rep. 2022, 26, 95–97. [Google Scholar] [CrossRef] [PubMed]
- García Sanchez, V.C.; Calle Romero, Y.; De La Peña Parra, E.; Lorenzo Borda, S. Penfigoide ampolloso inducido por hidroclorotiazida. SEMERGEN—Med. Fam. 2013, 39, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Cozzani, E.; Muracchioli, A.; Russo, R.; Burlando, M.; Parodi, A. Acetazolamide: A New Trigger for Bullous Pemphigoid? Eur. J. Dermatol. 2020, 30, 321–322. [Google Scholar] [CrossRef]
- Varpuluoma, O.; Jokelainen, J.; Försti, A.-K.; Turpeinen, M.; Timonen, M.; Huilaja, L.; Tasanen, K. Drugs Used for Neurologic and Psychiatric Conditions Increase the Risk for Bullous Pemphigoid: A Case–Control Study. J. Am. Acad. Dermatol. 2019, 81, 250–253. [Google Scholar] [CrossRef]
- Arslan, D.; Aksakal, A.B.; Erdem, Ö.; Tuncer, M.A. A Case of Drug-Induced Bullous Pemphigoid Associated with Teriflunomide: A Patient with Relapsing Multiple Sclerosis. Mult. Scler. Relat. Disord. 2020, 43, 102157. [Google Scholar] [CrossRef]
- Kridin, K.; Zelber-Sagi, S.; Kridin, M.; Cohen, A.D. Bullous Pemphigoid and Neuropsychiatric Medications: An Influence of Drugs or of Underlying Conditions? J. Am. Acad. Dermatol. 2023, 88, e137. [Google Scholar] [CrossRef]
- Ballout, R.A.; Musharrafieh, U.; Khattar, J. Lisinopril-associated Bullous Pemphigoid in an Elderly Woman: A Case Report of a Rare Adverse Drug Reaction. Br. J. Clin. Pharmacol. 2018, 84, 2678–2682. [Google Scholar] [CrossRef]
- Angelis, E.; Lombardi, M.L.; Grassi, M.; Ruocco, V. Enalapril: A Powerful in Vitro Non-Thiol Acantholytic Agent. Int. J. Dermatol. 1992, 31, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Mallet, L.; Cooper, J.W.; Thomas, J. Bullous Pemphigoid Associated with Captopril. DICP 1989, 23, 63. [Google Scholar] [CrossRef]
- Saraceno, R.; Citarella, L.; Spallone, G.; Chimenti, S. A Biological Approach in a Patient with Psoriasis and Bullous Pemphigoid Associated with Losartan Therapy. Clin. Exp. Dermatol. 2008, 33, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Cao, Y.; Zeng, X.; Song, X. Valsartan-associated Bullous Pemphigoid Initially Presenting as Erythema Multiforme. Health Sci. Rep. 2021, 4, e452. [Google Scholar] [CrossRef] [PubMed]
- Park, K.Y.; Kim, B.J.; Kim, M.N. Amlodipine-Associated Bullous Pemphigoid with Erythema Multiforme-like Clinical Features: Correspondence. Int. J. Dermatol. 2011, 50, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Ameen, M.; Harman, K.E.; Black, M.M. Pemphigoid Nodularis Associated with Nifedipine. Br. J. Dermatol. 2000, 142, 575–577. [Google Scholar] [CrossRef]
- Brenner, S.; Ruocco, V.; Bialy-Golan, A.; Tur, E.; Flaminio, C.; Ruocco, E.; Lombardi, M.L. Pemphigus and Pemphigoid-like Effects of Nifedipine on in Vitro Cultured Normal Human Skin Explants: Pemphigus and Pemphigoid-like Effects of Nifedipine. Int. J. Dermatol. 1999, 38, 36–40. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Zafiriou, E.; Koukoulis, G.K.; Roussaki-Schulze, A.V. Drugs Associated with Bullous Pemphigoid: Role of HΜG- CO a Reductase Inhibitors. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e269–e270. [Google Scholar] [CrossRef]
- Chang, T.-H.; Wu, C.-Y.; Chang, Y.-T.; Lin, Y.-H.; Wu, C.-Y. The Association between Statins and Subsequent Risk of Bullous Pemphigoid: A Population-Based Cohort Study. JAAD Int. 2021, 3, 23–25. [Google Scholar] [CrossRef]
- Ma, H.-J.; Hu, R.; Jia, C.-Y.; Yang, Y.; Song, L.-J. Case of Drug-Induced Bullous Pemphigoid by Levofloxacin. J. Dermatol. 2012, 39, 1086–1087. [Google Scholar] [CrossRef]
- Cozzani, E.; Chinazzo, C.; Burlando, M.; Romagnoli, M.; Parodi, A. Ciprofloxacin as a Trigger for Bullous Pemphigoid: The Second Case in the Literature. Am. J. Ther. 2016, 23, e1202–e1204. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs Associated with Bullous Pemphigoid Development | |||
|---|---|---|---|
| Dipeptidyl peptidase 4 inhibitors | Diuretics | Immune checkpoint inhibitors | Biologic agents |
| Alogliptin | Acetazolamide | Atezolizumab | Adalimumab |
| Anagliptin | Bumetanide | Cemiplimab | Efalizumab |
| Linagliptin | Furosemide | Durvalumab | Etanercept |
| Saxagliptin | Hydrochlorothiazide | Ipilimumab | Guselkumab |
| Sitagliptin | Spironolactone | Nivolumab | Infliximab |
| Teneligliptin | Torsemide | Pembrolizumab | Secukinumab |
| Vildagliptin | Ustekinumab | ||
| Cardiovascular drugs | Neurological drugs | Antimicrobial agents | Anti-inflammatory drugs and salicylates |
| Amiodarone | Amantadine | Actinomycin | Aspirin |
| Amlodipine | Doxepin | Amoxicilin | Azapropazone |
| Atenolol | Escitalopram | Ampicilin | Celecoxib |
| Captopril | Fluoxetin | Cephalexin | Ibuprofen |
| Enalapril | Flupenthixol | Ciprofloxacin | Mefenamic acid |
| Lisinopril | Gabapentin | Chloroquine | Mesalazine |
| Losartan | Galantamine | Dactinomycin | Metamizole |
| Nadolol | Levetiracetam | Griseofulvin | Phenacetin |
| Nifedipine | Risperidone | Levofloxacin | Sulfasalazine |
| Practolol | Teriflunomide | Metronidazole | Salicylazosulphapyridine |
| Rosuvastatin | Penicillin | ||
| Valsartan | Rifampicin | ||
| Sulfonamide | |||
| Terbinafine | |||
| Other drugs | Topical drugs | ||
| Aldesleukin | Palbociclib | 5-Fluorouracil | |
| Arsenic | Potassium iodide | Anthralin | |
| D-Penicillamine | Psoralens with ultraviolet A (PUVA) | Benzyl benzoate | |
| Dabrafenib | Coal tar | ||
| Enoxaparin | Serratiopeptidase | Diclofenac | |
| Erlotinib | Sirolimus | Dorzolamide | |
| Everolimus | Tiobutarit | Iodophor adhesive band | |
| Omeprazole | Timolol | ||
| Drug-Induced BP | Drug-Triggered BP | |
|---|---|---|
| Different clinical entity | Similar to classic BP | |
| Clinical phenotype | Non-inflammatory BP | Inflammatory BP |
| Latency period after DPP4i initiation | Shorter; higher risk in the first 3 months | Longer; even more than 6 years after initiation |
| Autoantibody profile | Anti-BP180 full length, associated or not with anti-BP180 LAD-1 and LABD97 | Anti-BP180 NC16A |
| Eosinophils in skin biopsies | Decreased | Moderate infiltrate |
| Eosinophils in serum | Decreased | Augmented |
| DPP4i withdrawal | Mandatory | Recommendable |
| Clinical course | Less likely to persist after drug withdrawal | More likely to persist after drug withdrawal |
| Summary of Drugs Associated with Bullous Pemphigoid Development | |
|---|---|
| Strongly associated | Anecdotally associated |
| Dipeptidyl peptidase 4 inhibitors Immune checkpoint inhibitors Biologic agents (anti-TNF-α, anti-IL17/23) | Diuretics Neurological drugs Cardiovascular drugs Antimicrobial agents Anti-inflammatory drugs and salicylates Other drugs (D-Penicillamine, antineoplastic agents, topical drugs) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Nicolas-Ruanes, B.; Ballester-Martinez, A.; Garcia-Mouronte, E.; Berna-Rico, E.; Azcarraga-Llobet, C.; Fernandez-Guarino, M. From Molecular Insights to Clinical Perspectives in Drug-Associated Bullous Pemphigoid. Int. J. Mol. Sci. 2023, 24, 16786. https://doi.org/10.3390/ijms242316786
de Nicolas-Ruanes B, Ballester-Martinez A, Garcia-Mouronte E, Berna-Rico E, Azcarraga-Llobet C, Fernandez-Guarino M. From Molecular Insights to Clinical Perspectives in Drug-Associated Bullous Pemphigoid. International Journal of Molecular Sciences. 2023; 24(23):16786. https://doi.org/10.3390/ijms242316786
Chicago/Turabian Stylede Nicolas-Ruanes, Belen, Asuncion Ballester-Martinez, Emilio Garcia-Mouronte, Emilio Berna-Rico, Carlos Azcarraga-Llobet, and Montserrat Fernandez-Guarino. 2023. "From Molecular Insights to Clinical Perspectives in Drug-Associated Bullous Pemphigoid" International Journal of Molecular Sciences 24, no. 23: 16786. https://doi.org/10.3390/ijms242316786