
A Comparative Analysis of Selection Pressures Suffered by Mitochondrial Genomes in Two Planthopper Species with Divergent Climate Distributions


Abstract
:1. Introduction
2. Results
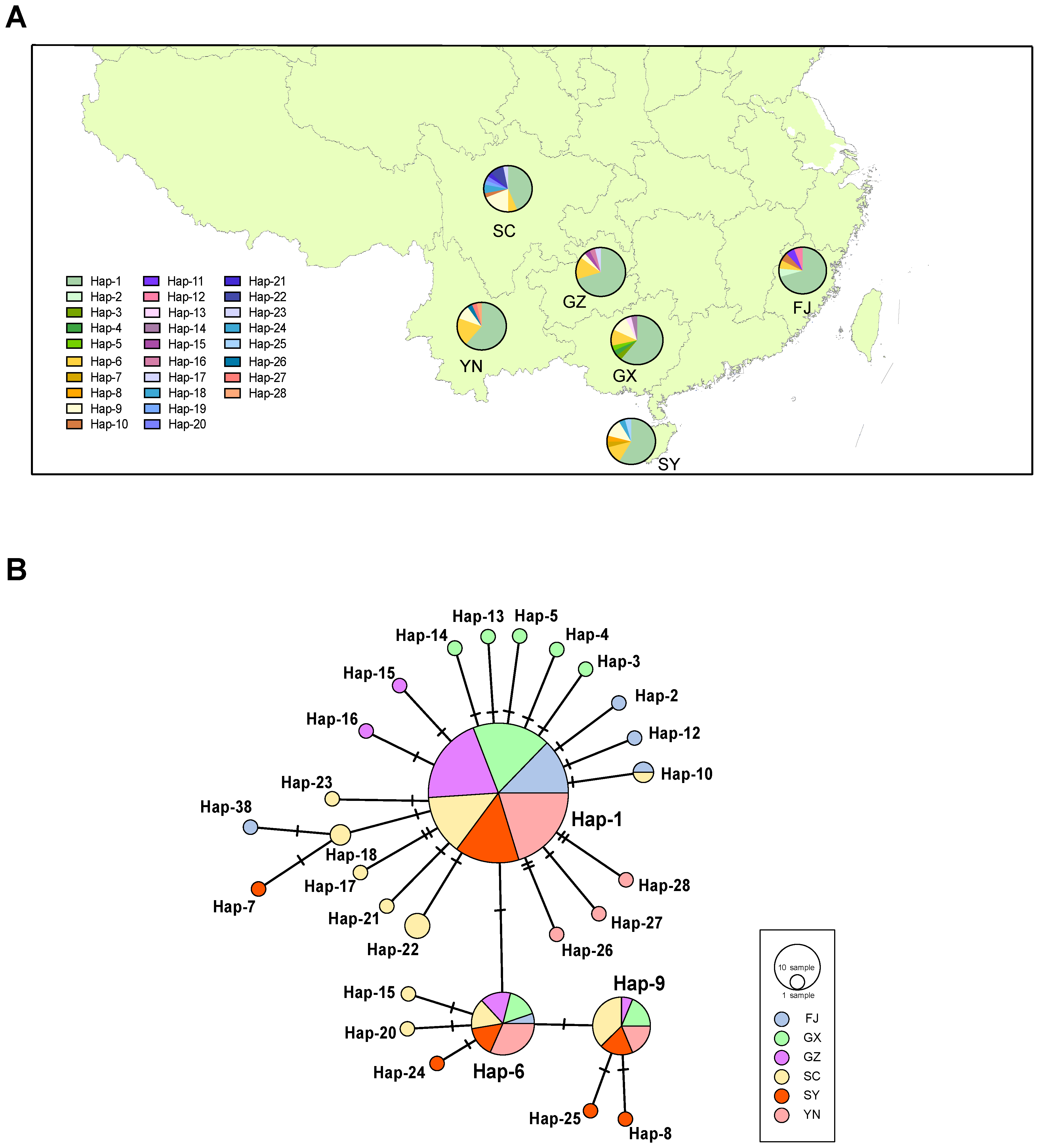
2.1. WBPH Population Genetic Structure
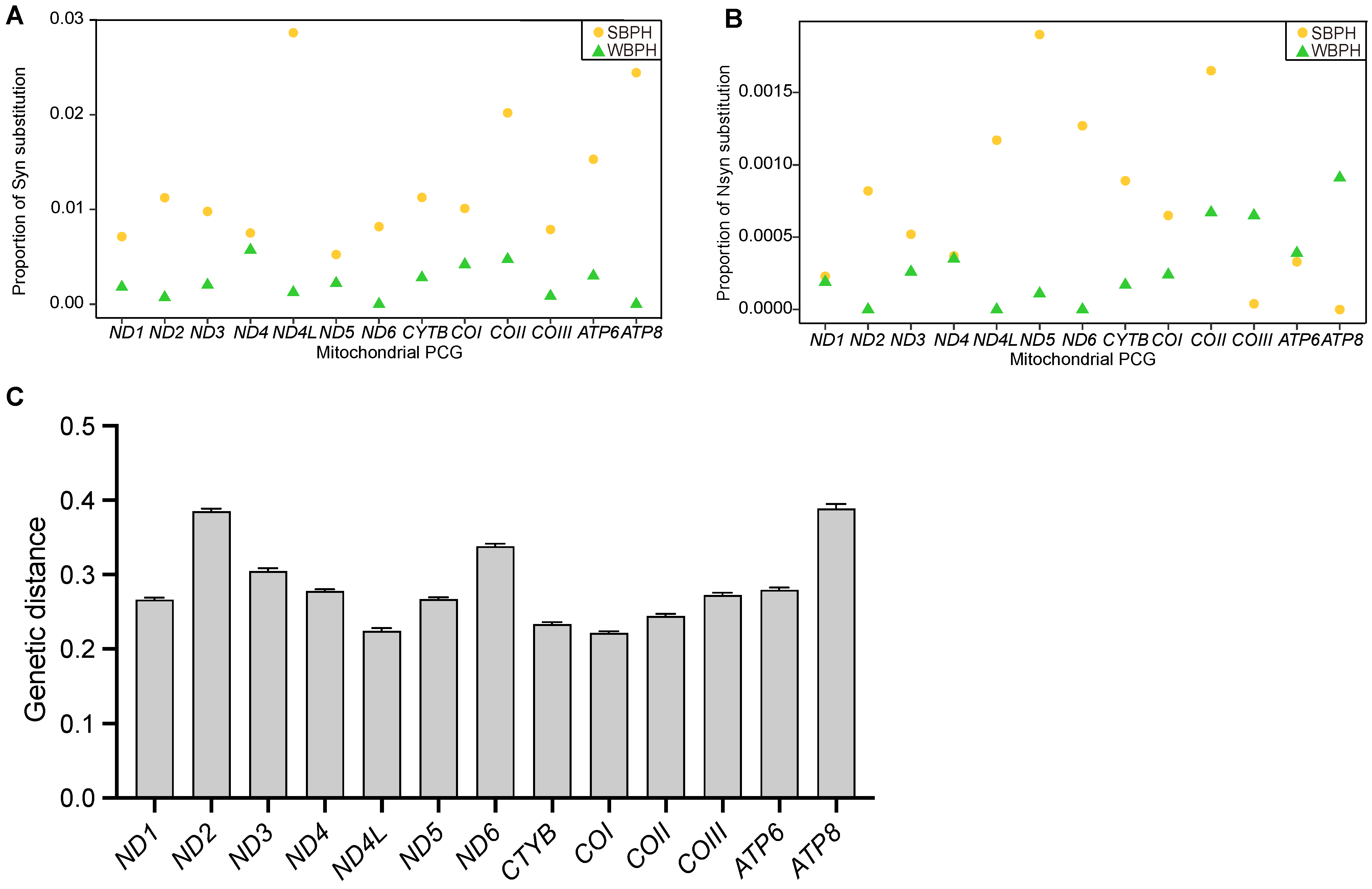
2.2. Mitogenome Genetic Diversity and Divergence
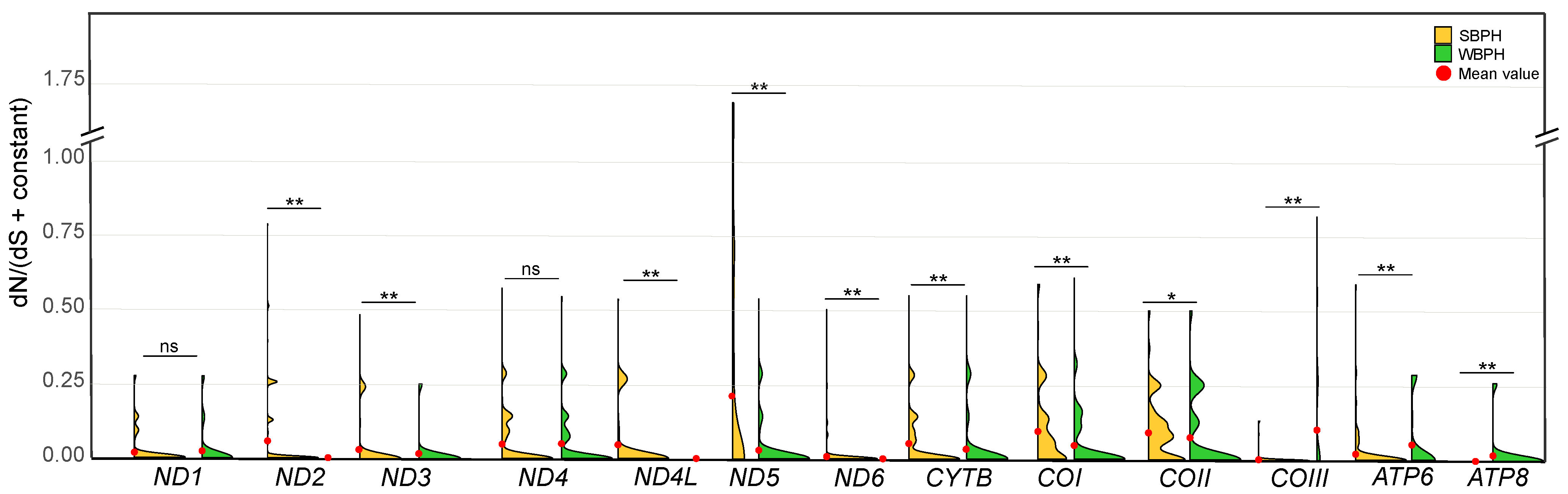
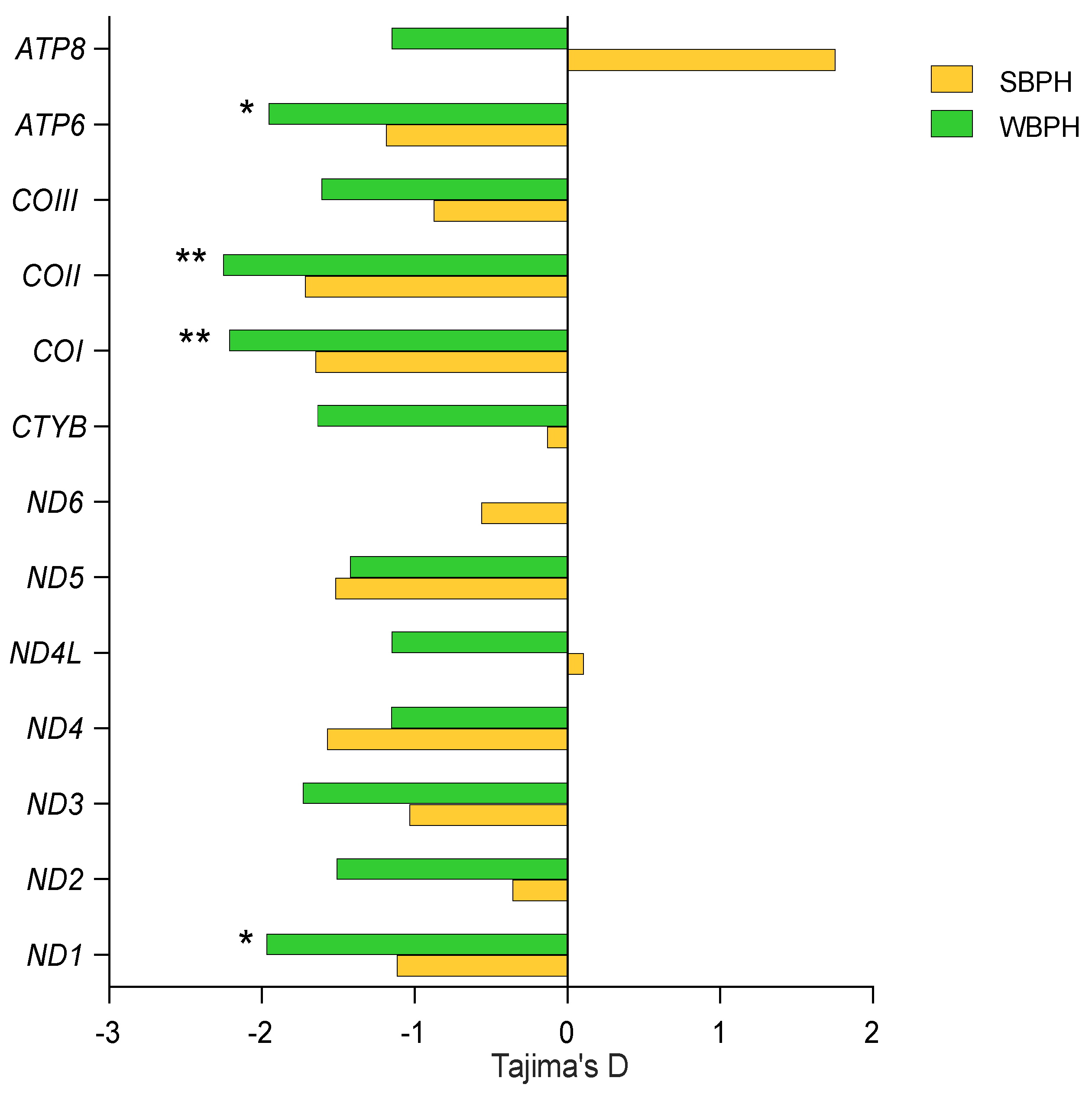
2.3. Selection Pressure Comparisons
3. Discussion
4. Materials and Methods
4.1. Sample Collection and DNA Extraction
4.2. Mitochondrial Haplotype Characterization
4.3. Mitogenome Sequencing and Annotations
4.4. WBPH Population Genetic Structure
4.5. Mitogenome Genetic Diversity Comparison
4.6. dN/dS Analysis
4.7. McDonald–Kreitman Test Analysis
4.8. Genetic Distance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ballard, J.W.; James, A.C. Differential fitness of mitochondrial DNA in perturbation cage studies correlates with global abundance and population history in Drosophila simulans. Proc. Biol. Sci./R. Soc. 2004, 271, 1197–1201. [Google Scholar] [CrossRef]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography—The mitochondrial-DNA bridge between population-genetics and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Dowling, D.K.; Wolff, J.N. Evolutionary genetics of the mitochondrial genome: Insights from Drosophila. Genetics 2023, 224, iyad036. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.W.O.; Pichaud, N. Mitochondrial DNA: More than an evolutionary bystander. Funct. Ecol. 2014, 28, 218–231. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.E. Mitonuclear Ecology. Mol. Biol. Evol. 2015, 32, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Bazin, E.; Glémin, S.; Galtier, N. Population size does not influence mitochondrial genetic diversity in animals. Science 2006, 312, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Garvin, M.R.; Bielawski, J.P.; Sazanov, L.A.; Gharrett, A.J. Review and meta-analysis of natural selection in mitochondrial complex I in metazoans. J. Zool. Syst. Evol. Res. 2015, 53, 1–17. [Google Scholar] [CrossRef]
- Sun, J.T.; Duan, X.Z.; Hoffmann, A.A.; Liu, Y.; Garvin, M.R.; Chen, L.; Hu, G.; Zhou, J.C.; Huang, H.J.; Xue, X.F.; et al. Mitochondrial variation in small brown planthoppers linked to multiple traits and probably reflecting a complex evolutionary trajectory. Mol. Ecol. 2019, 28, 3306–3323. [Google Scholar] [CrossRef]
- Camus, M.F.; Wolff, J.N.; Sgro, C.M.; Dowling, D.K. Experimental support that natural selection has shaped the latitudinal distribution of mitochondrial haplotypes in Australian Drosophila melanogaster. Mol. Biol. Evol. 2017, 34, 2600–2612. [Google Scholar] [CrossRef]
- Ballard, J.W.; Melvin, R.G.; Katewa, S.D.; Maas, K. Mitochondrial DNA variation is associated with measurable differences in life-history traits and mitochondrial metabolism in Drosophila simulans. Evolution 2007, 61, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.T.; Ingelmo, P.; Rand, D.M. GxGxE for lifespan in Drosophila: Mitochondrial, nuclear, and dietary interactions that modify longevity. PLoS Genet. 2014, 10, e1004354. [Google Scholar] [CrossRef] [PubMed]
- Zsengellér, Z.K.; Ellezian, L.; Brown, D.; Horváth, B.; Mukhopadhyay, P.; Kalyanaraman, B.; Parikh, S.M.; Karumanchi, S.A.; Stillman, I.E.; Pacher, P. Cisplatin Nephrotoxicity Involves Mitochondrial Injury with Impaired Tubular Mitochondrial Enzyme Activity. J. Histochem. Cytochem. 2012, 60, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Mun, J.H.; Song, Y.H.; Heong, K.L.; Roderick, G.K. Genetic variation among Asian populations of rice planthoppers, Nilaparvata lugens and Sogatella furcifera (Hemiptera: Delphacidae): Mitochondrial DNA sequences. Bull. Entomol. Res. 1999, 89, 245–253. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Matsumura, M.; Sanada-Morimura, S.; Hirai, Y.; Sato, Y.; Noda, H. Mitochondrial cox sequences of Nilaparvata lugens and Sogatella furcifera (Hemiptera, Delphacidae): Low specificity among Asian planthopper populations. Bull. Entomol. Res. 2013, 103, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Dong, Z.; Chen, A.; Yin, Y.; Chu, D. Migration of Sogatella furcifera between the Greater Mekong Subregion and northern China revealed by mtDNA and SNP. BMC Evol. Biol. 2020, 20, 154. [Google Scholar] [CrossRef]
- Sun, J.T.; Jiang, X.Y.; Wang, M.M.; Hong, X.Y. Development of microsatellite markers for, and a preliminary population genetic analysis of, the white-backed planthopper. Bull. Entomol. Res. 2014, 104, 765–773. [Google Scholar] [CrossRef]
- Morales, H.E.; Pavlova, A.; Joseph, L.; Sunnucks, P. Positive and purifying selection in mitochondrial genomes of a bird with mitonuclear discordance. Mol. Ecol. 2015, 24, 2820–2837. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Melvin, R.G. Linking the mitochondrial genotype to the organismal phenotype. Mol. Ecol. 2010, 19, 1523–1539. [Google Scholar] [CrossRef]
- Mustafa, M.F.; Fakurazi, S.; Abdullah, M.A.; Maniam, S. Pathogenic Mitochondria DNA Mutations: Current Detection Tools and Interventions. Genes 2020, 11, 192. [Google Scholar] [CrossRef]
- McDonald, J.H.; Kreitman, M. Adaptive protein evolution at the Adh locus in Drosophila. Nature 1991, 351, 652–654. [Google Scholar] [CrossRef]
- Hartley, A.M.; Lukoyanova, N.; Zhang, Y.; Cabrera-Orefice, A.; Arnold, S.; Meunier, B.; Pinotsis, N.; Maréchal, A. Structure of yeast cytochrome c oxidase in a supercomplex with cytochrome bc1. Nat. Struct. Mol. Biol. 2019, 26, 78–83. [Google Scholar] [CrossRef]
- Burton, R.S.; Barreto, F.S. A disproportionate role for mtDNA in Dobzhansky-Muller incompatibilities? Mol. Ecol. 2012, 21, 4942–4957. [Google Scholar] [CrossRef]
- Rand, D.M.; Haney, R.A.; Fry, A.J. Cytonuclear coevolution: The genomics of cooperation. Trends Ecol. Evol. 2004, 19, 645–653. [Google Scholar] [CrossRef]
- Dowling, D.K.; Friberg, U.; Hailer, F.; Arnqvist, G. Intergenomic epistasis for fitness: Within-population interactions between cytoplasmic and nuclear genes in Drosophila melanogaster. Genetics 2007, 175, 235–244. [Google Scholar] [CrossRef]
- Ellison, C.K.; Burton, R.S. Interpopulation hybrid breakdown maps to the mitochondrial genome. Evolution 2008, 62, 631–638. [Google Scholar] [CrossRef]
- Kazancıoğlu, E.; Arnqvist, G. The maintenance of mitochondrial genetic variation by negative frequency-dependent selection. Ecol. Lett. 2014, 17, 22–27. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, R.R.; Johnson, W.E.; O’Brien, S.J.; Ramos, M.J.; Antunes, A. The adaptive evolution of the mammalian mitochondrial genome. BMC Genom. 2008, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.T.; Jin, P.Y.; Hoffmann, A.A.; Duan, X.Z.; Dai, J.; Hu, G.; Xue, X.F.; Hong, X.Y. Evolutionary divergence of mitochondrial genomes in two Tetranychus species distributed across different climates. Insect Mol. Biol. 2018, 27, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Howe, D.K.; Denver, D.R. Muller’s Ratchet and compensatory mutation in Caenorhabditis briggsae mitochondrial genome evolution. BMC Evol. Biol. 2008, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.J.; Zhu, W.C.; Rong, X.; Liu, J.; Ding, X.L.; Hong, X.Y. The complete mitochondrial genome sequence of Sogatella furcifera (Horváth) and a comparative mitogenomic analysis of three predominant rice planthoppers. Gene 2014, 533, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Mishmar, D.; Ruiz-Pesini, E.; Golik, P.; Macaulay, V.; Clark, A.G.; Hosseini, S.; Brandon, M.; Easley, K.; Chen, E.; Brown, M.D.; et al. Natural selection shaped regional mtDNA variation in humans. Proc. Natl. Acad. Sci. USA 2003, 100, 171–176. [Google Scholar] [CrossRef]
- Egea, R.; Casillas, S.; Barbadilla, A. Standard and generalized McDonald-Kreitman test: A website to detect selection by comparing different classes of DNA sites. Nucleic Acids Res. 2008, 36, W157–W162. [Google Scholar] [CrossRef]
- Jukes, T.H.; Cantor, C.R. CHAPTER 24—Evolution of Protein Molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: Cambridge, MA, USA, 1969; pp. 21–132. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locality/Zone | Number of Haplotypes, Nh | Number of Private Haplotypes, Np | Haplotype Diversity, Hd | Nucleotide Diversity, π(%) |
|---|---|---|---|---|
| FJ | 6 | 3 | 0.51471 | 0.038 |
| GX | 8 | 5 | 0.62434 | 0.049 |
| GZ | 6 | 3 | 0.49573 | 0.037 |
| SC | 10 | 6 | 0.77621 | 0.074 |
| SY | 7 | 4 | 0.64855 | 0.068 |
| YN | 6 | 3 | 0.59355 | 0.050 |
| Gene | Syn Divergence | Syn Polymorphism | Nsyn Divergence | Nsyn Polymorphism | NI | p-Value |
|---|---|---|---|---|---|---|
| ND1 | 270.02 | 16 | 78.44 | 3 | 0.645 | 0.492 |
| ND2 | 361.84 | 12 | 166.08 | 6 | 1.089 | 0.866 |
| ND3 | 68.56 | 6 | 51.94 | 4 | 0.879 | 0.848 |
| ND4 | 196.47 | 24 | 197.25 | 14 | 0.581 | 0.118 |
| ND4L | 28.82 | 7 | 29.58 | 3 | 0.417 | 0.227 |
| ND5 | 251.93 | 26 | 229.73 | 23 | 0.97 | 0.919 |
| ND6 | 87.77 | 5 | 96.91 | 2 | 0.362 | 0.214 |
| CYTB | 387.62 | 16 | 52.04 | 9 | 4.189 | <0.001 |
| COI | 518.98 | 50 | 40.93 | 13 | 3.296 | <0.001 |
| COII | 250.13 | 30 | 26.64 | 20 | 4.55 | <0.001 |
| COIII | 245.5 | 11 | 67.76 | 4 | 1.317 | 0.644 |
| ATP6 | 144.28 | 22 | 71.26 | 5 | 0.46 | 0.124 |
| ATP8 | 23.57 | 1 | 21.47 | 1 | 1.097 | 0.948 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, K.-K.; Ding, Y.; Chen, L.; Sun, J.-T. A Comparative Analysis of Selection Pressures Suffered by Mitochondrial Genomes in Two Planthopper Species with Divergent Climate Distributions. Int. J. Mol. Sci. 2023, 24, 16847. https://doi.org/10.3390/ijms242316847
Sun K-K, Ding Y, Chen L, Sun J-T. A Comparative Analysis of Selection Pressures Suffered by Mitochondrial Genomes in Two Planthopper Species with Divergent Climate Distributions. International Journal of Molecular Sciences. 2023; 24(23):16847. https://doi.org/10.3390/ijms242316847
Chicago/Turabian StyleSun, Kang-Kang, Yi Ding, Lei Chen, and Jing-Tao Sun. 2023. "A Comparative Analysis of Selection Pressures Suffered by Mitochondrial Genomes in Two Planthopper Species with Divergent Climate Distributions" International Journal of Molecular Sciences 24, no. 23: 16847. https://doi.org/10.3390/ijms242316847
APA StyleSun, K.-K., Ding, Y., Chen, L., & Sun, J.-T. (2023). A Comparative Analysis of Selection Pressures Suffered by Mitochondrial Genomes in Two Planthopper Species with Divergent Climate Distributions. International Journal of Molecular Sciences, 24(23), 16847. https://doi.org/10.3390/ijms242316847