
Mutant-Huntingtin Molecular Pathways Elucidate New Targets for Drug Repurposing

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HD Mechanisms
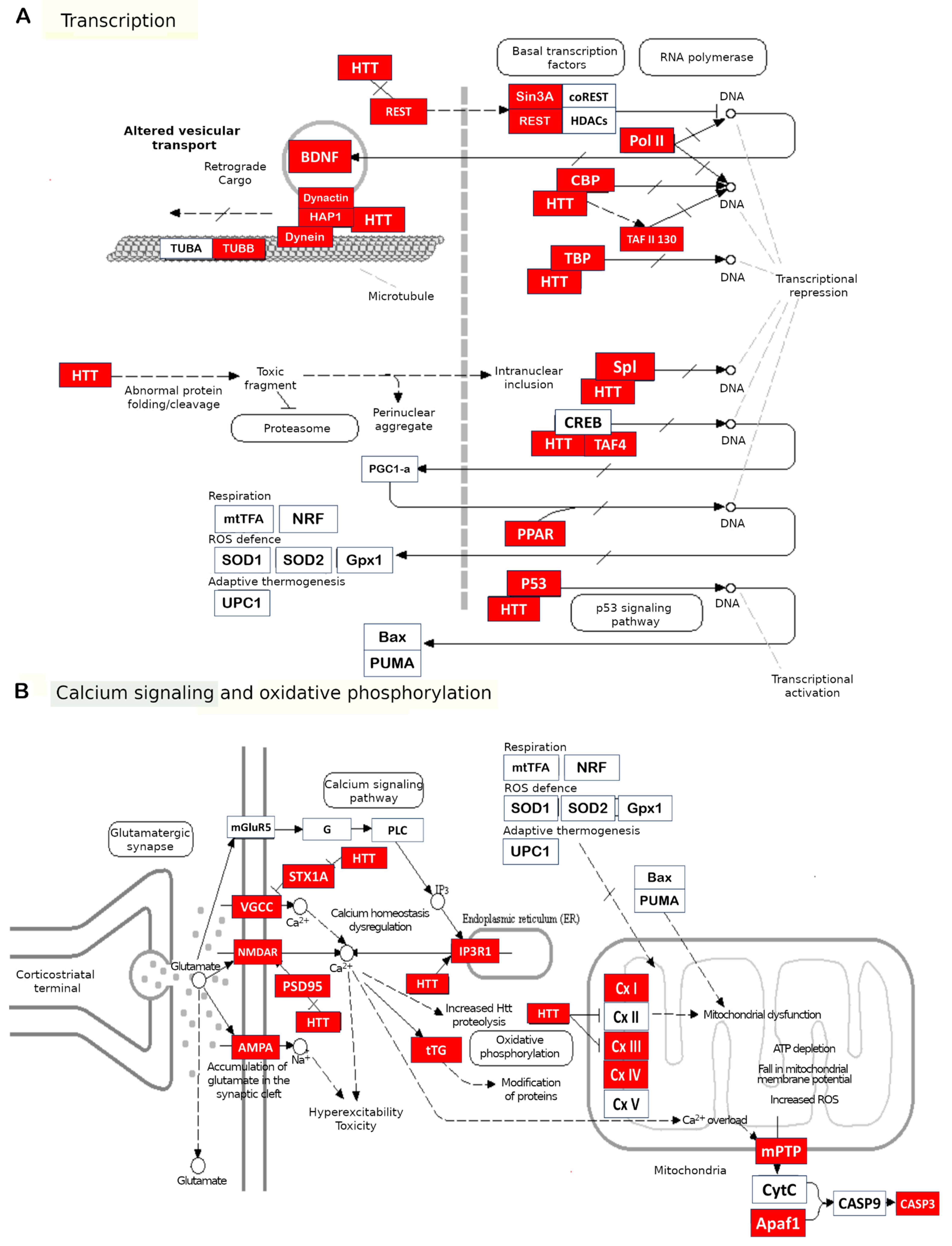
2.1. Transcription Dysfunction
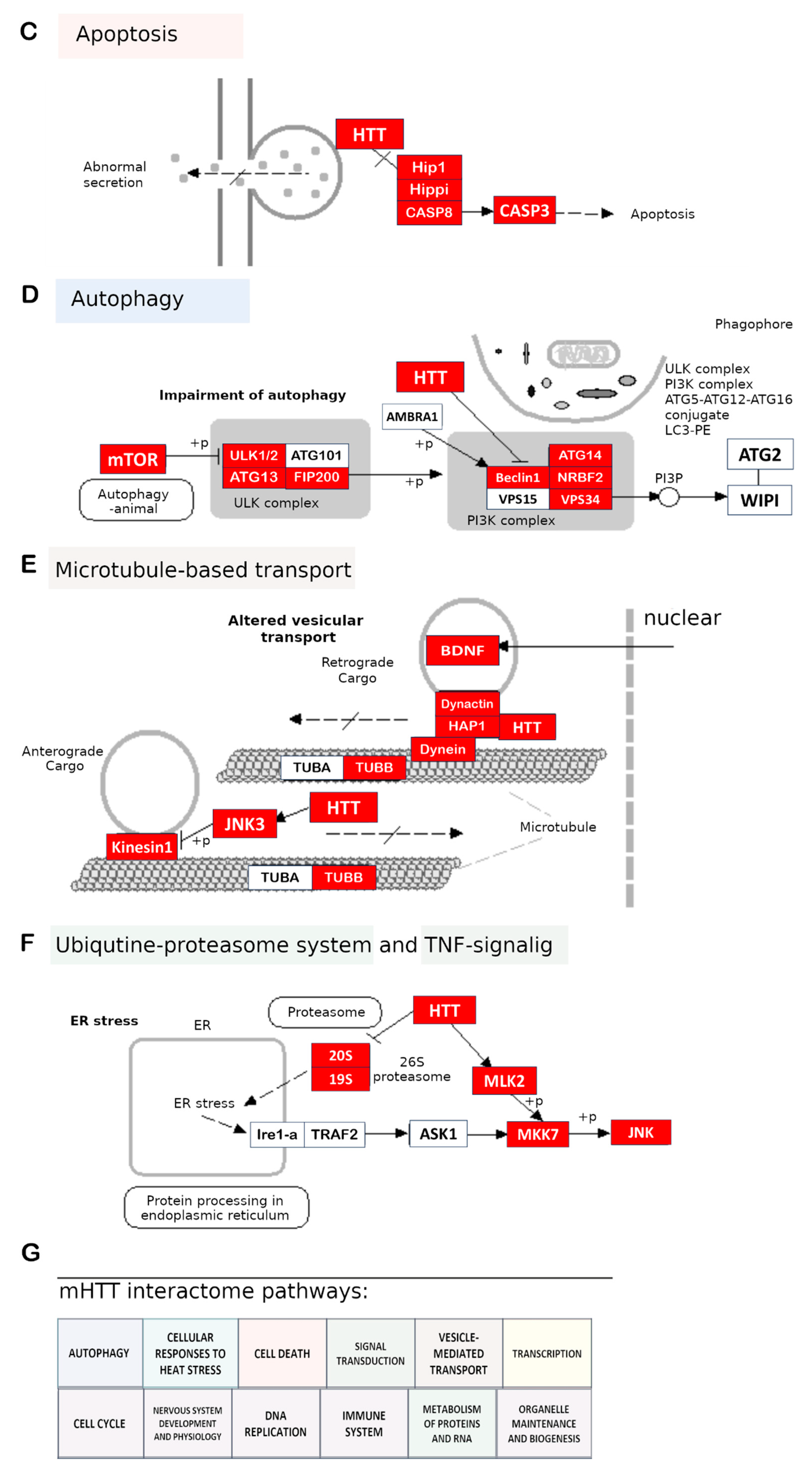
2.2. Systems of Clearance of Proteins and Other Cell Components
2.3. Cytoskeleton Impairment: Intracellular Transport and Synaptic Transmission
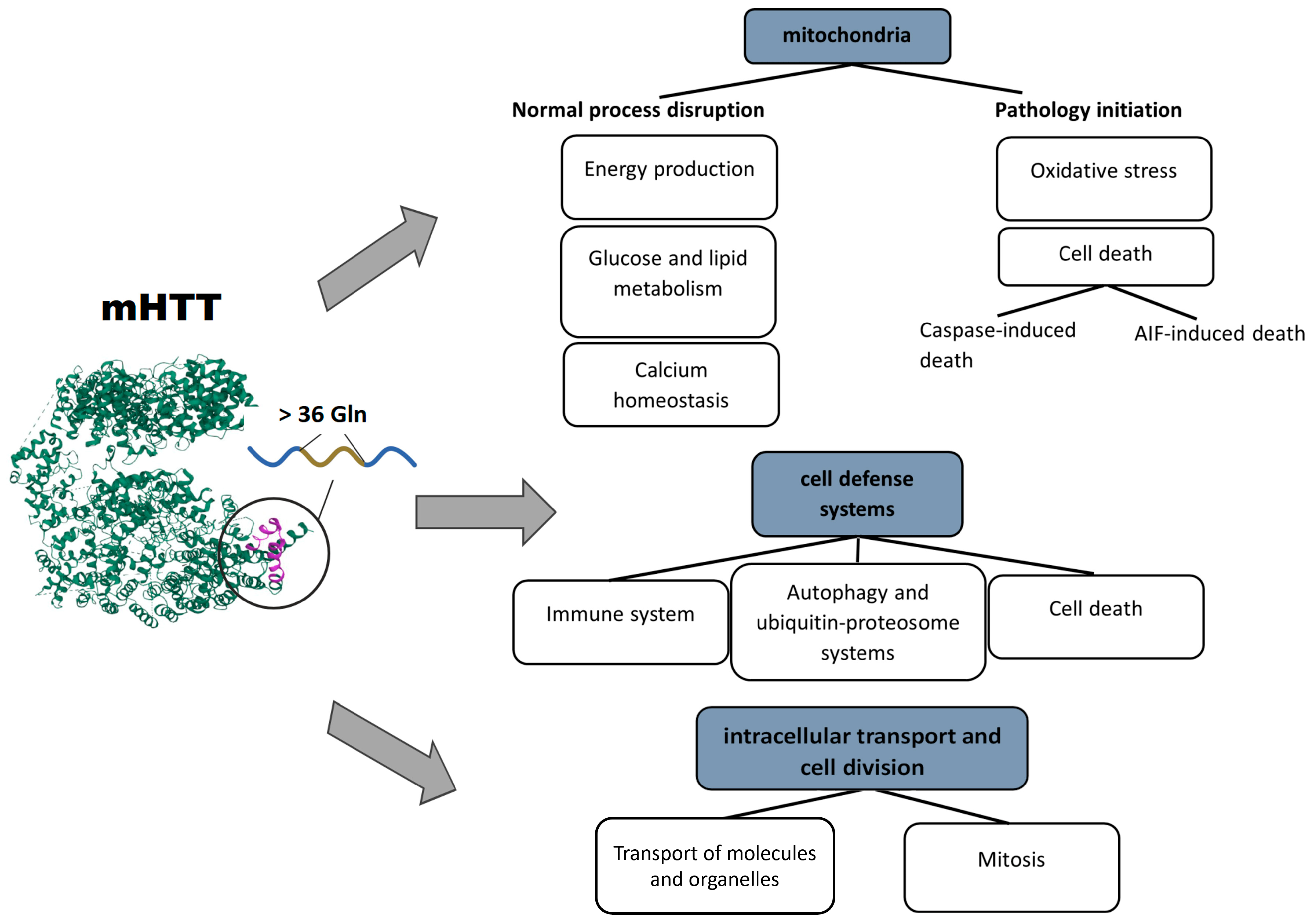
2.4. Mitochondrial Dysfunction
2.5. Cell Death
2.6. The Role of Astrocytes
2.7. Molecular Pathways of HD
2.8. HD Treatment
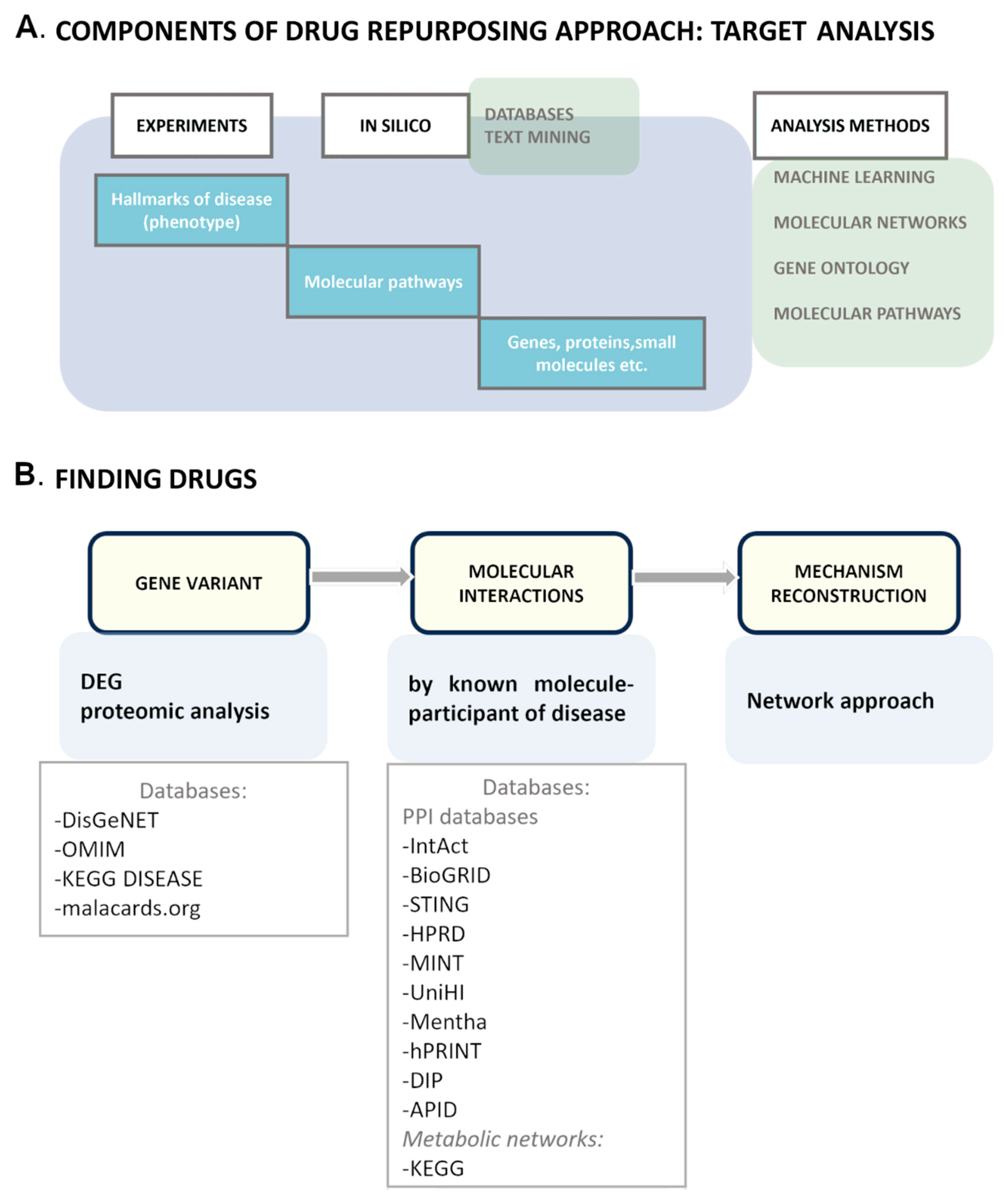
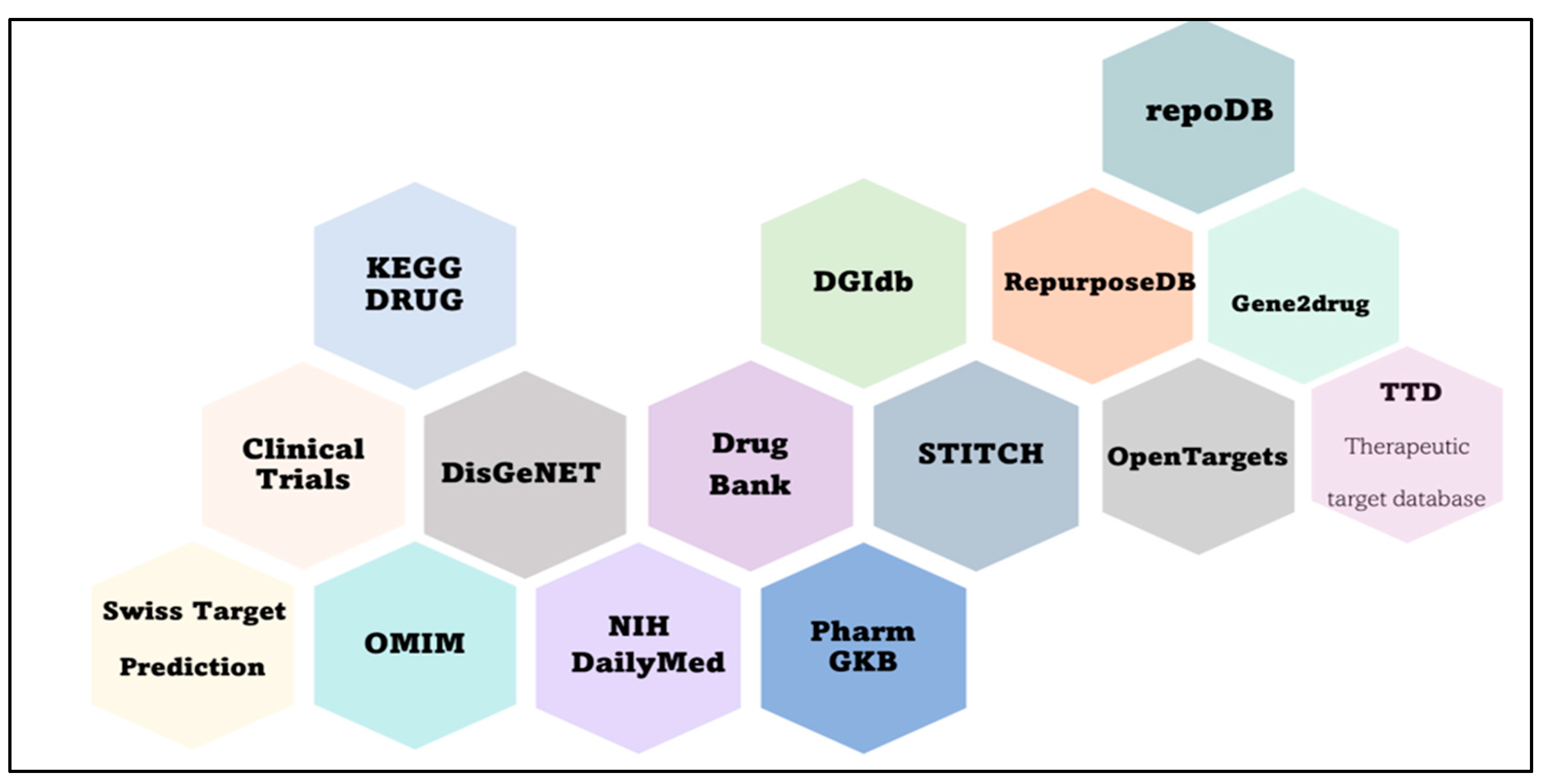
3. Drug Repurposing
- 1.
- Activity-based or experimental drug repurposing [107].
- 2.
- Computational drug repurposing.
- a database with drug data: phases of clinical trials, a mechanism of action, involvement in diseases, and physicochemical properties;
- a database with disease data: genes, molecular pathways, and mechanisms;
- tools for the analysis of molecular interactions (construction of gene, metabolic, and protein networks), Gene Ontology, and analysis of transcriptome data;
- tools of molecular dynamics and docking: construction and analysis of structures of target molecules and of the drug as well as interactions;
- text-mining tools, machine learning (ML), and neural networks.
- 3.
- Genome-wide association study (GWAS)-based methods.They identify a gene variant–disease relation, which subsequently helps with the selection of treatment targets.
- 4.
- Network-Based Methods.These procedures include system biology approaches for integrating and analyzing data on relations between various objects: an interaction of cellular structures with each other and with drugs under various conditions, including during the progression of a disease.
- 5.
- ML-based approaches and literature-based discovery methods.ML offers methods for regression analysis, clustering and classification, dimensionality reduction, neural networks, and other tools helping to analyze biological data and to infer new trends [112]. ML is now actively utilized at various stages of drug design: from investigating disease mechanisms, target identification, target validation, and compound screening to finding new markers of drug efficacy.
4. Drug Repurposing for HD Treatment
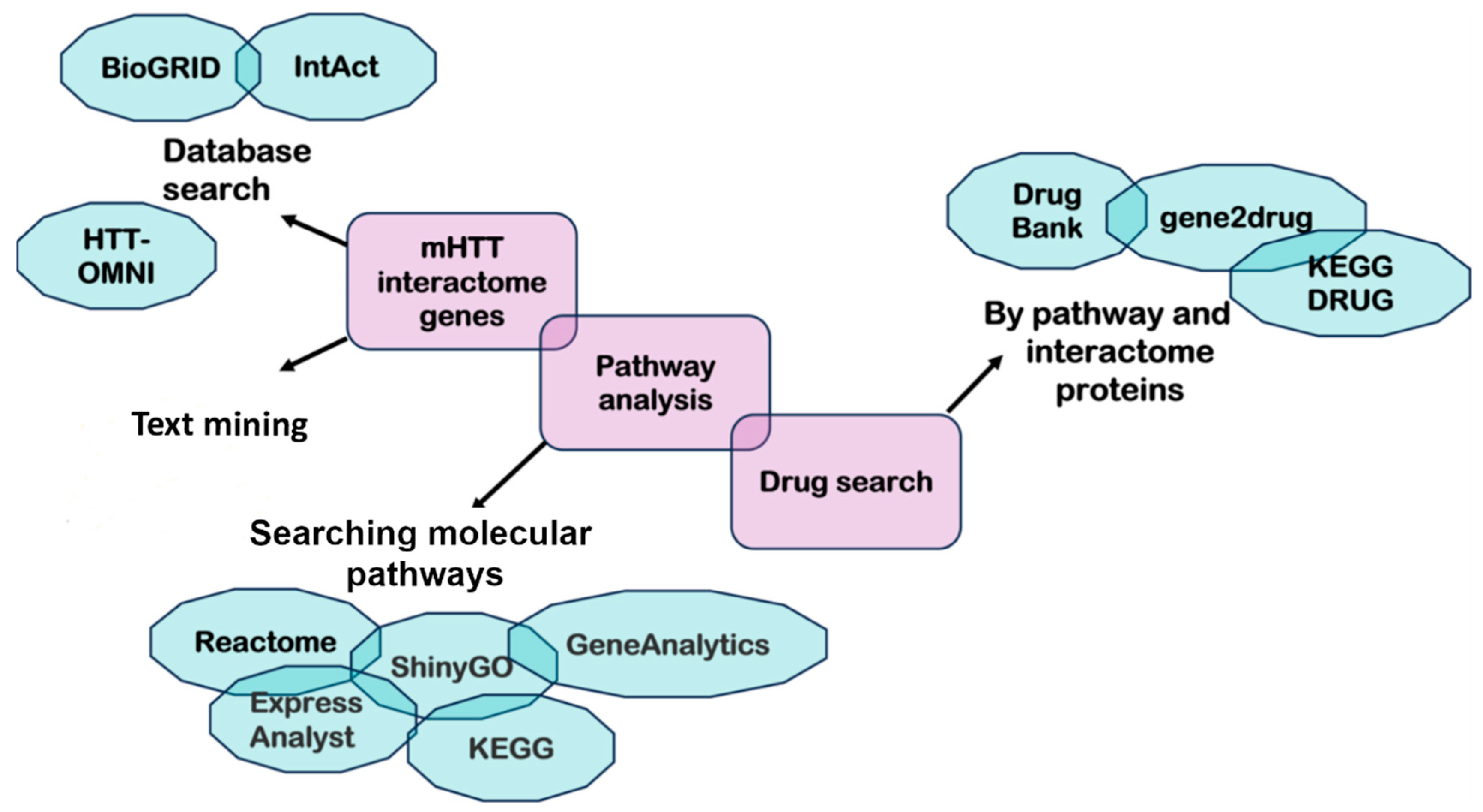
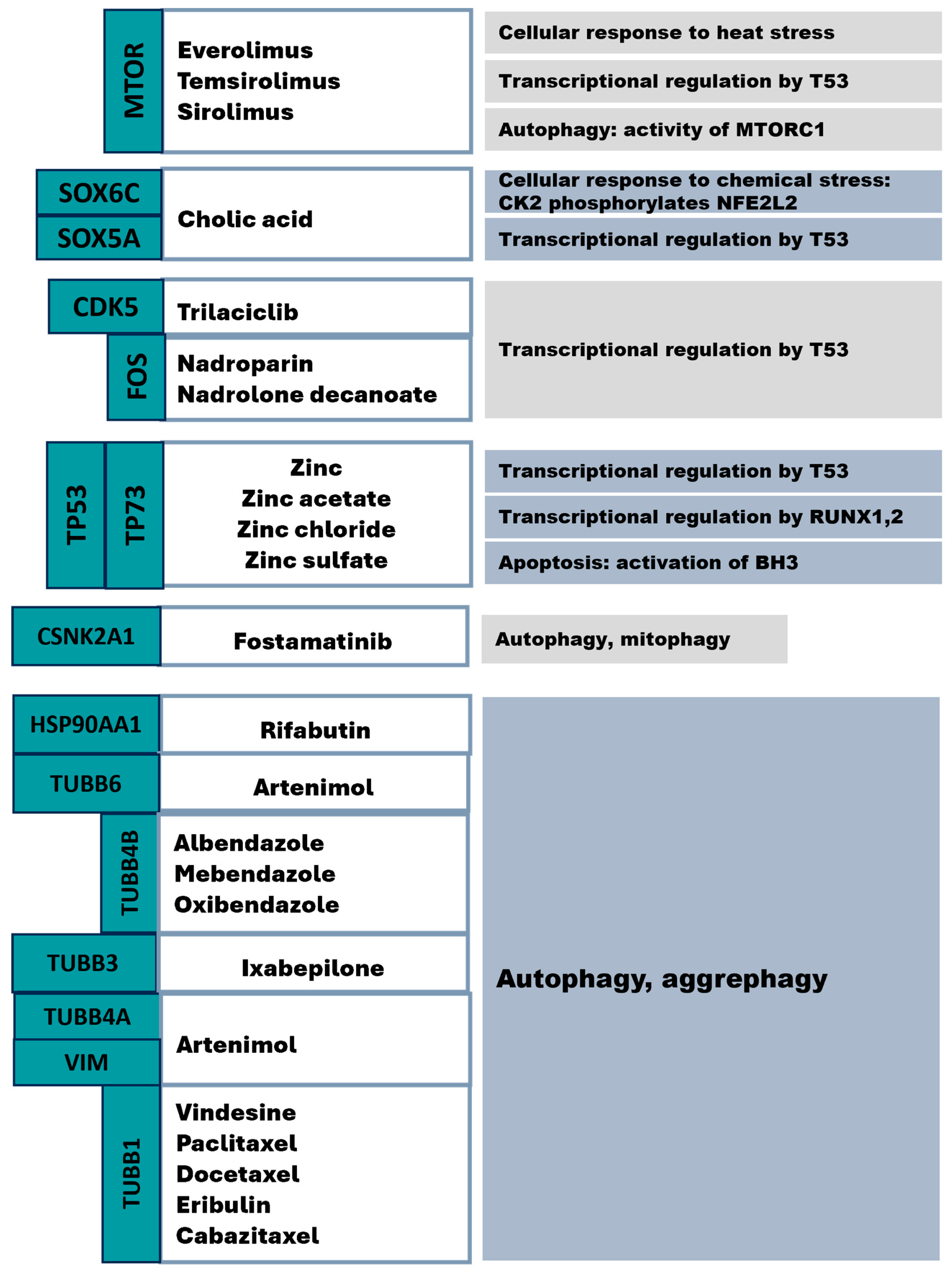
5. Drug Repurposing Targeting mHTT Interactome Pathways
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kakoti, B.B.; Bezbaruah, R.; Ahmed, N. Therapeutic Drug Repositioning with Special Emphasis on Neurodegenerative Diseases: Threats and Issues. Front. Pharmacol. 2022, 13, 1007315. [Google Scholar] [CrossRef] [PubMed]
- Mekhaeil, M.; Dev, K.K.; Conroy, M.J. Existing Evidence for the Repurposing of PARP-1 Inhibitors in Rare Demyelinating Diseases. Cancers 2022, 14, 687. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Zhang, G. The Role of PARP1 in Neurodegenerative Diseases and Aging. FEBS J. 2022, 289, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, S.-H.; Johnston, H.; Shelbourne, P.F.; Li, X.-J. Amino-Terminal Fragments of Mutant Huntingtin Show Selective Accumulation in Striatal Neurons and Synaptic Toxicity. Nat. Genet. 2000, 25, 385–389. [Google Scholar] [CrossRef]
- Wilson, D.M.; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of Neurodegenerative Diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Gonzalez Rojas, N.; Cesarini, M.E.; Peker, G.; Da Prat, G.A.; Etcheverry, J.L.; Gatto, E.M. Review of Huntington’s Disease: From Basics to Advances in Diagnosis and Treatment. J. Neurol. Res. 2022, 12, 93–113. [Google Scholar] [CrossRef]
- Vuono, R.; Winder-Rhodes, S.; de Silva, R.; Cisbani, G.; Drouin-Ouellet, J.; Spillantini, M.G.; Cicchetti, F.; Barker, R.A. The Role of Tau in the Pathological Process and Clinical Expression of Huntington’s Disease. Brain 2015, 138, 1907–1918. [Google Scholar] [CrossRef]
- Choudhary, S.; Kumar, P.; Malik, J. Plants and Phytochemicals for Huntington’s Disease. Pharmacogn. Rev. 2013, 7, 81–91. [Google Scholar] [CrossRef]
- Irfan, Z.; Khanam, S.; Karmakar, V.; Firdous, S.M.; El Khier, B.S.I.A.; Khan, I.; Rehman, M.U.; Khan, A. Pathogenesis of Huntington’s Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies. Brain Sci. 2022, 12, 1389. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A. Molecular Pathophysiological Mechanisms in Huntington’s Disease. Biomedicines 2022, 10, 1432. [Google Scholar] [CrossRef] [PubMed]
- Hervás-Corpión, I.; Guiretti, D.; Alcaraz-Iborra, M.; Olivares, R.; Campos-Caro, A.; Barco, Á.; Valor, L.M. Early Alteration of Epigenetic-Related Transcription in Huntington’s Disease Mouse Models. Sci. Rep. 2018, 8, 9925. [Google Scholar] [CrossRef] [PubMed]
- Neueder, A.; Gipson, T.A.; Batterton, S.; Lazell, H.J.; Farshim, P.P.; Paganetti, P.; Housman, D.E.; Bates, G.P. HSF1-Dependent and -Independent Regulation of the Mammalian in Vivo Heat Shock Response and Its Impairment in Huntington’s Disease Mouse Models. Sci. Rep. 2017, 7, 12556. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, N.; Cavalier, S.; Rybalchenko, V.; Kulkarni, A.; Anderson, A.G.; Konopka, G.; Gibson, J.R. FOXP1 Negatively Regulates Intrinsic Excitability in D2 Striatal Projection Neurons by Promoting Inwardly Rectifying and Leak Potassium Currents. Mol. Psychiatry 2021, 26, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.-I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. P53 Mediates Cellular Dysfunction and Behavioral Abnormalities in Huntington’s Disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Gines, S.; Seong, I.S.; Fossale, E.; Ivanova, E.; Trettel, F.; Gusella, J.F.; Wheeler, V.C.; Persichetti, F.; MacDonald, M.E. Specific Progressive CAMP Reduction Implicates Energy Deficit in Presymptomatic Huntington’s Disease Knock-in Mice. Hum. Mol. Genet. 2003, 12, 497–508. [Google Scholar] [CrossRef]
- Sugars, K.L.; Brown, R.; Cook, L.J.; Swartz, J.; Rubinsztein, D.C. Decreased CAMP Response Element-Mediated Transcription. J. Biol. Chem. 2004, 279, 4988–4999. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.-Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s Disease Protein Interacts with P53 and CREB-Binding Protein and Represses Transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef]
- Landles, C.; Bates, G.P. Huntingtin and the Molecular Pathogenesis of Huntington’s Disease. EMBO Rep. 2004, 5, 958–963. [Google Scholar] [CrossRef]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1alpha by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Di Cristo, F.; Finicelli, M.; Digilio, F.A.; Paladino, S.; Valentino, A.; Scialò, F.; D’Apolito, M.; Saturnino, C.; Galderisi, U.; Giordano, A.; et al. Meldonium Improves Huntington’s Disease Mitochondrial Dysfunction by Restoring Peroxisome Proliferator-activated Receptor γ Coactivator 1α Expression. J. Cell. Physiol. 2019, 234, 9233–9246. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Jeong, H.; Cui, L.; Krainc, D.; Tjian, R. In Vitro Analysis of Huntingtin-Mediated Transcriptional Repression Reveals Multiple Transcription Factor Targets. Cell 2005, 123, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-Y.; Zukin, R.S. REST, a Master Transcriptional Regulator in Neurodegenerative Disease. Curr. Opin. Neurobiol. 2018, 48, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Shahani, N.; Bae, B.-I.; Sbodio, J.I.; Chung, Y.; Nakaso, K.; Paul, B.D.; Sawa, A. Allele-Specific Regulation of Mutant Huntingtin by Wig1, a Downstream Target of P53. Hum. Mol. Genet. 2016, 25, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Ginty, D.D. CREB Family Transcription Factors Inhibit Neuronal Suicide. Nat. Med. 2002, 8, 450–451. [Google Scholar] [CrossRef]
- Choi, Y.-S.; Lee, B.; Cho, H.-Y.; Reyes, I.B.; Pu, X.-A.; Saido, T.C.; Hoyt, K.R.; Obrietan, K. CREB Is a Key Regulator of Striatal Vulnerability in Chemical and Genetic Models of Huntington’s Disease. Neurobiol. Dis. 2009, 36, 259–268. [Google Scholar] [CrossRef]
- Malla, B.; Guo, X.; Senger, G.; Chasapopoulou, Z.; Yildirim, F. A Systematic Review of Transcriptional Dysregulation in Huntington’s Disease Studied by RNA Sequencing. Front. Genet. 2021, 12, 751033. [Google Scholar] [CrossRef]
- Sakahira, H.; Breuer, P.; Hayer-Hartl, M.K.; Hartl, F.U. Molecular Chaperones as Modulators of Polyglutamine Protein Aggregation and Toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 16412–16418. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Molecular Chaperones in the Cytosol: From Nascent Chain to Folded Protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef]
- Smalle, J.; Vierstra, R.D. The Ubiquitin 26S Proteasome Proteolytic Pathway. Annu. Rev. Plant Biol. 2004, 55, 555–590. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.; Weiss, A.; Parker, C.N.; Bibel, M.; Paganetti, P.; Kaupmann, K. A Screen for Enhancers of Clearance Identifies Huntingtin as a Heat Shock Protein 90 (Hsp90) Client Protein. J. Biol. Chem. 2012, 287, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T. Analysis of a Multiqueue Model for an ISDN Access Interface. Perform. Eval. 1992, 15, 65–75. [Google Scholar] [CrossRef]
- Wacker, J.L.; Zareie, M.H.; Fong, H.; Sarikaya, M.; Muchowski, P.J. Hsp70 and Hsp40 Attenuate Formation of Spherical and Annular Polyglutamine Oligomers by Partitioning Monomer. Nat. Struct. Mol. Biol. 2004, 11, 1215–1222. [Google Scholar] [CrossRef]
- Li, H.; Li, S.-H.; Yu, Z.-X.; Shelbourne, P.; Li, X.-J. Huntingtin Aggregate-Associated Axonal Degeneration Is an Early Pathological Event in Huntington’s Disease Mice. J. Neurosci. 2001, 21, 8473–8481. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.M.; Yoshihara, M.; Littleton, J.T. Cytoplasmic Aggregates Trap Polyglutamine-Containing Proteins and Block Axonal Transport in a Drosophila Model of Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3224–3229. [Google Scholar] [CrossRef]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L.F. Huntingtin Facilitates Dynein/Dynactin-Mediated Vesicle Transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pagès, M.; Li, X.-J.; Saudou, F.; Humbert, S. Huntingtin Phosphorylation Acts as a Molecular Switch for Anterograde/Retrograde Transport in Neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef]
- Mandal, M.; Wei, J.; Zhong, P.; Cheng, J.; Duffney, L.J.; Liu, W.; Yuen, E.Y.; Twelvetrees, A.E.; Li, S.; Li, X.-J.; et al. Impaired α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid (AMPA) Receptor Trafficking and Function by Mutant Huntingtin. J. Biol. Chem. 2011, 286, 33719–33728. [Google Scholar] [CrossRef]
- Twelvetrees, A.E.; Yuen, E.Y.; Arancibia-Carcamo, I.L.; MacAskill, A.F.; Rostaing, P.; Lumb, M.J.; Humbert, S.; Triller, A.; Saudou, F.; Yan, Z.; et al. Delivery of GABAARs to Synapses Is Mediated by HAP1-KIF5 and Disrupted by Mutant Huntingtin. Neuron 2010, 65, 53–65. [Google Scholar] [CrossRef]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin Alters Retrograde Transport of TrkB Receptors in Striatal Dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef] [PubMed]
- Zsindely, N.; Siági, F.; Bodai, L. DNA Methylation in Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 12736. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, Calcium and Mitochondria: A Triad in Synaptic Neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, X.; Ye, K.; Orth, M.; Gu, Z. Accelerated Expansion of Pathogenic Mitochondrial DNA Heteroplasmies in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2014610118. [Google Scholar] [CrossRef] [PubMed]
- Jesse, S.; Bayer, H.; Alupei, M.C.; Zügel, M.; Mulaw, M.; Tuorto, F.; Malmsheimer, S.; Singh, K.; Steinacker, J.; Schumann, U.; et al. Ribosomal Transcription Is Regulated by PGC-1alpha and Disturbed in Huntington’s Disease. Sci. Rep. 2017, 7, 8513. [Google Scholar] [CrossRef]
- Bae, B.-I.; Hara, M.R.; Cascio, M.B.; Wellington, C.L.; Hayden, M.R.; Ross, C.A.; Ha, H.C.; Li, X.-J.; Snyder, S.H.; Sawa, A. Mutant Huntingtin: Nuclear Translocation and Cytotoxicity Mediated by GAPDH. Proc. Natl. Acad. Sci. USA 2006, 103, 3405–3409. [Google Scholar] [CrossRef]
- Theos, A.C.; Tenza, D.; Martina, J.A.; Hurbain, I.; Peden, A.A.; Sviderskaya, E.V.; Stewart, A.; Robinson, M.S.; Bennett, D.C.; Cutler, D.F.; et al. Functions of Adaptor Protein (AP)-3 and AP-1 in Tyrosinase Sorting from Endosomes to Melanosomes. Mol. Biol. Cell 2005, 16, 5356–5372. [Google Scholar] [CrossRef]
- Grewal, A.K.; Singh, T.G.; Sharma, D.; Sharma, V.; Singh, M.; Rahman, M.H.; Najda, A.; Walasek-Janusz, M.; Kamel, M.; Albadrani, G.M.; et al. Mechanistic Insights and Perspectives Involved in Neuroprotective Action of Quercetin. Biomed. Pharmacother. 2021, 140, 111729. [Google Scholar] [CrossRef]
- Xiang, Z.; Valenza, M.; Cui, L.; Leoni, V.; Jeong, H.-K.; Brilli, E.; Zhang, J.; Peng, Q.; Duan, W.; Reeves, S.A.; et al. Peroxisome-Proliferator-Activated Receptor Gamma Coactivator 1 Contributes to Dysmyelination in Experimental Models of Huntington’s Disease. J. Neurosci. 2011, 31, 9544–9553. [Google Scholar] [CrossRef]
- Machiela, E.; Rudich, P.D.; Traa, A.; Anglas, U.; Soo, S.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Targeting Mitochondrial Network Disorganization Is Protective in C. elegans Models of Huntington’s Disease. Aging Dis. 2021, 12, 1753–1772. [Google Scholar] [CrossRef]
- Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9521. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Jurcau, C. Mitochondria in Huntington’s Disease: Implications in Pathogenesis and Mitochondrial-Targeted Therapeutic Strategies. Neural Regen. Res. 2023, 18, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. In Polyglutamine Disorders; Springer: Cham, Switzerland, 2018; pp. 59–83. [Google Scholar]
- Schapira, A.H.V.; Olanow, C.W.; Greenamyre, J.T.; Bezard, E. Slowing of Neurodegeneration in Parkinson’s Disease and Huntington’s Disease: Future Therapeutic Perspectives. Lancet 2014, 384, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Narne, P.; Pandey, V.; Simhadri, P.K.; Phanithi, P.B. Poly(ADP-Ribose)Polymerase-1 Hyperactivation in Neurodegenerative Diseases: The Death Knell Tolls for Neurons. Semin. Cell Dev. Biol. 2017, 63, 154–166. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New Insights into the Molecular and Cellular Functions of Poly(ADP-Ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Jansen, A.H.P.; van Hal, M.; op den Kelder, I.C.; Meier, R.T.; de Ruiter, A.; Schut, M.H.; Smith, D.L.; Grit, C.; Brouwer, N.; Kamphuis, W.; et al. Frequency of Nuclear Mutant Huntingtin Inclusion Formation in Neurons and Glia Is Cell-type-specific. Glia 2017, 65, 50–61. [Google Scholar] [CrossRef]
- Tydlacka, S.; Wang, C.-E.; Wang, X.; Li, S.; Li, X.-J. Differential Activities of the Ubiquitin–Proteasome System in Neurons versus Glia May Account for the Preferential Accumulation of Misfolded Proteins in Neurons. J. Neurosci. 2008, 28, 13285–13295. [Google Scholar] [CrossRef]
- Shin, J.-Y.; Fang, Z.-H.; Yu, Z.-X.; Wang, C.-E.; Li, S.-H.; Li, X.-J. Expression of Mutant Huntingtin in Glial Cells Contributes to Neuronal Excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef]
- Bradford, J.; Shin, J.-Y.; Roberts, M.; Wang, C.-E.; Li, X.-J.; Li, S. Expression of Mutant Huntingtin in Mouse Brain Astrocytes Causes Age-Dependent Neurological Symptoms. Proc. Natl. Acad. Sci. USA 2009, 106, 22480–22485. [Google Scholar] [CrossRef]
- Chou, S.-Y.; Weng, J.-Y.; Lai, H.-L.; Liao, F.; Sun, S.H.; Tu, P.-H.; Dickson, D.W.; Chern, Y. Expanded-Polyglutamine Huntingtin Protein Suppresses the Secretion and Production of a Chemokine (CCL5/RANTES) by Astrocytes. J. Neurosci. 2008, 28, 3277–3290. [Google Scholar] [CrossRef]
- Tong, X.; Ao, Y.; Faas, G.C.; Nwaobi, S.E.; Xu, J.; Haustein, M.D.; Anderson, M.A.; Mody, I.; Olsen, M.L.; Sofroniew, M.V.; et al. Astrocyte Kir4.1 Ion Channel Deficits Contribute to Neuronal Dysfunction in Huntington’s Disease Model Mice. Nat. Neurosci. 2014, 17, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Ferrari Bardile, C.; Garcia-Miralles, M.; Caron, N.S.; Rayan, N.A.; Langley, S.R.; Harmston, N.; Rondelli, A.M.; Teo, R.T.Y.; Waltl, S.; Anderson, L.M.; et al. Intrinsic Mutant HTT-Mediated Defects in Oligodendroglia Cause Myelination Deficits and Behavioral Abnormalities in Huntington Disease. Proc. Natl. Acad. Sci. USA 2019, 116, 9622–9627. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.M.; Rojas, N.G.; Persi, G.; Etcheverry, J.L.; Cesarini, M.E.; Perandones, C. Huntington Disease: Advances in the Understanding of Its Mechanisms. Clin. Park. Relat. Disord. 2020, 3, 100056. [Google Scholar] [CrossRef] [PubMed]
- Croce, K.R.; Yamamoto, A. A Role for Autophagy in Huntington’s Disease. Neurobiol. Dis. 2019, 122, 16–22. [Google Scholar] [CrossRef]
- Chi, H.; Chang, H.-Y.; Sang, T.-K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kam, T.I.; Dawson, T.M.; Dawson, V.L. Poly (ADP-Ribose) (PAR)-Dependent Cell Death in Neurodegenerative Diseases. Int. Rev. Cell Mol. Biol. 2020, 353, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Andre, R.; Carty, L.; Tabrizi, S.J. Disruption of Immune Cell Function by Mutant Huntingtin in Huntington’s Disease Pathogenesis. Curr. Opin. Pharmacol. 2016, 26, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Leon, R.; Bhagavatula, N.; Ulukpo, O.; McCollum, M.; Wei, J. BimEL as a Possible Molecular Link between Proteasome Dysfunction and Cell Death Induced by Mutant Huntingtin. Eur. J. Neurosci. 2010, 31, 1915–1925. [Google Scholar] [CrossRef]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The Pathobiology of Perturbed Mutant Huntingtin Protein–Protein Interactions in Huntington’s Disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef]
- Greco, T.M.; Secker, C.; Silva Ramos, E.; Federspiel, J.D.; Liu, J.-P.; Perez, A.M.; Al-Ramahi, I.; Cantle, J.P.; Carroll, J.B.; Botas, J.; et al. Dynamics of Huntingtin Protein Interactions in the Striatum Identifies Candidate Modifiers of Huntington Disease. Cell Syst. 2022, 13, 304–320. [Google Scholar] [CrossRef]
- Podvin, S.; Rosenthal, S.B.; Poon, W.; Wei, E.; Fisch, K.M.; Hook, V. Mutant Huntingtin Protein Interaction Map Implicates Dysregulation of Multiple Cellular Pathways in Neurodegeneration of Huntington’s Disease. J. Huntington’s Dis. 2022, 11, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained Therapeutic Reversal of Huntington’s Disease by Transient Repression of Huntingtin Synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.B.; Warby, S.C.; Southwell, A.L.; Doty, C.N.; Greenlee, S.; Skotte, N.; Hung, G.; Bennett, C.F.; Freier, S.M.; Hayden, M.R. Potent and Selective Antisense Oligonucleotides Targeting Single-Nucleotide Polymorphisms in the Huntington Disease Gene/Allele-Specific Silencing of Mutant Huntingtin. Mol. Ther. 2011, 19, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Garriga-Canut, M.; Agustín-Pavón, C.; Herrmann, F.; Sánchez, A.; Dierssen, M.; Fillat, C.; Isalan, M. Synthetic Zinc Finger Repressors Reduce Mutant Huntingtin Expression in the Brain of R6/2 Mice. Proc. Natl. Acad. Sci. USA 2012, 109, E3136–E3145. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-L.; Liu, W.; Wada, E.; Murata, M.; Wada, K.; Kanazawa, I. Clinico-Pathological Rescue of a Model Mouse of Huntington’s Disease by SiRNA. Neurosci. Res. 2005, 53, 241–249. [Google Scholar] [CrossRef]
- Pfister, E.L.; DiNardo, N.; Mondo, E.; Borel, F.; Conroy, F.; Fraser, C.; Gernoux, G.; Han, X.; Hu, D.; Johnson, E.; et al. Artificial MiRNAs Reduce Human Mutant Huntingtin Throughout the Striatum in a Transgenic Sheep Model of Huntington’s Disease. Hum. Gene Ther. 2018, 29, 663–673. [Google Scholar] [CrossRef]
- Franich, N.R.; Fitzsimons, H.L.; Fong, D.M.; Klugmann, M.; During, M.J.; Young, D. AAV Vector–Mediated RNAi of Mutant Huntingtin Expression Is Neuroprotective in a Novel Genetic Rat Model of Huntington’s Disease. Mol. Ther. 2008, 16, 947–956. [Google Scholar] [CrossRef]
- Bailus, B.J.; Scheeler, S.M.; Simons, J.; Sanchez, M.A.; Tshilenge, K.-T.; Creus-Muncunill, J.; Naphade, S.; Lopez-Ramirez, A.; Zhang, N.; Lakshika Madushani, K.; et al. Modulating FKBP5/FKBP51 and Autophagy Lowers HTT (Huntingtin) Levels. Autophagy 2021, 17, 4119–4140. [Google Scholar] [CrossRef]
- Amaro, I.A.; Henderson, L.A. An Intrabody Drug (RAAV6-INT41) Reduces the Binding of N-Terminal Huntingtin Fragment(s) to DNA to Basal Levels in PC12 Cells and Delays Cognitive Loss in the R6/2 Animal Model. J. Neurodegener. Dis. 2016, 2016, 7120753. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. Huntington’s Disease: Underlying Molecular Mechanisms and Emerging Concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-T.; Chu, K.; Park, J.-E.; Kang, L.; Ko, S.-Y.; Jung, K.-H.; Kim, M. Memantine Reduces Striatal Cell Death with Decreasing Calpain Level in 3-Nitropropionic Model of Huntington’s Disease. Brain Res. 2006, 1118, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Chabrier, P.E.; Auguet, M. Pharmacological Properties of BN82451: A Novel Multitargeting Neuroprotective Agent. CNS Drug Rev. 2007, 13, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Yero, T.; Rey, J.A. Tetrabenazine (Xenazine), An FDA-Approved Treatment Option For Huntington’s Disease-Related Chorea. Pharm. Ther. 2008, 33, 690–694. [Google Scholar]
- Furr Stimming, E.; Claassen, D.O.; Kayson, E.; Goldstein, J.; Mehanna, R.; Zhang, H.; Liang, G.S.; Haubenberger, D.; Adams, J.; Beck, C.; et al. Safety and Efficacy of Valbenazine for the Treatment of Chorea Associated with Huntington’s Disease (KINECT-HD): A Phase 3, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2023, 22, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Lansita, J.A.; Mease, K.M.; Qiu, H.; Yednock, T.; Sankaranarayanan, S.; Kramer, S. Nonclinical Development of ANX005: A Humanized Anti-C1q Antibody for Treatment of Autoimmune and Neurodegenerative Diseases. Int. J. Toxicol. 2017, 36, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Shen, Y.; Zhao, S.; Zhang, R.; Dong, W.; Lei, X. Protective Effect of Resveratrol on Mitochondrial Biogenesis during Hyperoxia-Induced Brain Injury in Neonatal Pups. BMC Neurosci. 2023, 24, 27. [Google Scholar] [CrossRef]
- Bhateja, D.K.; Dhull, D.K.; Gill, A.; Sidhu, A.; Sharma, S.; Reddy, B.V.K.; Padi, S.S.V. Peroxisome Proliferator-Activated Receptor-α Activation Attenuates 3-Nitropropionic Acid Induced Behavioral and Biochemical Alterations in Rats: Possible Neuroprotective Mechanisms. Eur. J. Pharmacol. 2012, 674, 33–43. [Google Scholar] [CrossRef]
- Todd, D.; Gowers, I.; Dowler, S.J.; Wall, M.D.; McAllister, G.; Fischer, D.F.; Dijkstra, S.; Fratantoni, S.A.; van de Bospoort, R.; Veenman-Koepke, J.; et al. A Monoclonal Antibody TrkB Receptor Agonist as a Potential Therapeutic for Huntington’s Disease. PLoS ONE 2014, 9, e87923. [Google Scholar] [CrossRef]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC Inhibitor 4b Ameliorates the Disease Phenotype and Transcriptional Abnormalities in Huntington’s Disease Transgenic Mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef]
- Prins, N.D.; Harrison, J.E.; Chu, H.-M.; Blackburn, K.; Alam, J.J.; Scheltens, P. A Phase 2 Double-Blind Placebo-Controlled 24-Week Treatment Clinical Study of the P38 Alpha Kinase Inhibitor Neflamapimod in Mild Alzheimer’s Disease. Alzheimer’s Res. Ther. 2021, 13, 106. [Google Scholar] [CrossRef]
- Chen, M.; Ona, V.O.; Li, M.; Ferrante, R.J.; Fink, K.B.; Zhu, S.; Bian, J.; Guo, L.; Farrell, L.A.; Hersch, S.M.; et al. Minocycline Inhibits Caspase-1 and Caspase-3 Expression and Delays Mortality in a Transgenic Mouse Model of Huntington Disease. Nat. Med. 2000, 6, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Al-Gharaibeh, A.; Culver, R.; Stewart, A.N.; Srinageshwar, B.; Spelde, K.; Frollo, L.; Kolli, N.; Story, D.; Paladugu, L.; Anwar, S.; et al. Induced Pluripotent Stem Cell-Derived Neural Stem Cell Transplantations Reduced Behavioral Deficits and Ameliorated Neuropathological Changes in YAC128 Mouse Model of Huntington’s Disease. Front. Neurosci. 2017, 11, 628. [Google Scholar] [CrossRef] [PubMed]
- Tarsy, D.; Baldessarini, R.J.; Tarazi, F.I. Effects of Newer Antipsychotics on Extrapyramidal Function. CNS Drugs 2002, 16, 23–45. [Google Scholar] [CrossRef]
- Pallier, P.N.; Morton, A.J. Management of Sleep/Wake Cycles Improves Cognitive Function in a Transgenic Mouse Model of Huntington’s Disease. Brain Res. 2009, 1279, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef]
- Tulloch, L.B.; Menzies, S.K.; Coron, R.P.; Roberts, M.D.; Florence, G.J.; Smith, T.K. Direct and Indirect Approaches to Identify Drug Modes of Action. IUBMB Life 2018, 70, 9–22. [Google Scholar] [CrossRef]
- Katsila, T.; Spyroulias, G.A.; Patrinos, G.P.; Matsoukas, M.-T. Computational Approaches in Target Identification and Drug Discovery. Comput. Struct. Biotechnol. J. 2016, 14, 177–184. [Google Scholar] [CrossRef]
- Schenone, M.; Dančík, V.; Wagner, B.K.; Clemons, P.A. Target Identification and Mechanism of Action in Chemical Biology and Drug Discovery. Nat. Chem. Biol. 2013, 9, 232–240. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Parvatikar, P.P.; Patil, S.; Khaparkhuntikar, K.; Patil, S.; Singh, P.K.; Sahana, R.; Kulkarni, R.V.; Raghu, A.V. Artificial Intelligence: Machine Learning Approach for Screening Large Database and Drug Discovery. Antivir. Res. 2023, 220, 105740. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.S.; Alagarsamy, V.; Solomon, V.R.; Jose, P.A.; Murugesan, S. Drug Repurposing: An Effective Tool in Modern Drug Discovery. Russ. J. Bioorg. Chem. 2023, 49, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Parisi, D.; Adasme, M.F.; Sveshnikova, A.; Bolz, S.N.; Moreau, Y.; Schroeder, M. Drug Repositioning or Target Repositioning: A Structural Perspective of Drug-Target-Indication Relationship for Available Repurposed Drugs. Comput. Struct. Biotechnol. J. 2020, 18, 1043–1055. [Google Scholar] [CrossRef]
- Zheng, W.; Sun, W.; Simeonov, A. Drug Repurposing Screens and Synergistic Drug-combinations for Infectious Diseases. Br. J. Pharmacol. 2018, 175, 181–191. [Google Scholar] [CrossRef]
- Jin, G.; Wong, S.T.C. Toward Better Drug Repositioning: Prioritizing and Integrating Existing Methods into Efficient Pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed Drugs Targeting EIF2α-P-Mediated Translational Repression Prevent Neurodegeneration in Mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; Koplev, S.; He, E.; Torre, D.; Wang, Z.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A.; et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6, 13–24. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of Machine Learning in Drug Discovery and Development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.R.; Acencio, M.L.; Lemke, N. A Machine Learning Approach for Genome-Wide Prediction of Morbid and Druggable Human Genes Based on Systems-Level Data. BMC Genom. 2010, 11, S9. [Google Scholar] [CrossRef] [PubMed]
- Bravo, À.; Piñero, J.; Queralt-Rosinach, N.; Rautschka, M.; Furlong, L.I. Extraction of Relations between Genes and Diseases from Text and Large-Scale Data Analysis: Implications for Translational Research. BMC Bioinform. 2015, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.T.; Ju, J.H.; Wong, Y.T.; Shmulevich, I.; Chiang, J.H. Literature-Based Discovery of New Candidates for Drug Repurposing. Brief. Bioinform. 2017, 18, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Tanoli, Z.; Seemab, U.; Scherer, A.; Wennerberg, K.; Tang, J.; Vähä-Koskela, M. Exploration of Databases and Methods Supporting Drug Repurposing: A Comprehensive Survey. Brief. Bioinform. 2021, 22, 1656–1678. [Google Scholar] [CrossRef]
- Masoudi-Sobhanzadeh, Y.; Omidi, Y.; Amanlou, M.; Masoudi-Nejad, A. Drug Databases and Their Contributions to Drug Repurposing. Genomics 2020, 112, 1087–1095. [Google Scholar] [CrossRef]
- van Vugt, J.P.P.; Siesling, S.; Vergeer, M.; van der Velde, E.A.; Roos, R.A.C. Clozapine versus Placebo in Huntington’s Disease: A Double Blind Randomised Comparative Study. J. Neurol. Neurosurg. Psychiatry 1997, 63, 35–39. [Google Scholar] [CrossRef]
- Paleacu, D.; Anca, M.; Giladi, N. Olanzapine in Huntington’s Disease. Acta Neurol. Scand. 2002, 105, 441–444. [Google Scholar] [CrossRef]
- Beister, A.; Kraus, P.; Kuhn, W.; Dose, M.; Weindl, A.; Gerlach, M. The N-Methyl-D-Aspartate Antagonist Memantine Retards Progression of Huntington’s Disease. In Focus on Extrapyramidal Dysfunction; Springer: Vienna, Austria, 2004; pp. 117–122. [Google Scholar]
- Paleacu, D. Tetrabenazine in the Treatment of Huntington’s Disease. Neuropsychiatr. Dis. Treat. 2007, 3, 545–551. [Google Scholar]
- Duff, K.; Beglinger, L.J.; O’Rourke, M.E.; Nopoulos, P.; Paulson, H.L.; Paulsen, J.S. Risperidone and the Treatment of Psychiatric, Motor, and Cognitive Symptoms in Huntington’s Disease. Ann. Clin. Psychiatry 2008, 20, 1–3. [Google Scholar] [CrossRef]
- Jankovic, J.; Clarence-Smith, K. Tetrabenazine for the Treatment of Chorea and Other Hyperkinetic Movement Disorders. Expert Rev. Neurother. 2011, 11, 1509–1523. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Ondo, W.; Fahn, S.; Hunter, C.; Oakes, D.; Plumb, S.; Marshall, F.; Shoulson, I.; Eberly, S.; Walker, F.; et al. A Study of Chorea After Tetrabenazine Withdrawal in Patients with Huntington Disease. Clin. Neuropharmacol. 2008, 31, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group. Tetrabenazine as Antichorea Therapy in Huntington Disease: A Randomized Controlled Trial. Neurology 2006, 66, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Arbez, N.; Roby, E.; Akimov, S.; Eddings, C.; Ren, M.; Wang, X.; Ross, C.A. Cysteamine Protects Neurons from Mutant Huntingtin Toxicity1. J. Huntington’s Dis. 2019, 8, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Verny, C.; Bachoud-Lévi, A.-C.; Durr, A.; Goizet, C.; Azulay, J.-P.; Simonin, C.; Tranchant, C.; Calvas, F.; Krystkowiak, P.; Charles, P.; et al. A Randomized, Double-Blind, Placebo-Controlled Trial Evaluating Cysteamine in Huntington’s Disease. Mov. Disord. 2017, 32, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Borrell-Pages, M. Cystamine and Cysteamine Increase Brain Levels of BDNF in Huntington Disease via HSJ1b and Transglutaminase. J. Clin. Investig. 2006, 116, 1410–1424. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.D.C.; Johnson, G.V.W. The Protective Effects of Cystamine in the R6/2 Huntington’s Disease Mouse Involve Mechanisms Other than the Inhibition of Tissue Transglutaminase. Neurobiol. Aging 2006, 27, 871–879. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Pearson, J.; Bailey, C.D.C.; Rogers, D.A.; Johnson, G.V.W.; Hayden, M.R.; Leavitt, B.R. Cystamine Treatment Is Neuroprotective in the YAC128 Mouse Model of Huntington Disease. J. Neurochem. 2005, 95, 210–220. [Google Scholar] [CrossRef]
- Karpuj, M.V.; Becher, M.W.; Steinman, L. Evidence for a Role for Transglutaminase in Huntington’s Disease and the Potential Therapeutic Implications. Neurochem. Int. 2002, 40, 31–36. [Google Scholar] [CrossRef]
- Danivas, V.; Moily, N.; Thimmaiah, R.; Muralidharan, K.; Purushotham, M.; Muthane, U.; Jain, S. Off Label Use of Lithium in the Treatment of Huntington’s Disease: A Case Series. Indian J. Psychiatry 2013, 55, 81–83. [Google Scholar] [CrossRef]
- Mattsson, B.; Persson, S.Å. Huntington’s Chorea, Lithium, and G.A.B.A. Lancet 1973, 2, 684. [Google Scholar] [CrossRef]
- Dalén, P. Lithium Therapy in Huntington’s Chorea and Tardive Dyskinesia. Lancet 1973, 1, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Andén, N.-E.; Dalén, P.; Johansson, B. Baclofen and Lithium in Huntington’s Chorea. Lancet 1973, 302, 93. [Google Scholar] [CrossRef]
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic Approaches to Huntington Disease: From the Bench to the Clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Miralles, M.; Hong, X.; Tan, L.J.; Caron, N.S.; Huang, Y.; To, X.V.; Lin, R.Y.; Franciosi, S.; Papapetropoulos, S.; Hayardeny, L.; et al. Laquinimod Rescues Striatal, Cortical and White Matter Pathology and Results in Modest Behavioural Improvements in the YAC128 Model of Huntington Disease. Sci. Rep. 2016, 6, 31652. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Perlstein, E.O.; Imarisio, S.; Pineau, S.; Cordenier, A.; Maglathlin, R.L.; Webster, J.A.; Lewis, T.A.; O’Kane, C.J.; Schreiber, S.L.; et al. Small Molecules Enhance Autophagy and Reduce Toxicity in Huntington’s Disease Models. Nat. Chem. Biol. 2007, 3, 331–338. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium Induces Autophagy by Inhibiting Inositol Monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a Novel MTOR-Independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and α-Synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of MTOR Induces Autophagy and Reduces Toxicity of Polyglutamine Expansions in Fly and Mouse Models of Huntington Disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-Prone Proteins with Polyglutamine and Polyalanine Expansions Are Degraded by Autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel Targets for Huntington’s Disease in an MTOR-Independent Autophagy Pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s Disease and Dementia with Lewy Bodies. J. Park. Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Anjum, M.; Arellano, J.; et al. Nilotinib Effects on Safety, Tolerability, and Potential Biomarkers in Parkinson Disease. JAMA Neurol. 2020, 77, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Lyketsos, C.G.; Peskind, E.R.; Porsteinsson, A.P.; Mintzer, J.E.; Scharre, D.W.; De La Gandara, J.E.; Agronin, M.; Davis, C.S.; Nguyen, U.; et al. Effect of Dextromethorphan-Quinidine on Agitation in Patients with Alzheimer Disease Dementia. JAMA 2015, 314, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; Paldino, E.; Giampà, C.; Bernardi, G.; Fusco, F.R. PARP-1 Inhibition Is Neuroprotective in the R6/2 Mouse Model of Huntington’s Disease. PLoS ONE 2015, 10, e0134482. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Dooms, M.M.; Amaral-Garcia, S.; Igoillo-Esteve, M. Current Drug Repurposing Strategies for Rare Neurodegenerative Disorders. Front. Pharmacol. 2021, 12, 768023. [Google Scholar] [CrossRef] [PubMed]
- Shen, V.; Clarence-Smith, K.; Hunter, C.; Jankovic, J. Safety and Efficacy of Tetrabenazine and Use of Concomitant Medications During Long-Term, Open-Label Treatment of Chorea Associated with Huntington’s and Other Diseases. Tremor Other Hyperkinetic Mov. 2013, 3, tre-03-191-4337-1. [Google Scholar] [CrossRef]
- Gupta, H.; Perkins, W.; Stark, C.; Kikkeri, S.; Kakazu, J.; Kaye, A.D.; Kaye, A.D. Deutetrabenazine for the Treatment of Chorea Associated with Huntington’s Disease. Health Psychol. Res. 2022, 10, 36040. [Google Scholar] [CrossRef]
- Wei, P.F.; Zhang, L.; Nethi, S.K.; Barui, A.K.; Lin, J.; Zhou, W.; Shen, Y.; Man, N.; Zhang, Y.J.; Xu, J.; et al. Accelerating the Clearance of Mutant Huntingtin Protein Aggregates through Autophagy Induction by Europium Hydroxide Nanorods. Biomaterials 2014, 35, 899–907. [Google Scholar] [CrossRef]
- Scheuing, L.; Chiu, C.-T.; Liao, H.-M.; Linares, G.R.; Chuang, D.-M. Preclinical and Clinical Investigations of Mood Stabilizers for Huntington’s Disease: What Have We Learned? Int. J. Biol. Sci. 2014, 10, 1024–1038. [Google Scholar] [CrossRef]
- Yatham, L.N.; Kennedy, S.H.; Parikh, S.V.; Schaffer, A.; Bond, D.J.; Frey, B.N.; Sharma, V.; Goldstein, B.I.; Rej, S.; Beaulieu, S.; et al. Canadian Network for Mood and Anxiety Treatments (canmat) and International Society for Bipolar Disorders (isbd) 2018 Guidelines for the Management of Patients with Bipolar Disorder. Bipolar Disord. 2018, 20, 97–170. [Google Scholar] [CrossRef] [PubMed]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A Novel Pathogenic Pathway of Immune Activation Detectable before Clinical Onset in Huntington’s Disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Roussakis, A.A.; Gennaro, M.; Gordon, M.F.; Reilmann, R.; Borowsky, B.; Rynkowski, G.; Lao-Kaim, N.P.; Papoutsou, Z.; Savola, J.M.; Hayden, M.R.; et al. A PET-CT Study on Neuroinflammation in Huntington’s Disease Patients Participating in a Randomized Trial with Laquinimod. Brain Commun. 2023, 5, fcad084. [Google Scholar] [CrossRef] [PubMed]
- Durães, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla, C.; Mahía, J.; Bernal, A.; Puerto, A. The D 2/D 3-Receptor Antagonist Tiapride Impairs Concurrent but Not Sequential Taste Aversion Learning. Brain Res. Bull. 2012, 87, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, V.M.; Rajkumar, A.P.; Jacob, K.S.; Jacob, M. Gene-Gene Interaction between DRD4 and COMT Modulates Clinical Response to Clozapine in Treatment-Resistant Schizophrenia. Pharmacogenet. Genom. 2018, 28, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Coppen, E.M.; Roos, R.A.C. Current Pharmacological Approaches to Reduce Chorea in Huntington’s Disease. Drugs 2017, 77, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Anitha, M.; Nandhu, M.S.; Anju, T.R.; Jes, P.; Paulose, C.S. Targeting Glutamate Mediated Excitotoxicity in Huntington’s Disease: Neural Progenitors and Partial Glutamate Antagonist--Memantine. Med. Hypotheses 2011, 76, 138–140. [Google Scholar] [CrossRef]
- Ferguson, M.W.; Kennedy, C.J.; Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Current and Possible Future Therapeutic Options for Huntington’s Disease. J. Cent. Nerv. Syst. Dis. 2022, 14, 11795735221092517. [Google Scholar] [CrossRef]
- Maiuri, T.; Suart, C.E.; Hung, C.L.K.; Graham, K.J.; Barba Bazan, C.A.; Truant, R. DNA Damage Repair in Huntington’s Disease and Other Neurodegenerative Diseases. Neurotherapeutics 2019, 16, 948–956. [Google Scholar] [CrossRef]
- Massey, T.H.; Jones, L. The Central Role of DNA Damage and Repair in CAG Repeat Diseases. Dis. Models Mech. 2018, 11, dmm031930. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Gao, R.; Bush, K.; Zhang, N.; Wairkar, Y.P.; Sarkar, P.S. Polyglutamine Expansion in Huntingtin and Mechanism of DNA Damage Repair Defects in Huntington’s Disease. Front. Cell. Neurosci. 2022, 16, 837576. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt, C.; Hensman-Moss, D.; Flower, M.; Wiethoff, S.; Brice, A.; Goizet, C.; Stevanin, G.; Koutsis, G.; Karadima, G.; Panas, M.; et al. DNA Repair Pathways Underlie a Common Genetic Mechanism Modulating Onset in Polyglutamine Diseases. Ann. Neurol. 2016, 79, 983–990. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-Linked Mechanisms and Therapeutic Opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, B.; Ahel, I.; Altmeyer, M.; Ashworth, A.; Bai, P.; Chang, P.; Cohen, M.; Corda, D.; Dantzer, F.; Daugherty, M.D.; et al. ADP-ribosyltransferases, an Update on Function and Nomenclature. FEBS J. 2022, 289, 7399–7410. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Poly(ADP-Ribose) Polymerase Inhibition: Past, Present and Future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Mojsilovic-Petrovic, J.; Van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear Poly(ADP-Ribose) Activity Is a Therapeutic Target in Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2018, 6, 84. [Google Scholar] [CrossRef]
- Lavrik, O.I. PARPs’ Impact on Base Excision DNA Repair. DNA Repair 2020, 93, 102911. [Google Scholar] [CrossRef]
- Moor, N.A.; Vasil’eva, I.A.; Kuznetsov, N.A.; Lavrik, O.I. Human Apurinic/Apyrimidinic Endonuclease 1 Is Modified in Vitro by Poly(ADP-Ribose) Polymerase 1 under Control of the Structure of Damaged DNA. Biochimie 2020, 168, 144–155. [Google Scholar] [CrossRef]
- Love, S.; Barber, R.; Wilcock, G.K. Increased Poly(ADP-Ribosyl)Ation of Nuclear Proteins in Alzheimer’s Disease. Brain 1999, 122 Pt 2, 247–253. [Google Scholar] [CrossRef]
- Hoch, N.C.; Hanzlikova, H.; Rulten, S.L.; Tétreault, M.; Komulainen, E.; Ju, L.; Hornyak, P.; Zeng, Z.; Gittens, W.; Rey, S.A.; et al. XRCC1 Mutation Is Associated with PARP1 Hyperactivation and Cerebellar Ataxia. Nature 2017, 541, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, T.; Mocle, A.J.; Hung, C.L.; Xia, J.; van Roon-Mom, W.M.C.; Truant, R. Huntingtin Is a Scaffolding Protein in the ATM Oxidative DNA Damage Response Complex. Hum. Mol. Genet. 2016, 26, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, T.; Hung, C.L.K.; Suart, C.; Begeja, N.; Barba-Bazan, C.; Peng, Y.; Savic, N.; Wong, T.; Truant, R. DNA Repair in Huntington’s Disease and Spinocerebellar Ataxias: Somatic Instability and Alternative Hypotheses. J. Huntington’s Dis. 2021, 10, 165–173. [Google Scholar] [CrossRef]
- Paldino, E.; D’Angelo, V.; Laurenti, D.; Angeloni, C.; Sancesario, G.; Fusco, F.R. Modulation of Inflammasome and Pyroptosis by Olaparib, a PARP-1 Inhibitor, in the R6/2 Mouse Model of Huntington’s Disease. Cells 2020, 9, 2286. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Greco, T.M.; Song, B.; Cristea, I.M. HTT-OMNI: A Web-Based Platform for Huntingtin Interaction Exploration and Multi-Omics Data Integration. Mol. Cell. Proteom. 2022, 21, 100275. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Kejariwal, A.; Campbell, M.J.; Mi, H.; Diemer, K.; Guo, N.; Ladunga, I.; Ulitsky-Lazareva, B.; Muruganujan, A.; Rabkin, S.; et al. PANTHER: A Browsable Database of Gene Products Organized by Biological Function, Using Curated Protein Family and Subfamily Classification. Nucleic Acids Res. 2003, 31, 334–341. [Google Scholar] [CrossRef]
- Pepe, G.; Capocci, L.; Marracino, F.; Realini, N.; Lenzi, P.; Martinello, K.; Bovier, T.F.; Bichell, T.J.; Scarselli, P.; Di Cicco, C.; et al. Treatment with THI, an Inhibitor of Sphingosine-1-Phosphate Lyase, Modulates Glycosphingolipid Metabolism and Results Therapeutically Effective in Experimental Models of Huntington’s Disease. Mol. Ther. 2023, 31, 282–299. [Google Scholar] [CrossRef]
- Lacombe, L.; Hovington, H.; Brisson, H.; Mehdi, S.; Beillevaire, D.; Émond, J.-P.; Wagner, A.; Villeneuve, L.; Simonyan, D.; Ouellet, V.; et al. UGT2B28 Accelerates Prostate Cancer Progression through Stabilization of the Endocytic Adaptor Protein HIP1 Regulating AR and EGFR Pathways. Cancer Lett. 2023, 553, 215994. [Google Scholar] [CrossRef]
- Tak, Y.J.; Kang, S. The E2 Ubiquitin-Conjugating Enzyme HIP2 Is a Crucial Regulator of Quality Control against Mutant SOD1 Proteotoxicity. Biochim. Biophys. Acta-Mol. Basis Dis. 2022, 1868, 166316. [Google Scholar] [CrossRef]
- Churkina, A.S.; Shakhov, A.S.; Kotlobay, A.A.; Alieva, I.B. Huntingtin and Other Neurodegeneration-Associated Proteins in the Development of Intracellular Pathologies: Potential Target Search for Therapeutic Intervention. Int. J. Mol. Sci. 2022, 23, 15533. [Google Scholar] [CrossRef]
- Gutiérrez-Garcia, R.; Koyuncu, S.; Hommen, F.; Bilican, S.; Lee, H.J.; Fatima, A.; Vilchez, D. G3BP1-Dependent Mechanism Suppressing Protein Aggregation in Huntington’s Models and Its Demise upon Stress Granule Assembly. Hum. Mol. Genet. 2023, 32, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Dionisio, L.E.; Yang, X.W. Group Dynamics Goes Awry: PolyQ-Expanded Huntingtin Gains Unwanted Partners. Cell Syst. 2022, 13, 268–270. [Google Scholar] [CrossRef]
- Phillips, G.R.; Saville, J.T.; Hancock, S.E.; Brown, S.H.J.; Jenner, A.M.; McLean, C.; Fuller, M.; Newell, K.A.; Mitchell, T.W. The Long and the Short of Huntington’s Disease: How the Sphingolipid Profile Is Shifted in the Caudate of Advanced Clinical Cases. Brain Commun. 2022, 4, fcab303. [Google Scholar] [CrossRef] [PubMed]
- Bonavita, R.; Scerra, G.; Di Martino, R.; Nuzzo, S.; Polishchuk, E.; Di Gennaro, M.; Williams, S.V.; Caporaso, M.G.; Caiazza, C.; Polishchuk, R.; et al. The HSPB1-P62/SQSTM1 Functional Complex Regulates the Unconventional Secretion and Transcellular Spreading of the HD-Associated Mutant Huntingtin Protein. Hum. Mol. Genet. 2023, 32, 2269–2291. [Google Scholar] [CrossRef] [PubMed]
- Ayala Mariscal, S.M.; Pigazzini, M.L.; Richter, Y.; Özel, M.; Grothaus, I.L.; Protze, J.; Ziege, K.; Kulke, M.; ElBediwi, M.; Vermaas, J.V.; et al. Identification of a HTT-Specific Binding Motif in DNAJB1 Essential for Suppression and Disaggregation of HTT. Nat. Commun. 2022, 13, 4692. [Google Scholar] [CrossRef] [PubMed]
- deGruyter, J.N.; Malins, L.R.; Baran, P.S. Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry 2017, 56, 3863–3873. [Google Scholar] [CrossRef]
- Li, Y.; Duan, P.; Guan, Y.; Chen, Q.; Grenda, A.; Christopoulos, P.; Denis, M.G.; Guo, Q. High Efficacy of Alectinib in a Patient with Advanced Lung Adenocarcinoma with 2 Rare ALK Fusion Sites: A Case Report. Transl. Lung Cancer Res. 2022, 11, 100–110. [Google Scholar] [CrossRef]
- Boler, M. Introduction to Classical and New Testament Greek; Catholic University of America Press: Washington, DC, USA, 2020; Chapter 36; pp. 293–298. ISBN 9781949822038. [Google Scholar]
- Guan, J.; Song, Z.; Wei, G.; Qiao, Q. Distinct Binding Interactions Trigger Opposite Conformational Modulations on Pathogenic and Wildtype Huntingtin Exon 1 Proteins. Phys. Chem. Chem. Phys. 2022, 24, 24959–24974. [Google Scholar] [CrossRef]
- Lemarié, F.L.; Sanders, S.S.; Nguyen, Y.; Martin, D.D.O.; Hayden, M.R. Full-Length Huntingtin Is Palmitoylated at Multiple Sites and Post-Translationally Myristoylated Following Caspase-Cleavage. Front. Physiol. 2023, 14, 1086112. [Google Scholar] [CrossRef]
- Trujillo-Del Río, C.; Tortajada-Pérez, J.; Gómez-Escribano, A.P.; Casterá, F.; Peiró, C.; Millán, J.M.; Herrero, M.J.; Vázquez-Manrique, R.P. Metformin to Treat Huntington Disease: A Pleiotropic Drug against a Multi-System Disorder. Mech. Ageing Dev. 2022, 204, 111670. [Google Scholar] [CrossRef]
- Yang, N.; Liang, Y.; Yang, P.; Jiang, L. Flurbiprofen Inhibits Cell Proliferation in Thyroid Cancer through Interrupting HIP1R-Induced Endocytosis of PTEN. Eur. J. Med. Res. 2022, 27, 29. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Jarquín, U.N.; Sharma, M.; Zhou, W.; Shahani, N.; Subramaniam, S. Deletion of SUMO1 Attenuates Behavioral and Anatomical Deficits by Regulating Autophagic Activities in Huntington Disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2107187119. [Google Scholar] [CrossRef]
- Chivet, M.; McCluskey, M.; Nicot, A.S.; Brocard, J.; Beaufils, M.; Giovannini, D.; Giannesini, B.; Poreau, B.; Brocard, J.; Humbert, S.; et al. Huntingtin Regulates Calcium Fluxes in Skeletal Muscle. J. Gen. Physiol. 2023, 155, e202213103. [Google Scholar] [CrossRef]
- Nazarov, S.; Chiki, A.; Boudeffa, D.; Lashuel, H.A. Structural Basis of Huntingtin Fibril Polymorphism Revealed by Cryogenic Electron Microscopy of Exon 1 HTT Fibrils. J. Am. Chem. Soc. 2022, 144, 10723–10735. [Google Scholar] [CrossRef] [PubMed]
- Mees, I.; Nisbet, R.M.; Hannan, A.J.; Renoir, T. Implications of Tau Dysregulation in Huntington’s Disease and Potential for New Therapeutics. J. Huntington’s Dis. 2023, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Arredondo, J.M.; Venkataraman, R.; Varkey, J.; Isas, J.M.; Situ, A.J.; Xu, H.; Chen, J.; Ulmer, T.S.; Langen, R. Molecular Basis of Q-Length Selectivity for the MW1 Antibody–Huntingtin Interaction. J. Biol. Chem. 2023, 299, 104616. [Google Scholar] [CrossRef] [PubMed]
- Sap, K.A.; Geijtenbeek, K.W.; Schipper-Krom, S.; Guler, A.T.; Reits, E.A. Ubiquitin-Modifying Enzymes in Huntington’s Disease. Front. Mol. Biosci. 2023, 10, 1107323. [Google Scholar] [CrossRef]
- Kim, H.-N.; Park, H.-J.; Lin, Y.; Cho, T.; Ryu, K.-S.; Won, H.-S.; Jin, H.-E.; Kim, J.-H.; Baek, S.-H.; Lee, Y.-H.; et al. Coiled-Coil Structure Mediated Inhibition of the Cytotoxic Huntingtin Amyloid Fibrils by an IP3 Receptor Fragment. Int. J. Biol. Macromol. 2023, 232, 123412. [Google Scholar] [CrossRef]
- Wen, Y.; Zhang, G.; Liu, L.; Zhang, P.; Lin, L.; Mei, R.; Zhang, F.; Chen, Y.; Li, R. HAP1 Interacts with 14–3-3 to Regulate Epileptic Seizure via GABAAR-Mediated Inhibitory Synaptic Transmission in Pentylenetetrazole Rat Model. Neurosci. Res. 2022, 182, 7–14. [Google Scholar] [CrossRef]
- Krzystek, T.J.; White, J.A.; Rathnayake, R.; Thurston, L.; Hoffmar-Glennon, H.; Li, Y.; Gunawardena, S. HTT (Huntingtin) and RAB7 Co-Migrate Retrogradely on a Signaling LAMP1-Containing Late Endosome during Axonal Injury. Autophagy 2023, 19, 1199–1220. [Google Scholar] [CrossRef]
- Vagiona, A.-C.; Mier, P.; Petrakis, S.; Andrade-Navarro, M.A. Analysis of Huntington’s Disease Modifiers Using the Hyperbolic Mapping of the Protein Interaction Network. Int. J. Mol. Sci. 2022, 23, 5853. [Google Scholar] [CrossRef]
- Paldino, E.; Fusco, F.R. Emerging Role of NLRP3 Inflammasome/Pyroptosis in Huntington’s Disease. Int. J. Mol. Sci. 2022, 23, 8363. [Google Scholar] [CrossRef] [PubMed]
- Matlahov, I.; Boatz, J.C.; van der Wel, P.C.A. Selective Observation of Semi-Rigid Non-Core Residues in Dynamically Complex Mutant Huntingtin Protein Fibrils. J. Struct. Biol. X 2022, 6, 100077. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, C.; Zhu, C.; Wang, Z.; Yang, H.; Wu, P.; Cui, X.; Botas, J.; Dang, Y.; Ding, Y.; et al. Suppression of Toxicity of the Mutant Huntingtin Protein by Its Interacting Compound, Desonide. Proc. Natl. Acad. Sci. USA 2022, 119, e2114303119. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, T.; Jiang, M.; O’Meally, R.N.; Rauniyar, P.; Chighladze, E.; Faragó, A.; Kamath, S.V.; Jin, J.; Shevelkin, A.V.; Cole, R.N.; et al. Interaction of Huntingtin with PRMTs and Its Subsequent Arginine Methylation Affects HTT Solubility, Phase Transition Behavior and Neuronal Toxicity. Hum. Mol. Genet. 2022, 31, 1651–1672. [Google Scholar] [CrossRef] [PubMed]
- Abjean, L.; Ben Haim, L.; Riquelme-Perez, M.; Gipchtein, P.; Derbois, C.; Palomares, M.-A.; Petit, F.; Hérard, A.-S.; Gaillard, M.-C.; Guillermier, M.; et al. Reactive Astrocytes Promote Proteostasis in Huntington’s Disease through the JAK2-STAT3 Pathway. Brain 2023, 146, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Latoszek, E.; Wiweger, M.; Ludwiczak, J.; Dunin-Horkawicz, S.; Kuznicki, J.; Czeredys, M. Siah-1-Interacting Protein Regulates Mutated Huntingtin Protein Aggregation in Huntington’s Disease Models. Cell Biosci. 2022, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, A.; Wang, Z.; Xu, X.-H.; Tao, Y. Biological Functions and Potential Therapeutic Applications of Huntingtin-Associated Protein 1: Progress and Prospects. Clin. Transl. Oncol. 2022, 24, 203–214. [Google Scholar] [CrossRef]
- Seefelder, M.; Klein, F.A.C.; Landwehrmeyer, B.; Fernández-Busnadiego, R.; Kochanek, S. Huntingtin and Its Partner Huntingtin-Associated Protein 40: Structural and Functional Considerations in Health and Disease. J. Huntington’s Dis. 2022, 11, 227–242. [Google Scholar] [CrossRef]
- Lee, Y.H.; Tsai, Y.; Chang, C.; Ho, C.; Shih, H.; Chen, H.; Lai, H.; Lee, C.; Lee, Y.; Liao, Y.; et al. A PIAS1 Protective Variant S510G Delays PolyQ Disease Onset by Modifying Protein Homeostasis. Mov. Disord. 2022, 37, 767–777. [Google Scholar] [CrossRef]
- Yang, J.; Xu, H.; Zhang, C.; Yang, X.; Cai, W.; Chen, X. A Prion-like Domain of TFEB Mediates the Co-Aggregation of TFEB and MHTT. Autophagy 2023, 19, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, K.; Trojniak, A.E.; Guzmán Rodríguez, K.B.; Klus, N.J.; Huntley, C.; McDonald, P.; Roy, A.; Frankowski, K.J.; Aubé, J.; Muma, N.A. Small-Molecule Disruptors of Mutant Huntingtin–Calmodulin Protein–Protein Interaction Attenuate Deleterious Effects of Mutant Huntingtin. ACS Chem. Neurosci. 2022, 13, 2315–2337. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lu, C.; Chen, Y.; Li, S.; Ma, N.; Tao, X.; Li, Y.; Wang, J.; Zhou, M.; Yan, Y.-B.; et al. CCT2 Is an Aggrephagy Receptor for Clearance of Solid Protein Aggregates. Cell 2022, 185, 1325–1345. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Wu, Y.-Y.; Feng, Y.; Hsieh, W.-C.; Song, J.-S.; Lin, Y.-S.; Tseng, Y.-H.; Liao, W.-J.; Chu, Y.-F.; Liu, Y.-C.; et al. Chemical Interference with DSIF Complex Formation Lowers Synthesis of Mutant Huntingtin Gene Products and Curtails Mutant Phenotypes. Proc. Natl. Acad. Sci. USA 2022, 119, e2204779119. [Google Scholar] [CrossRef] [PubMed]
- Díaz Casas, A.; Cordoba, J.J.; Ferrer, B.J.; Balakrishnan, S.; Wurm, J.E.; Pastrana-Ríos, B.; Chazin, W.J. Binding by Calmodulin Is Coupled to Transient Unfolding of the Third ff Domain of prp40a. Protein Sci. 2023, 32, e4606. [Google Scholar] [CrossRef]
- Prowse, E.N.P.; Chaudhary, A.R.; Sharon, D.; Hendricks, A.G. Huntingtin S421 Phosphorylation Increases Kinesin and Dynein Engagement on Early Endosomes and Lysosomes. Biophys. J. 2023, 122, 1168–1184. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Bensalel, J.; Raju, S.; Capobianco, E.; Lu, M.L.; Wei, J. Characterization of Huntingtin Interactomes and Their Dynamic Responses in Living Cells by Proximity Proteomics. J. Neurochem. 2023, 164, 512–528. [Google Scholar] [CrossRef] [PubMed]
- Beasley, M.; Frazee, N.; Groover, S.; Valentine, S.J.; Mertz, B.; Legleiter, J. Physicochemical Properties Altered by the Tail Group of Lipid Membranes Influence Huntingtin Aggregation and Lipid Binding. J. Phys. Chem. B 2022, 126, 3067–3081. [Google Scholar] [CrossRef]
- Moharir, S.C.; Raghawan, A.K.; Ramaswamy, R.; Swarup, G. Autophagy-Independent Cytoprotection by Optineurin from Toxicity of Aggregates Formed by Mutant Huntingtin and Mutant Ataxin-3. J. Biochem. 2022, 171, 555–565. [Google Scholar] [CrossRef]
- Baines, K.; Yoshioka, K.; Takuwa, Y.; Lane, J.D. The ATG5 Interactome Links Clathrin-Mediated Vesicular Trafficking with the Autophagosome Assembly Machinery. Autophagy Rep. 2022, 1, 88–118. [Google Scholar] [CrossRef]
- Fatima, N.; Alomari, M.; Belov, L.; Shen, Y.; Christopherson, R.I. Adenylated Proteins in Mouse B16-F10 Melanoma Cells Cluster in Functional Categories: A New Paradigm for Cellular Regulation? Nucleosides Nucleotides Nucleic Acids 2022, 41, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Li, G.; Ye, X.; Chen, D.; Chen, Z.; Xu, Z.; Daniele, M.; Tambone, S.; Ceccacci, A.; Tomei, L.; et al. HAP40 Is a Conserved Central Regulator of Huntingtin and a Potential Modulator of Huntington’s Disease Pathogenesis. PLoS Genet. 2022, 18, e1010302. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The Reactome Pathway Knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Różycka, A.; Oczkowska, A.; Florczak-Wyspiańska, J.; Prendecki, M.; Dezor, M.; Postrach, I.; Jagodzinski, P.P.; Kozubski, W. Mutations of TP53 Gene and Oxidative Stress in Alzheimer’s Disease Patients. Adv. Alzheimer’s Dis. 2014, 3, 24–32. [Google Scholar] [CrossRef]
- Fukui, H.; Rünker, A.; Fabel, K.; Buchholz, F.; Kempermann, G. Transcription Factor Runx1 Is Pro-Neurogenic in Adult Hippocampal Precursor Cells. PLoS ONE 2018, 13, e0190789. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, H.; Zelenin, P.; Fontanet, P.; Wanderoy, S.; Petitpré, C.; Comai, G.; Bellardita, C.; Xue-Franzén, Y.; Huettl, R.-E.; et al. Muscle-Selective RUNX3 Dependence of Sensorimotor Circuit Development. Development 2019, 146, dev181750. [Google Scholar] [CrossRef] [PubMed]
- Kortuem, K.M.; Stewart, A.K. Carfilzomib. Blood 2013, 121, 893–897. [Google Scholar] [CrossRef]
- Pawaskar, D.K.; Straubinger, R.M.; Fetterly, G.J.; Hylander, B.H.; Repasky, E.A.; Ma, W.W.; Jusko, W.J. Synergistic Interactions between Sorafenib and Everolimus in Pancreatic Cancer Xenografts in Mice. Cancer Chemother. Pharmacol. 2013, 71, 1231–1240. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Napolitano, F.; Carrella, D.; Mandriani, B.; Pisonero-Vaquero, S.; Sirci, F.; Medina, D.L.; Brunetti-Pierri, N.; di Bernardo, D. Gene2drug: A Computational Tool for Pathway-Based Rational Drug Repositioning. Bioinformatics 2018, 34, 1498–1505. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makeeva, V.S.; Dyrkheeva, N.S.; Lavrik, O.I.; Zakian, S.M.; Malakhova, A.A. Mutant-Huntingtin Molecular Pathways Elucidate New Targets for Drug Repurposing. Int. J. Mol. Sci. 2023, 24, 16798. https://doi.org/10.3390/ijms242316798
Makeeva VS, Dyrkheeva NS, Lavrik OI, Zakian SM, Malakhova AA. Mutant-Huntingtin Molecular Pathways Elucidate New Targets for Drug Repurposing. International Journal of Molecular Sciences. 2023; 24(23):16798. https://doi.org/10.3390/ijms242316798
Chicago/Turabian StyleMakeeva, Vladlena S., Nadezhda S. Dyrkheeva, Olga I. Lavrik, Suren M. Zakian, and Anastasia A. Malakhova. 2023. "Mutant-Huntingtin Molecular Pathways Elucidate New Targets for Drug Repurposing" International Journal of Molecular Sciences 24, no. 23: 16798. https://doi.org/10.3390/ijms242316798
APA StyleMakeeva, V. S., Dyrkheeva, N. S., Lavrik, O. I., Zakian, S. M., & Malakhova, A. A. (2023). Mutant-Huntingtin Molecular Pathways Elucidate New Targets for Drug Repurposing. International Journal of Molecular Sciences, 24(23), 16798. https://doi.org/10.3390/ijms242316798