
Role of Conformational Dynamics of Sulfotransferases SULT1A1 and SULT1A3 in Substrate Specificity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
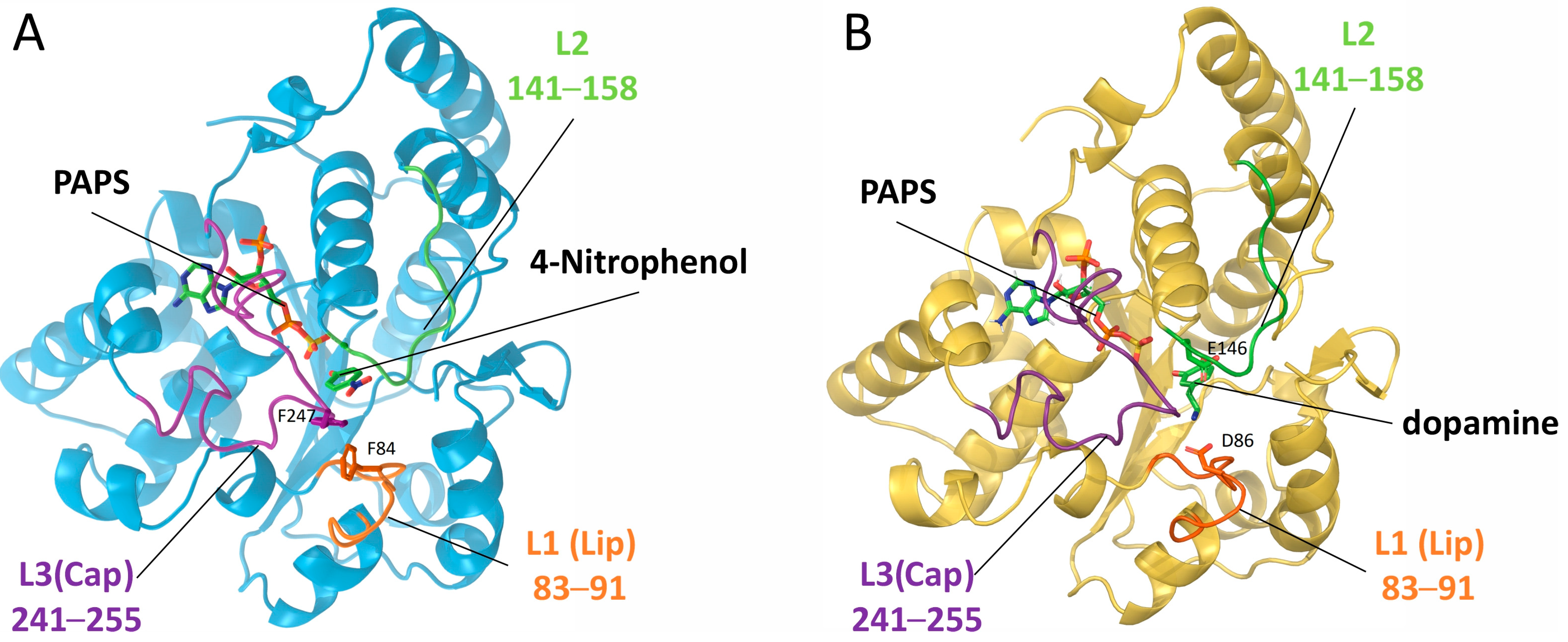
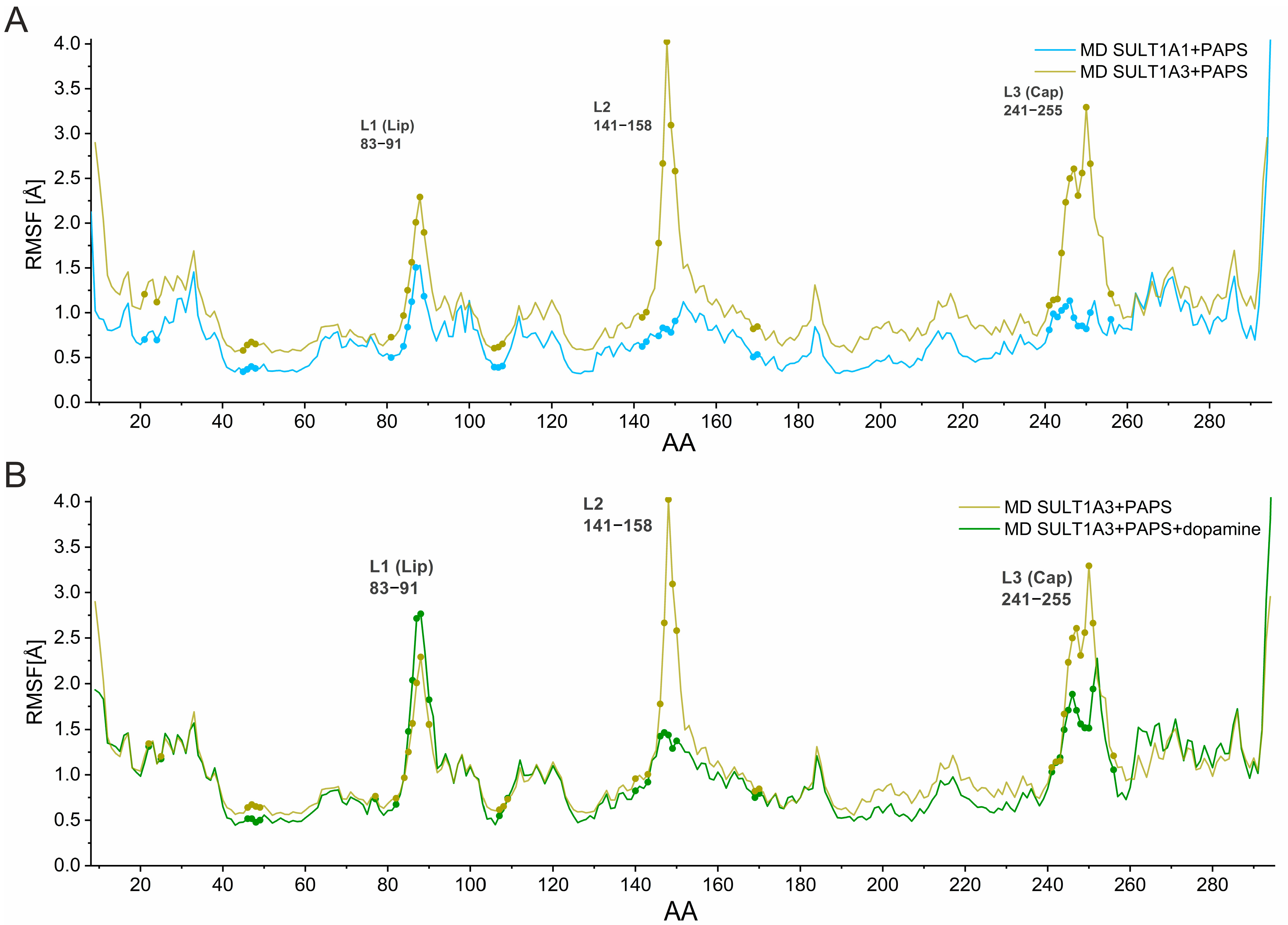
2.1. Structural Analysis of SULT1A1 and SULT1A3
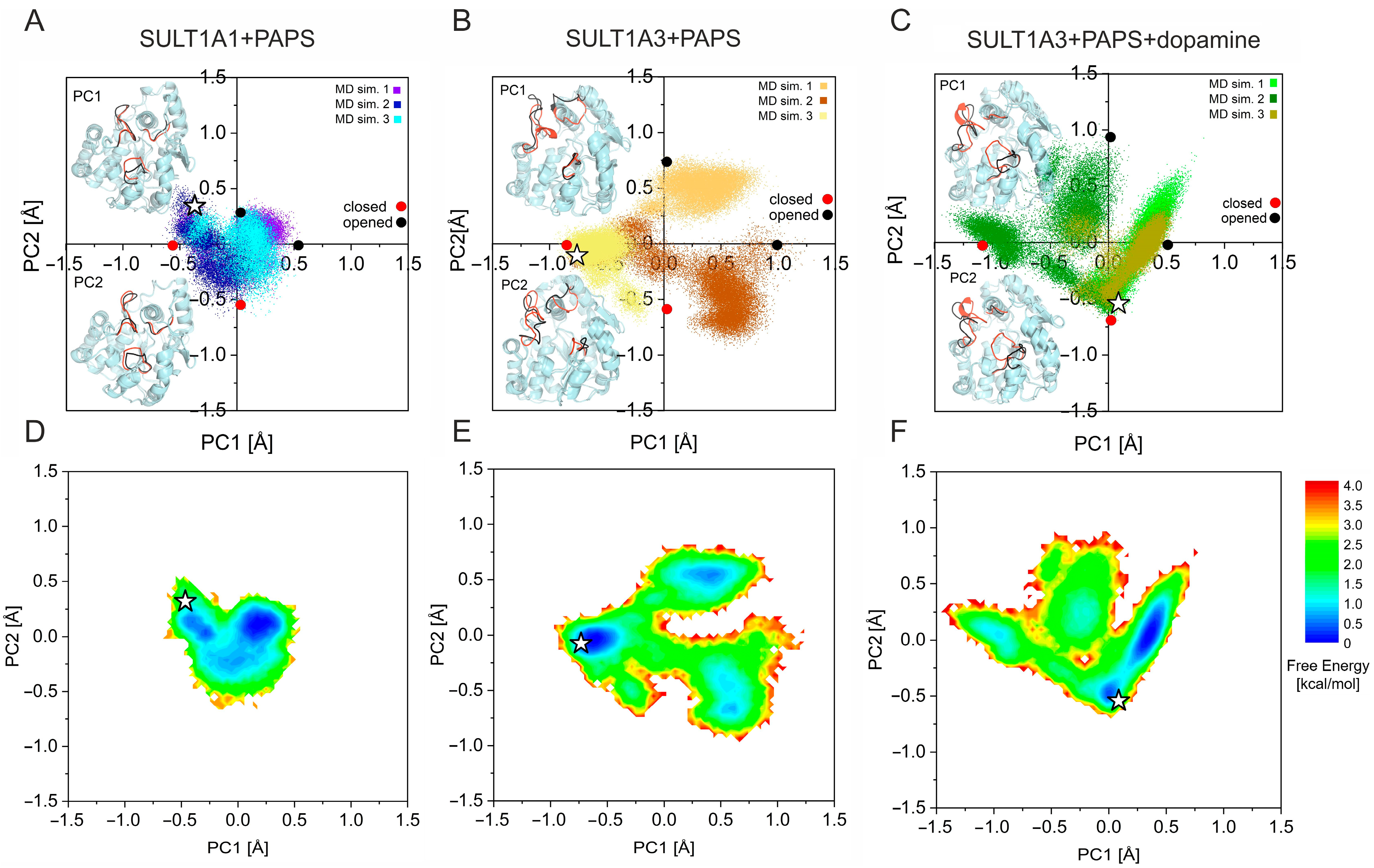
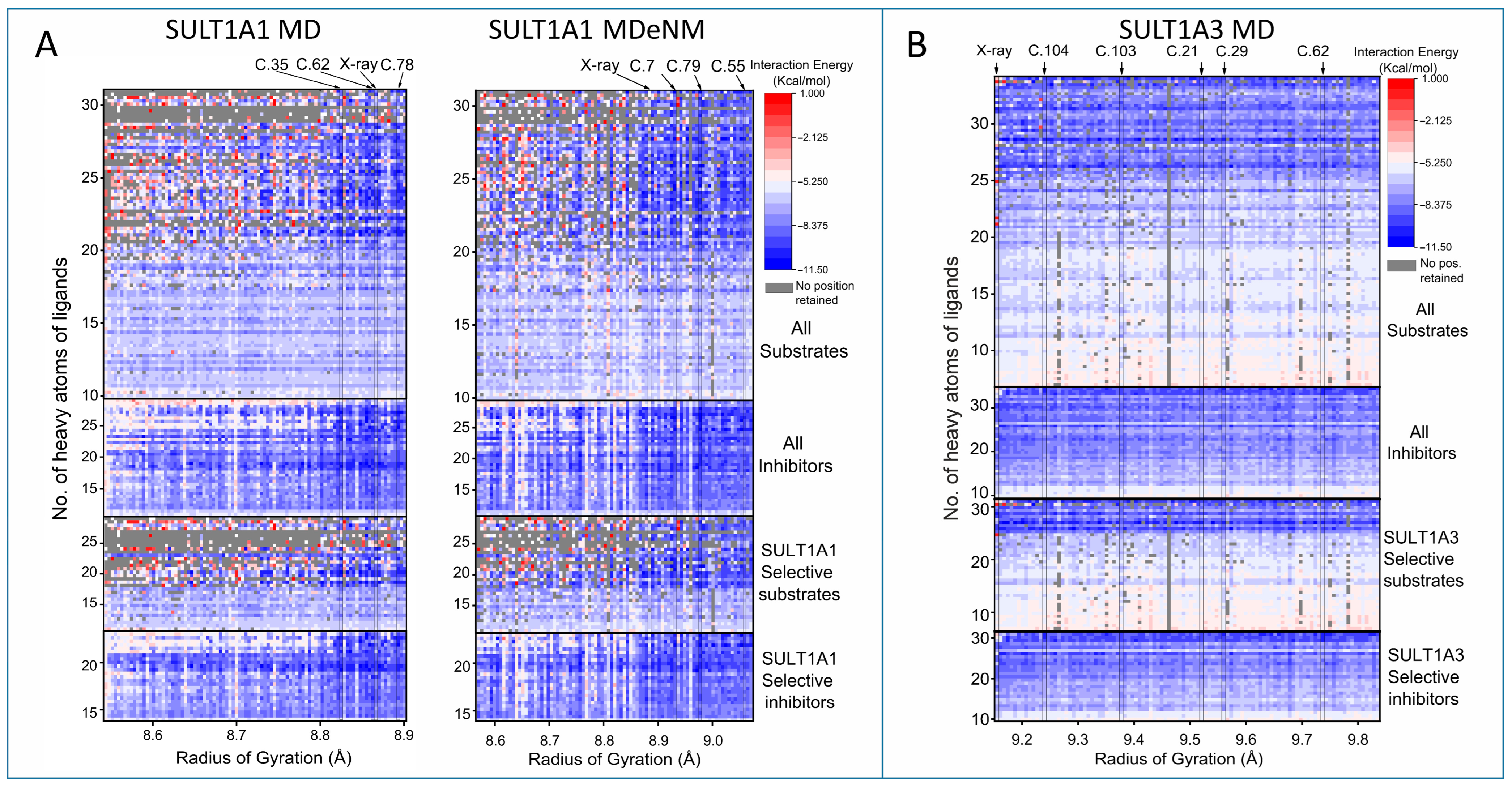
2.2. Clustering and Ensemble Docking
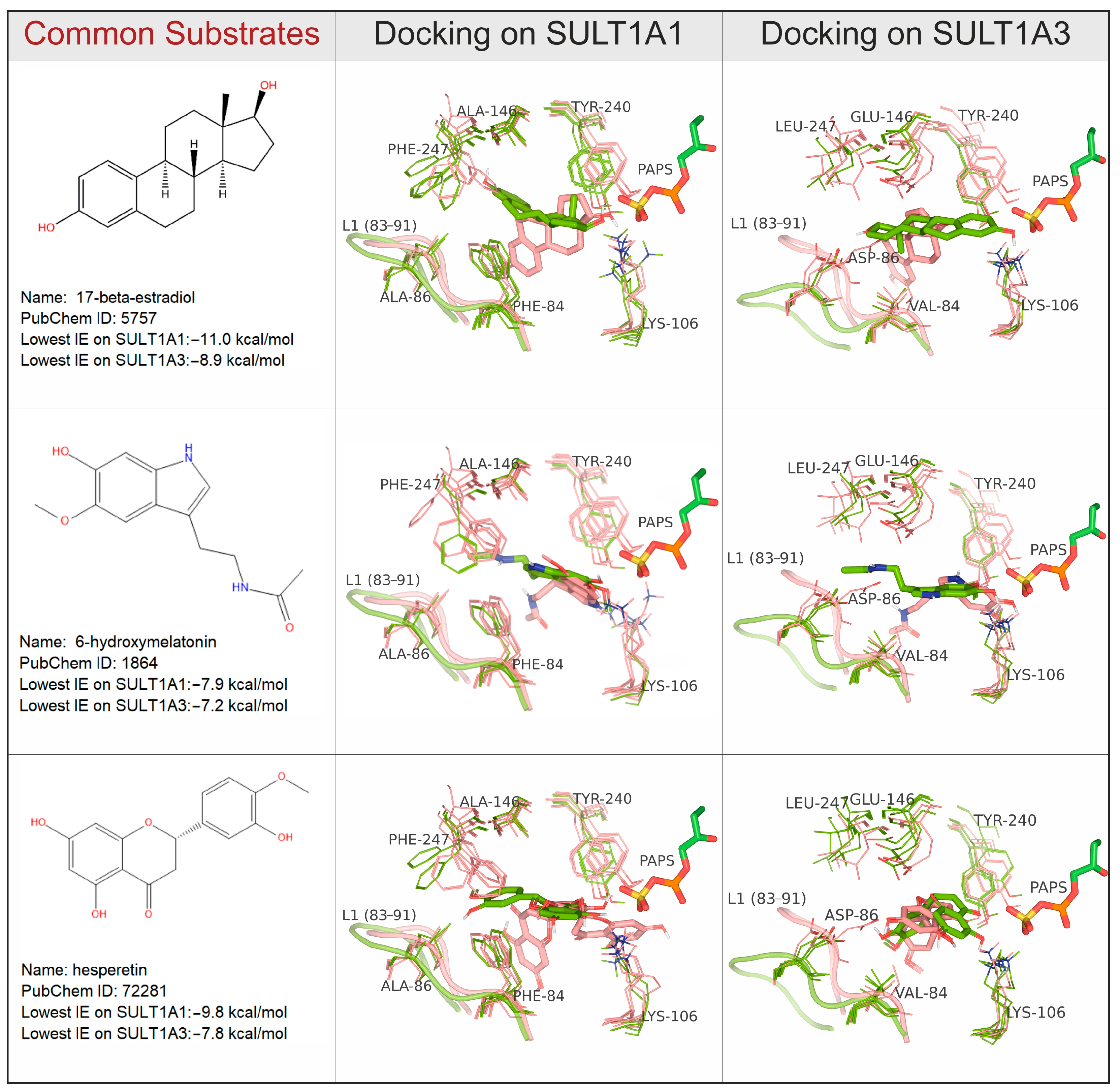
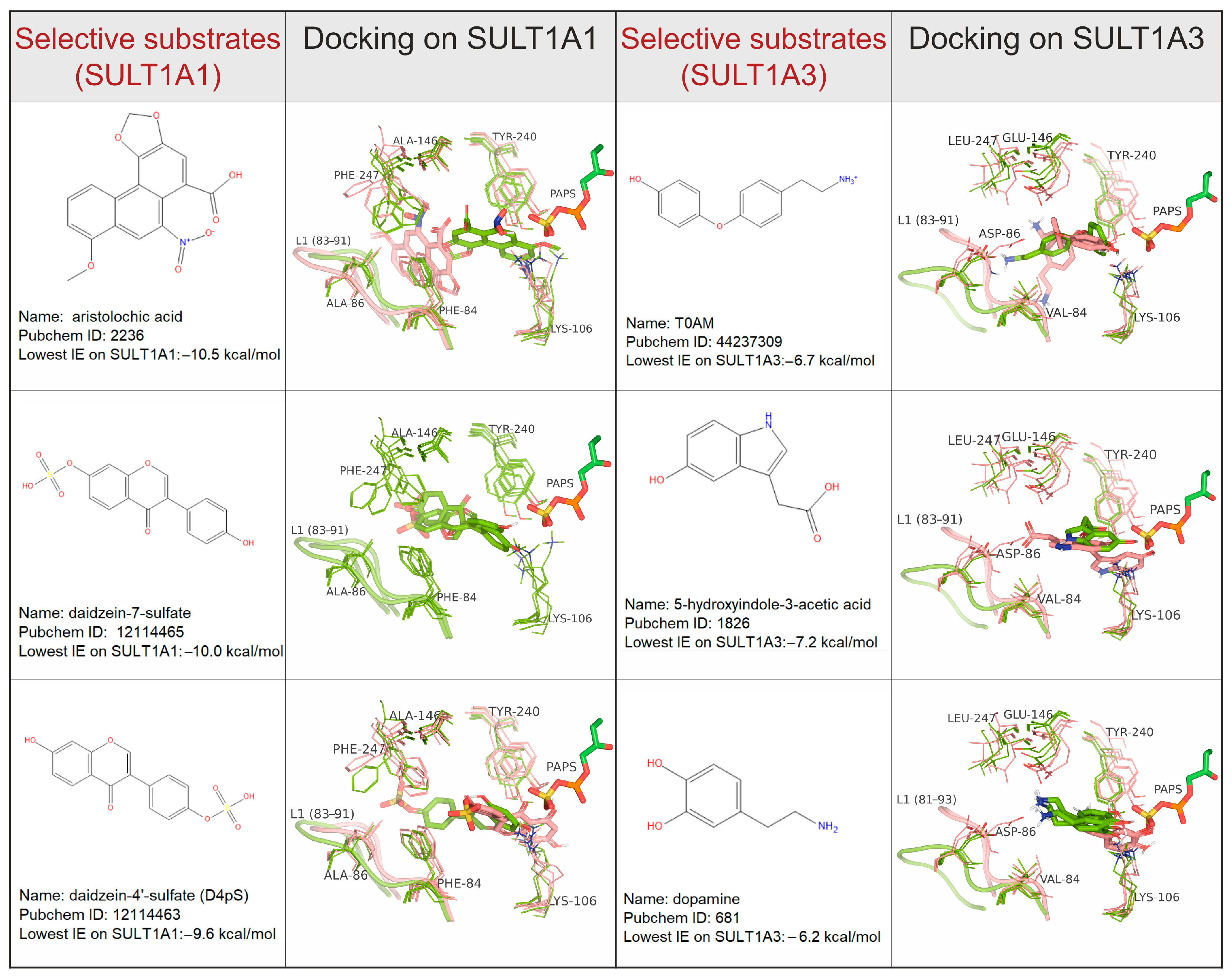
2.3. Specific Interactions with Substrates
3. Materials and Methods
3.1. Preparation of the Systems
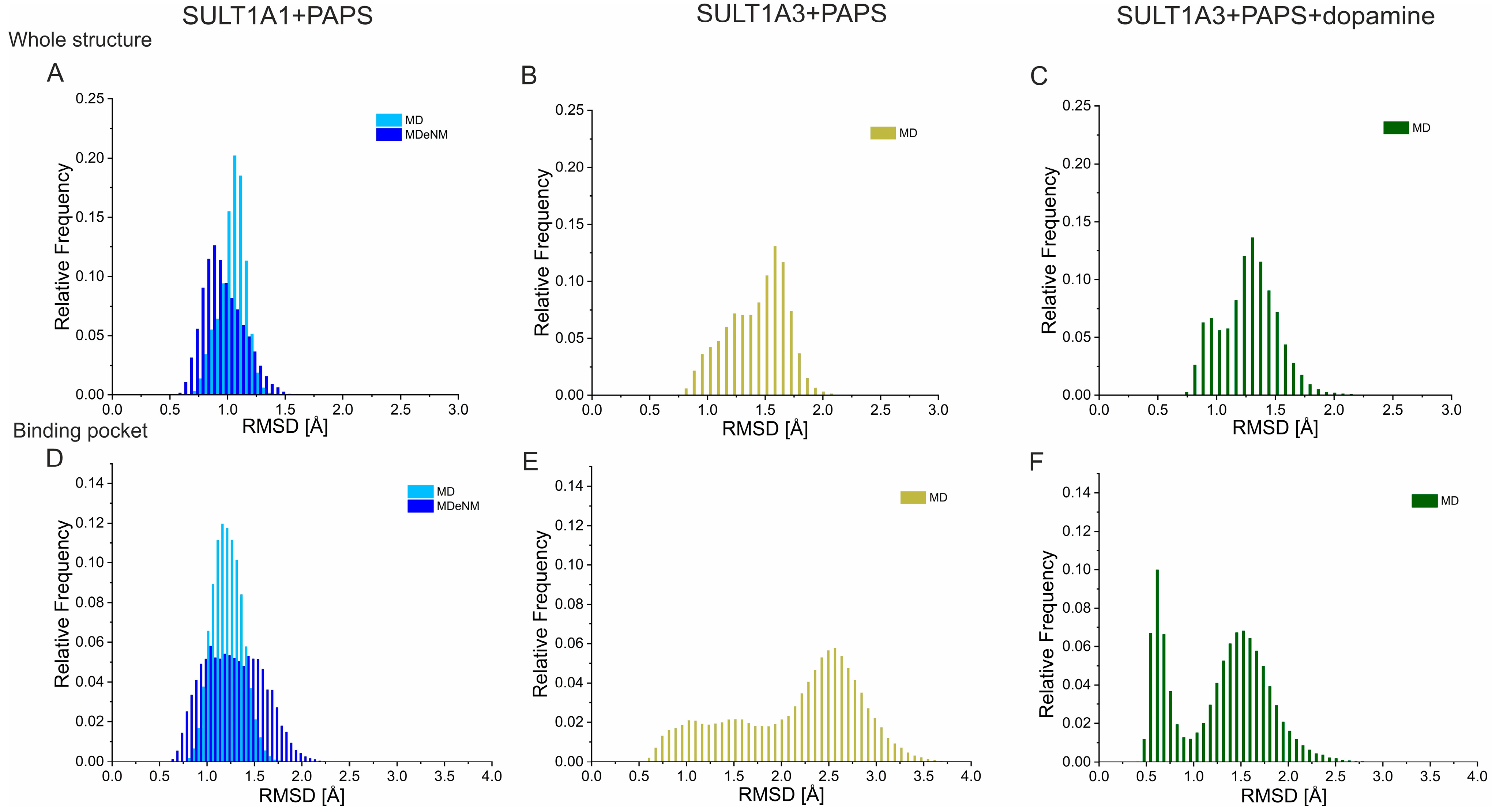
3.2. MD Simulations and Analysis
3.3. Clustering
3.4. Ligand Collection and Preparation
3.5. Ensemble Docking
3.6. Substrate Clustering
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strott, C.A. Sulfonation and Molecular Action. Endocr. Rev. 2002, 23, 703–732. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ako, R.; Wu, B. Crystal structures of human sulfotransferases: Insights into the mechanisms of action and substrate selectivity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human Sulfotransferases and Their Role in Chemical Metabolism. Toxicol. Sci. 2005, 90, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Bojarová, P.; Williams, S.J. Sulfotransferases, sulfatases and formylglycine-generating enzymes: A sulfation fascination. Curr. Opin. Chem. Biol. 2008, 12, 573–581. [Google Scholar] [CrossRef]
- Lessigiarska, I.; Peng, Y.; Tsakovska, I.; Alov, P.; Lagarde, N.; Jereva, D.; Villoutreix, B.O.; Nicot, A.B.; Pajeva, I.; Pencheva, T.; et al. Computational Analysis of Chemical Space of Natural Compounds Interacting with Sulfotransferases. Molecules 2021, 26, 6360. [Google Scholar] [CrossRef]
- Sun, H.; Scott, D.O. Structure-based Drug Metabolism Predictions for Drug Design. Chem. Biol. Drug Des. 2009, 75, 3–17. [Google Scholar] [CrossRef]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef]
- Allali-Hassani, A.; Pan, P.W.; Dombrovski, L.; Najmanovich, R.; Tempel, W.; Dong, A.; Loppnau, P.; Martin, F.; Thornton, J.; Edwards, A.M.; et al. Structural and Chemical Profiling of the Human Cytosolic Sulfotransferases. PLoS Biol. 2007, 5, e97. [Google Scholar]
- Gamage, N.U.; Tsvetanov, S.; Duggleby, R.G.; McManus, M.E.; Martin, J.L. The Structure of Human Sult1a1 Crystallized with Estradiol. An Insight into Active Site Plasticity and Substrate Inhibition with Multi-Ring Substrates. J. Biol. Chem. 2005, 280, 41482–41486. [Google Scholar] [CrossRef]
- Martiny, V.Y.; Carbonell, P.; Lagorce, D.; Villoutreix, B.O.; Moroy, G.; Miteva, M.A. In Silico Mechanistic Profiling to Probe Small Molecule Binding to Sulfotransferases. PLoS ONE 2013, 8, e73587. [Google Scholar] [CrossRef]
- Coughtrie, M.W. Function and organization of the human cytosolic sulfotransferase (SULT) family. Chem. Biol. Interact. 2016, 259, 2–7. [Google Scholar] [CrossRef]
- Kurogi, K.; Rasool, M.I.; Alherz, F.A.; El Daibani, A.A.; Bairam, A.F.; Abunnaja, M.S.; Yasuda, S.; Wilson, L.J.; Hui, Y.; Liu, M.C. Sult Genetic Polymorphisms: Physiological, Pharmacological and Clinical Implications. Expert Opin. Drug Metab. Toxicol. 2021, 17, 767–784. [Google Scholar] [CrossRef]
- Isvoran, A.; Peng, Y.; Ceauranu, S.; Schmidt, L.; Nicot, A.B.; Miteva, M.A. Pharmacogenetics of human sulfotransferases and impact of amino acid exchange on Phase II drug metabolism. Drug Discov. Today 2022, 27, 103349. [Google Scholar] [CrossRef]
- Berger, I.; Guttman, C.; Amar, D.; Zarivach, R.; Aharoni, A. The Molecular Basis for the Broad Substrate Specificity of Human Sulfotransferase 1A1. PLoS ONE 2011, 6, e26794. [Google Scholar] [CrossRef]
- Basit, A.; Neradugomma, N.K.; Wolford, C.; Fan, P.W.; Murray, B.; Takahashi, R.H.; Khojasteh, S.C.; Smith, B.J.; Heyward, S.; Totah, R.A.; et al. Characterization of Differential Tissue Abundance of Major Non-CYP Enzymes in Human. Mol. Pharm. 2020, 17, 4114–4124. [Google Scholar] [CrossRef]
- Yasuda, S.; Suiko, M.; Sakakibara, Y.; Liu, M. Hydroxylated serotonin and dopamine as substrates and inhibitors for human cytosolic SULT1A3. J. Neurochem. 2007, 103, 2679–2689. [Google Scholar] [CrossRef]
- Dajani, R.; Hood, A.M.; Coughtrie, M.W.H. A Single Amino Acid, Glu146, Governs the Substrate Specificity of a Human Dopamine Sulfotransferase, SULT1A3. Mol. Pharmacol. 1998, 54, 942–948. [Google Scholar] [CrossRef]
- Cook, I.; Wang, T.; Almo, S.C.; Kim, J.; Falany, C.N.; Leyh, T.S. The Gate That Governs Sulfotransferase Selectivity. Biochemistry 2013, 52, 415–424. [Google Scholar] [CrossRef]
- Zhu, J.; Qi, R.; Liu, Y.; Zhao, L.; Han, W. Mechanistic Insights into the Effect of Ligands on Structural Stability and Selectivity of Sulfotransferase 2A1 (SULT2A1). ACS Omega 2019, 4, 22021–22034. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cook, I.; Leyh, T.S. Design and Interpretation of Human Sulfotransferase A1 Assays. Drug Metab. Dispos. 2016, 44, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Cook, I.; Wang, T.; Leyh, T.S. Sulfotransferase 1A1 Substrate Selectivity: A Molecular Clamp Mechanism. Biochemistry 2015, 54, 6114–6122. [Google Scholar] [CrossRef]
- Dash, R.; Ali, M.C.; Dash, N.; Azad, M.A.K.; Hosen, S.M.Z.; Hannan, M.A.; Moon, I.S. Structural and Dynamic Characterizations Highlight the Deleterious Role of Sult1a1 R213h Polymorphism in Substrate Binding. Int. J. Mol. Sci. 2019, 20, 6256. [Google Scholar] [CrossRef]
- Dudas, B.; Toth, D.; Perahia, D.; Nicot, A.B.; Balog, E.; Miteva, M.A. Insights into the substrate binding mechanism of SULT1A1 through molecular dynamics with excited normal modes simulations. Sci. Rep. 2021, 11, 13129. [Google Scholar] [CrossRef]
- Costa, M.G.S.; Batista, P.R.; Bisch, P.M.; Perahia, D. Exploring Free Energy Landscapes of Large Conformational Changes: Molecular Dynamics with Excited Normal Modes. J. Chem. Theory Comput. 2015, 11, 2755–2767. [Google Scholar] [CrossRef]
- Bidwell, L.M.; McManus, M.E.; Gaedigk, A.; Kakuta, Y.; Negishi, M.; Pedersen, L.; Martin, J.L. Crystal structure of human catecholamine sulfotransferase. J. Mol. Biol. 1999, 293, 521–530. [Google Scholar] [CrossRef]
- Paitz, R.T.; Bowden, R.M. Sulfonation of Maternal Steroids is a Conserved Metabolic Pathway in Vertebrates. Integr. Comp. Biol. 2013, 53, 895–901. [Google Scholar] [CrossRef]
- Cook, I.; Wang, T.; Falany, C.N.; Leyh, T.S. A Nucleotide-Gated Molecular Pore Selects Sulfotransferase Substrates. Biochemistry 2012, 51, 5674–5683. [Google Scholar] [CrossRef]
- Robinson, P.K. Enzymes: Principles and biotechnological applications. Essays Biochem. 2015, 59, 1–41. [Google Scholar] [CrossRef]
- Coughtrie, M.W.; Johnston, L.E. Interactions between Dietary Chemicals and Human Sulfotransferases-Molecular Mechanisms and Clinical Significance. Drug Metab. Dispos. 2001, 29, 522–528. [Google Scholar] [PubMed]
- Cook, I.; Wang, T.; Falany, C.N.; Leyh, T.S. The Allosteric Binding Sites of Sulfotransferase 1A1. Drug Metab. Dispos. 2014, 43, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Cook, I.; Wang, T.; Girvin, M.; Leyh, T.S. The Structure of the Catechin-Binding Site of Human Sulfotransferase 1a1. Proc. Natl. Acad. Sci. USA 2016, 113, 14312–14317. [Google Scholar] [CrossRef]
- Brix, L.A.; Barnett, A.C.; Duggleby, R.G.; Leggett, B.; McManus, M.E. Analysis of the Substrate Specificity of Human Sulfotransferases SULT1A1 and SULT1A3: Site-Directed Mutagenesis and Kinetic Studies. Biochemistry 1999, 38, 10474–10479. [Google Scholar] [CrossRef]
- Liu, M.-C.; Suiko, M.; Sakakibara, Y. Mutational Analysis of the Substrate Binding/Catalytic Domains of Human M Form and P Form Phenol Sulfotransferases. J. Biol. Chem. 2000, 275, 13460–13464. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Lu, J.H.; Li, H.T.; Liu, M.C.; Zhang, J.P.; Li, M.; An, X.M.; Chang, W.R. Crystal Structure of Human Sulfotransferase Sult1a3 in Complex with Dopamine and 3′-Phosphoadenosine 5′-Phosphate. Biochem. Biophys. Res. Commun. 2005, 335, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. Charmm-Gui: A Web-Based Graphical User Interface for Charmm. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. Charmm-Gui Input Generator for Namd, Gromacs, Amber, Openmm, and Charmm/Openmm Simulations Using the Charmm36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. Charmm: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Huang, J.; Mackerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Lokhande, K.B.; Shrivastava, A.; Singh, A. In silico discovery of potent inhibitors against monkeypox’s major structural proteins. J. Biomol. Struct. Dyn. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- More-Adate, P.; Lokhande, K.B.; Shrivastava, A.; Doiphode, S.; Nagar, S.; Singh, A.; Baheti, A. Pharmacoinformatics approach for the screening of Kovidra (Bauhinia variegata) phytoconstituents against tumor suppressor protein in triple negative breast cancer. J. Biomol. Struct. Dyn. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Heyer, L.J.; Kruglyak, S.; Yooseph, S. Exploring Expression Data: Identification and Analysis of Coexpressed Genes. Genome Res. 1999, 9, 1106–1115. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toth, D.; Dudas, B.; Miteva, M.A.; Balog, E. Role of Conformational Dynamics of Sulfotransferases SULT1A1 and SULT1A3 in Substrate Specificity. Int. J. Mol. Sci. 2023, 24, 16900. https://doi.org/10.3390/ijms242316900
Toth D, Dudas B, Miteva MA, Balog E. Role of Conformational Dynamics of Sulfotransferases SULT1A1 and SULT1A3 in Substrate Specificity. International Journal of Molecular Sciences. 2023; 24(23):16900. https://doi.org/10.3390/ijms242316900
Chicago/Turabian StyleToth, Daniel, Balint Dudas, Maria A. Miteva, and Erika Balog. 2023. "Role of Conformational Dynamics of Sulfotransferases SULT1A1 and SULT1A3 in Substrate Specificity" International Journal of Molecular Sciences 24, no. 23: 16900. https://doi.org/10.3390/ijms242316900