
Transcriptome and Metabolome Analyses Reveal Sugar and Acid Accumulation during Apricot Fruit Development

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
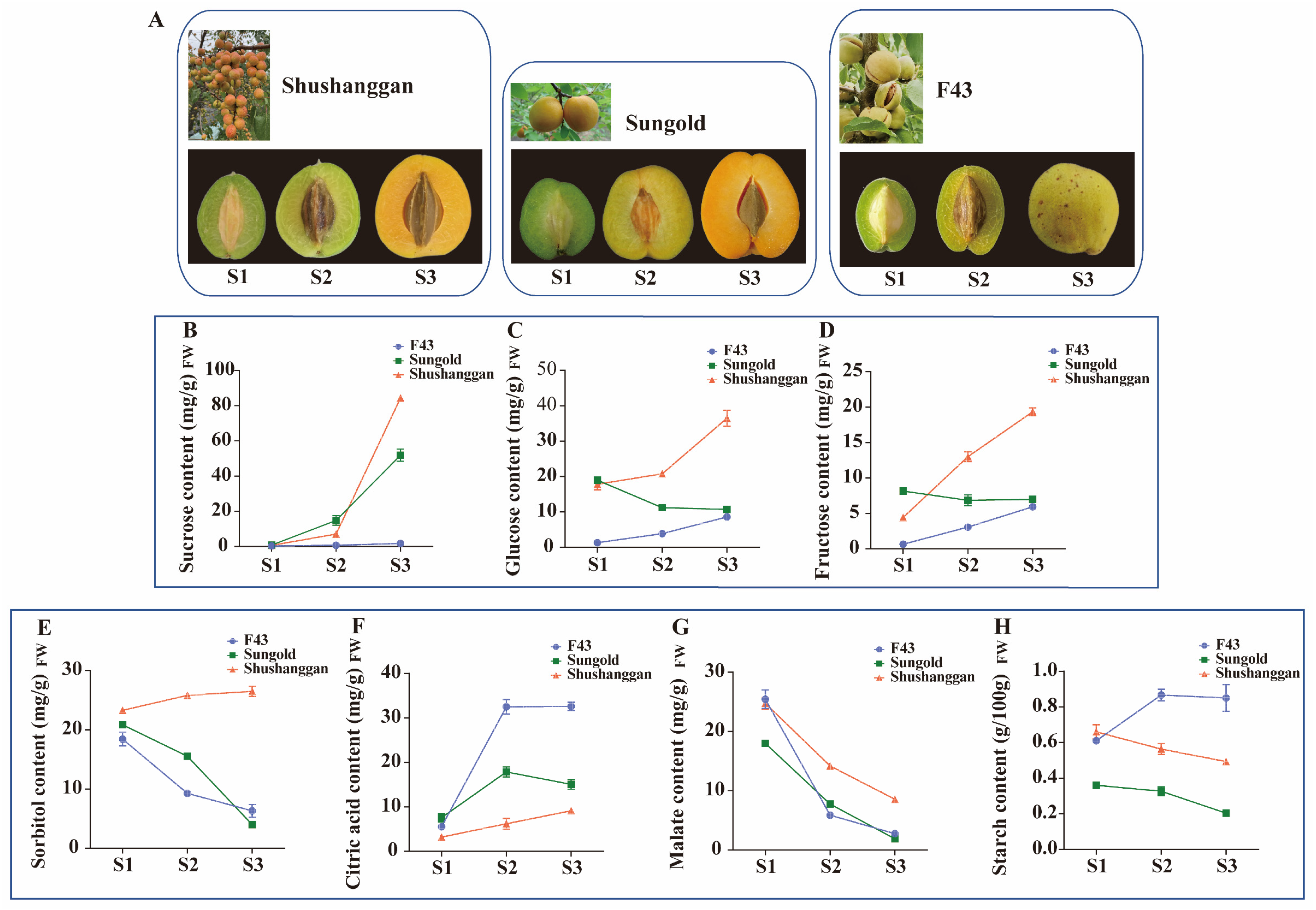
2.1. Soluble Sugar and Organic Acid Content during Three Apricot Cultivars’ Fruit Development
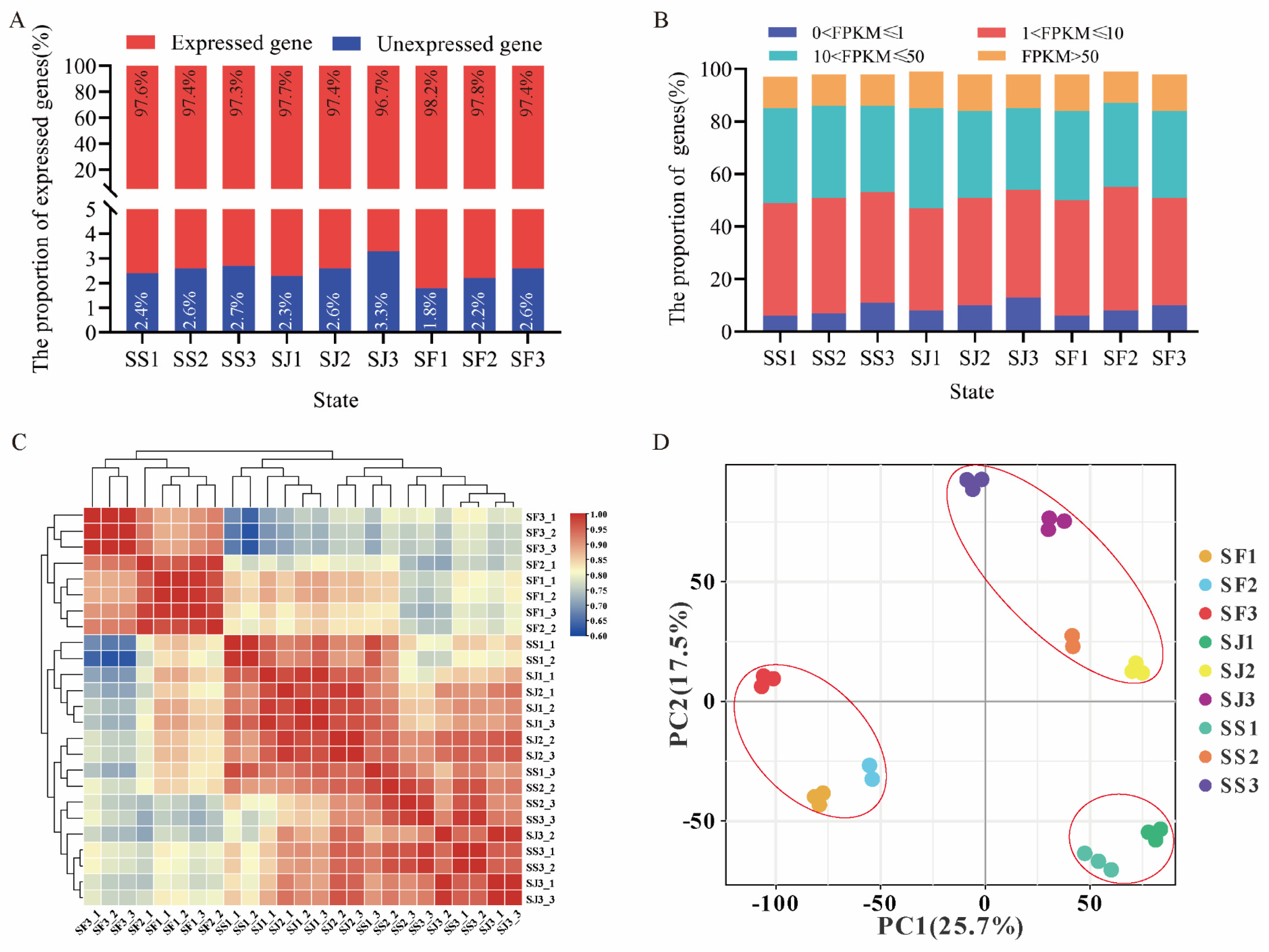
2.2. Transcriptome Sequencing and Gene Expression in Fruit Development of Three Apricot Cultivars
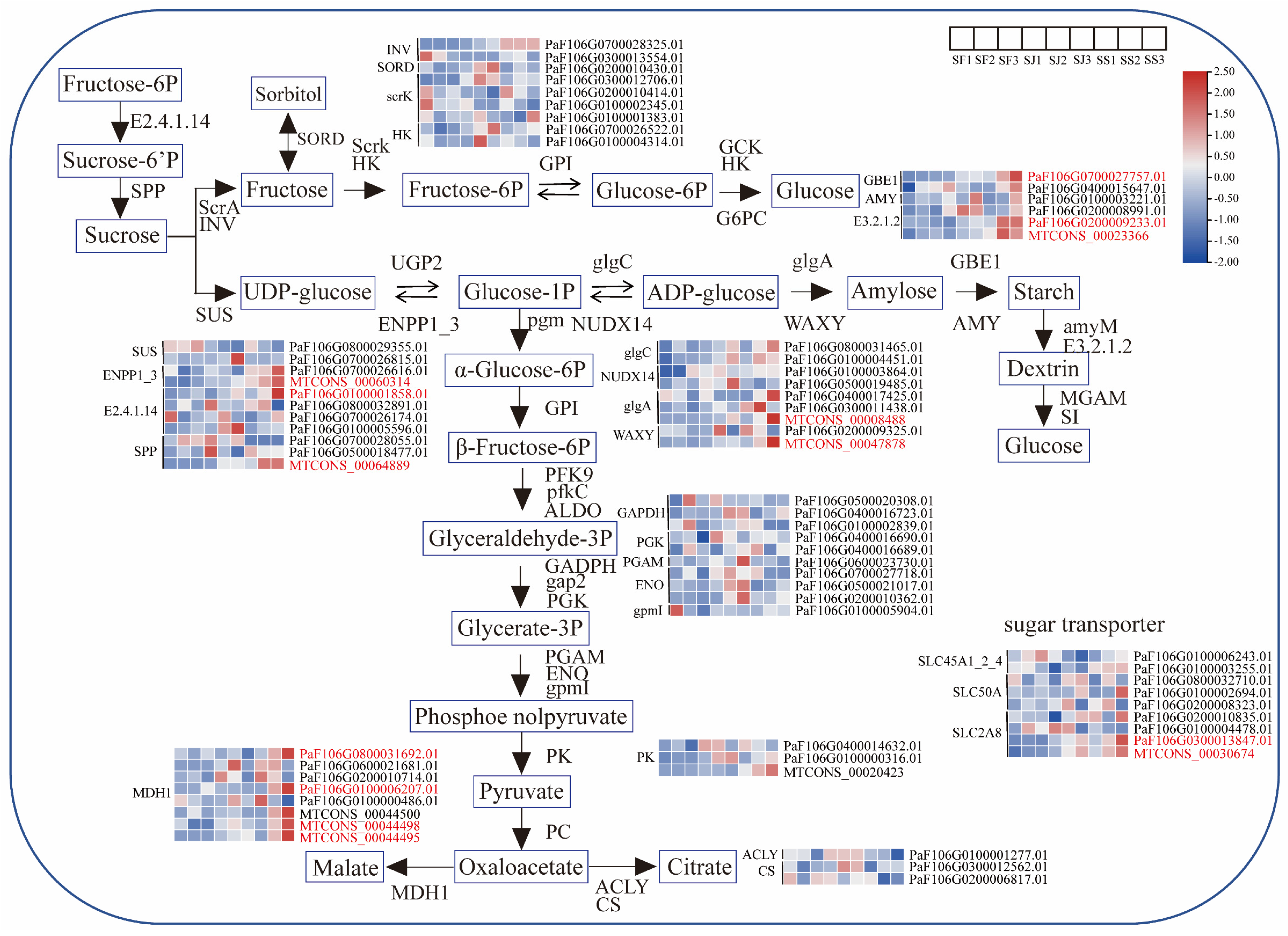
2.3. Identification of Sugar and Acid Metabolism Genes in Three Apricot Cultivars
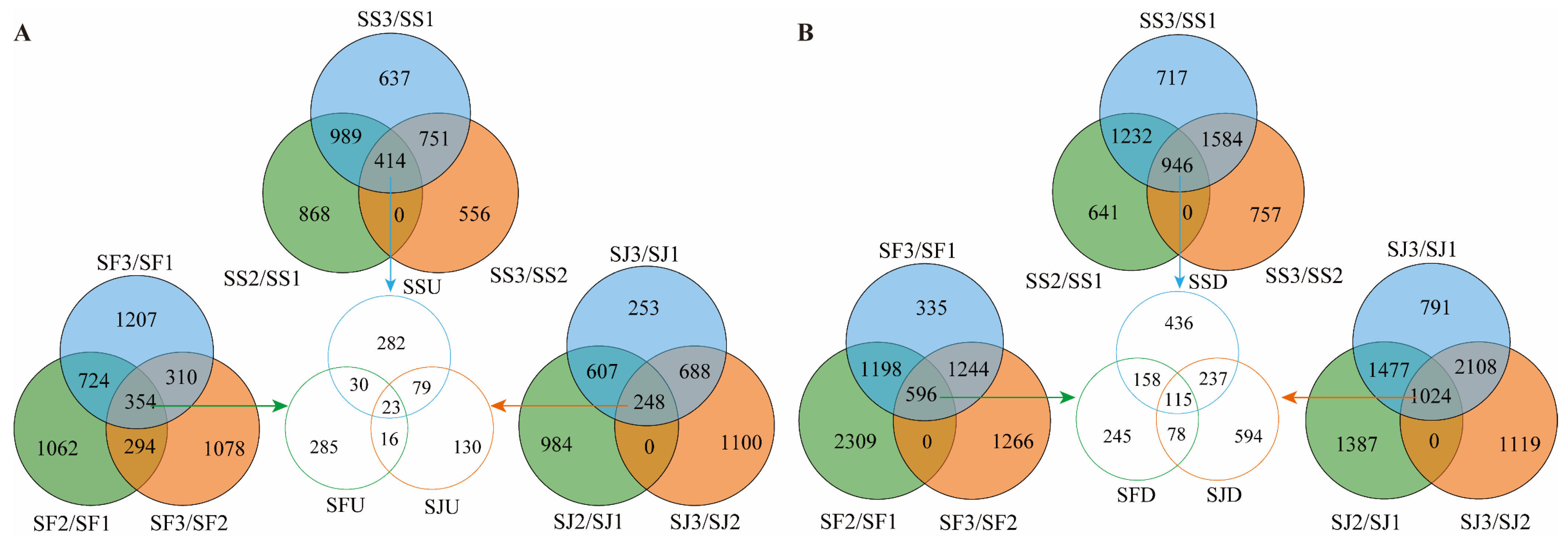
2.4. Identification of DEGs Associated with Sugar and Acid Metabolism Pathways
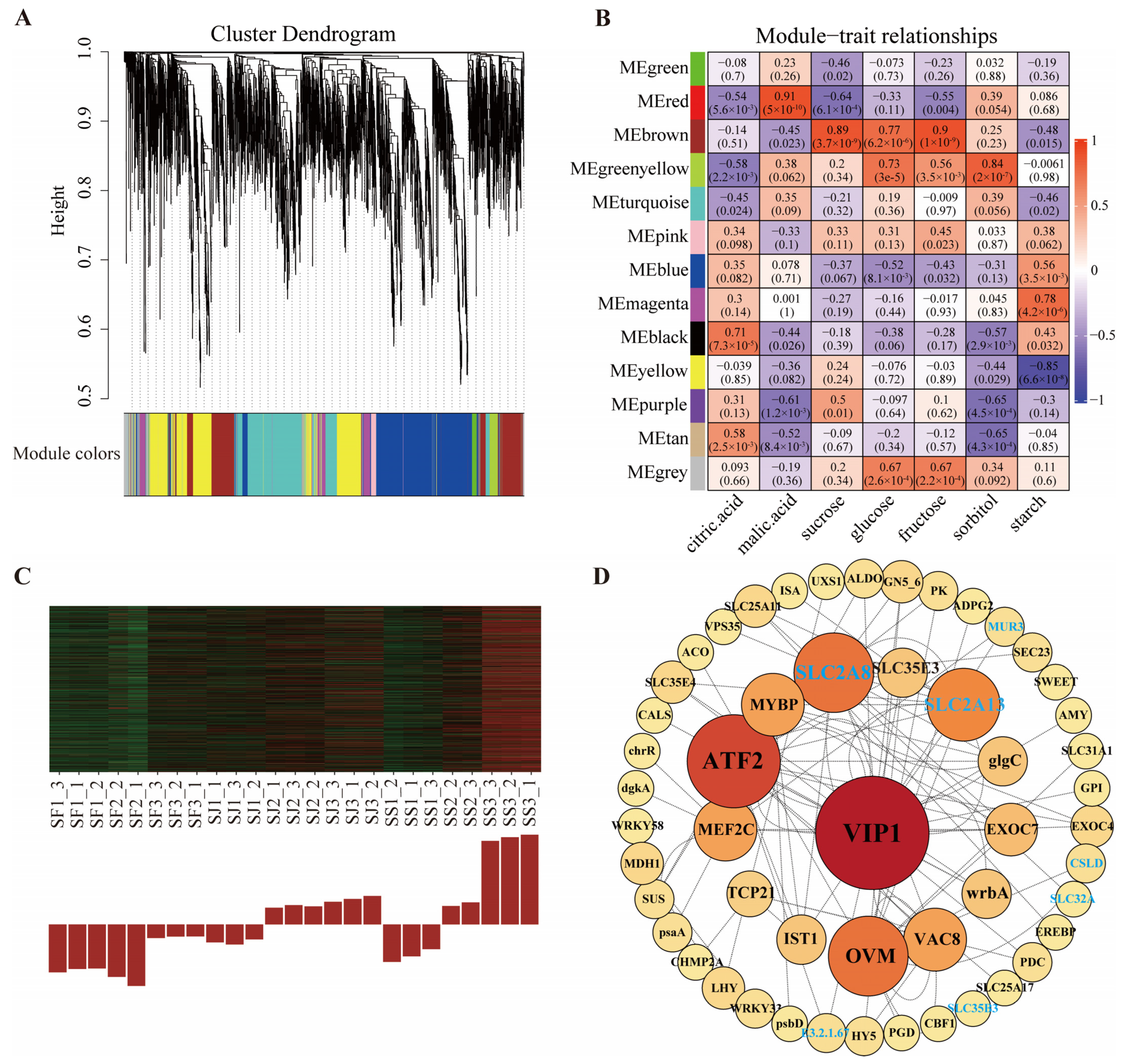
2.5. Identification of Co-Expression Network and Hub Genes Related to Sugar and Acid Metabolism
2.6. Identification of DEmRNAs and Their Corresponding lncRNAs and miRNAs Involved in Sugar and Acid Metabolism Pathways
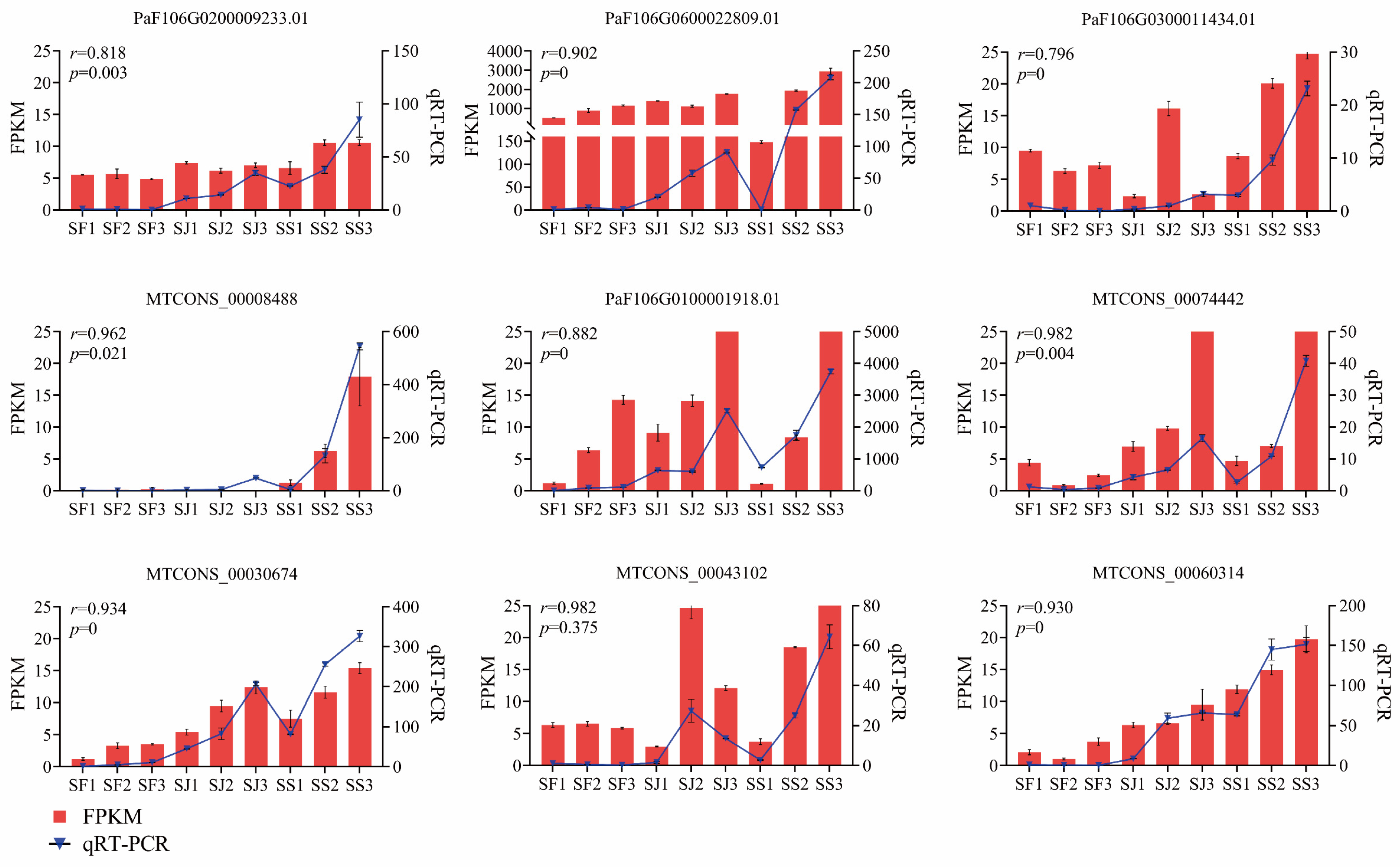
2.7. qRT−PCR Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Sugar and Organic Acid Measurements
4.3. Total RNA Extraction, lncRNA and Small RNA Library Construction, and Sequencing
4.4. Differentially Expressed Genes (DEGs) and Enrichment Analysis
4.5. Target Gene Prediction of lncRNAs and miRNAs
4.6. WGCNA and Gene Network Visualization
4.7. Real-Time Quantitative PCR
4.8. Statistical Analysis and Plotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Erdogan-Orhan, I.; Kartal, M. Insights into research on phytochemistry and biological activities of Prunus armeniaca L. (apricot). Food Res. Int. 2011, 44, 1238–1243. [Google Scholar] [CrossRef]
- Hegedues, A.; Engel, R.; Abranko, L.; Balogh, E.; Blazovics, A.; Herman, R.; Halasz, J.; Ercisli, S.; Pedryc, A.; Stefanovits-Banyai, E. Antioxidant and antiradical capacities in apricot (Prunus armeniaca L.) fruits: Variations from genotypes, years, and analytical methods. J. Food Sci. 2010, 75, C722–C730. [Google Scholar] [CrossRef]
- Ruiz, D.; Egea, J.; Tomás-Barberán, F.; Gil, M. Carotenoids from new apricot (Prunus armeniaca L.) varieties and their relationship with flesh and skin color. J. Agric. Food Chem. 2005, 53, 6368–6374. [Google Scholar] [CrossRef] [PubMed]
- Bureau, S.; Renard, C.; Reich, M.; Ginies, C.; Audergon, J.M. Change in anthocyanin concentrations in red apricot fruits during ripening. LWT-Food Sci. Technol. 2009, 42, 372–377. [Google Scholar] [CrossRef]
- Semra, T.; Topcu, A.; Karabulut, I.; Vural, H.; Hayaloglu, A. Fatty acid, triacylglycerol, phytosterol, and tocopherol variations in kernel oil of Malatya apricots from Turkey. J. Agric. Food Chem. 2007, 55, 10787–10794. [Google Scholar] [CrossRef]
- Dirlewanger, E.; Moing, A.; Rothan, C.; Svanella, L.; Pronier, V.; Guye, A.; Plomion, C.; Monet, R. Mapping QTLs controlling fruit quality in peach (Prunus persica (L.) Batsch). Theor. Appl. Genet. 1999, 98, 18–31. [Google Scholar] [CrossRef]
- Cirilli, M.; Bassi, D.; Ciacciulli, A. Sugars in peach fruit: A breeding perspective. Hortic. Res. 2016, 3, 15067. [Google Scholar] [CrossRef]
- Forcada, C.; Reig, G.; Gimenez, R.; Mignard, P.; Mestre, L.; Moreno, M. Sugars and organic acids profile and antioxidant compounds of nectarine fruits influenced by different rootstocks. Sci. Hortic. 2019, 248, 145–153. [Google Scholar] [CrossRef]
- Brooks, S.; James, N.; Moore, J.; Brad, M. Quantitative and qualitative changes in sugar content of peach genotypes [Prunus persica (L.) Batsch.]. J. Am. Soc. Hortic. Sci. 1993, 118, 97–100. [Google Scholar] [CrossRef]
- Yu, Y.; Guan, J.; Xu, Y.; Ren, F.; Zhang, Z.; Yan, J.; Fu, J.; Guo, J.; Shen, Z.; Zhao, J.; et al. Population-scale peach genome analyses unravel selection patterns and biochemical basis underlying fruit flavor. Nat. Commun. 2021, 12, 3604. [Google Scholar] [CrossRef]
- Ruiz, D.; Lambert, P.; Audergon, J.; Dondini, L.; Tartarini, S.; Adami, M.; Gennari, F.; Cervellati, C.; Franceschi, P.; Sansavini, S.; et al. Identification of QTLs for fruit quality traits in apricot. Acta Hortic. 2010, 862, 587–592. [Google Scholar] [CrossRef]
- Zhang, Q.; Feng, C.; Li, W.; Qu, Z.; Zeng, M.; Xi, W. Transcriptional regulatory networks controlling taste and aroma quality of apricot (Prunus armeniaca L.) fruit during ripening. BMC Genom. 2019, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Ni, X.; Bilal, M.; Shi, T.; Khalil-Ur-Rehman, M.; Pan, Z.; Gao, J.; Usman, M.; Gao, Z. Identification and expression profiling of sugar transporter genes during sugar accumulation at different stages of fruit development in apricot. Gene 2020, 742, 144584. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Yun, S.; Yoon, I.; Nam, E.; Kwon, J.; Jun, J. Assessment of organic acid and sugar composition in apricot, plumcot, plum, and peach during fruit development. J. Appl. Bot. Food Qual. 2014, 87, 24–29. [Google Scholar] [CrossRef]
- Farcuh, M.; Tajima, H.; Lerno, L.; Blumwald, E. Changes in ethylene and sugar metabolism regulate flavonoid composition in climacteric and non-climacteric plums during postharvest storage. Food Chem. Mol. Sci. 2022, 4, 100075. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Hong, C.; Wang, Q.; Lu, M.; An, H. Sugar composition and transcriptome analysis in developing ‘Fengtang’ plum (Prunus salicina Lindl.) reveal candidate genes regulating sugar accumulation. Plant Physiol. Biochem. 2023, 202, 107955. [Google Scholar] [CrossRef] [PubMed]
- Usenik, V.; Fabcic, J.; Stampar, F. Sugars, organic acids, phenolic composition and antioxidant activity of sweet cherry (Prunus avium L.). Food Chem. 2008, 107, 185–192. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, P.; Sun, L.; Li, Q.; Dai, S.; Sun, Y.; Kai, W.; Zhang, Y.; Liang, B.; Leng, P. Transcriptional regulation of PaPYLs, PaPP2Cs and PaSnRK2s during sweet cherry fruit development and in response to abscisic acid and auxin at onset of fruit ripening. Plant Growth Regul. 2015, 75, 455–464. [Google Scholar] [CrossRef]
- Teo, G.; Suziki, Y.; Uratsu, S.; Lampinen, B.; Ormonde, N.; Hu, W.; Dejong, T.; Dandekar, A. Silencing leaf sorbitol synthesis alters long-distance partitioning and apple fruit quality. Proc. Natl. Acad. Sci. USA 2006, 103, 18842–18847. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, W.; Qu, H.; Yan, L.; Li, L.; Zhao, Y.; Yang, H.; Zhang, H.; Yao, G.; Hu, K. Comparative physiological and transcriptomic analysis reveal MdWRKY75 associated with sucrose accumulation in postharvest ‘Honeycrisp’ apples with bitter pit. BMC Plant Biol. 2022, 22, 71. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Y.; Wu, B.; Yuan, Y.; Zhao, Z.Y. Plasma membrane-localized transporter MdSWEET12 is involved in sucrose unloading in apple fruit. J. Agric. Food Chem. 2022, 70, 15517–15530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, J.; Tao, S.; Wu, T.; Qi, K.; Zhang, S.; Wang, J.; Huang, W.; Wu, J.; Zhang, S. Evidence for apoplasmic phloem unloading in pear fruit. Plant Mol. Biol. Rep. 2014, 32, 931–939. [Google Scholar] [CrossRef]
- Zhang, M.; Xue, C.; Hu, H.; Li, J.; Xue, Y.; Wang, R.; Fan, J.; Zou, C.; Tao, S.; Qin, M.; et al. Genome-wide association studies provide insights into the genetic determination of fruit traits of pear. Nat. Commun. 2021, 12, 1144. [Google Scholar] [CrossRef]
- Li, J.; Chen, D.; Jiang, G.; Song, H.; Tu, M.; Sun, S. Molecular cloning and expression analysis of EjSWEET15, enconding for a sugar transporter from loquat. Sci. Hortic. 2020, 272, 0304–4238. [Google Scholar] [CrossRef]
- Hubbard, N.; Pharr, D.; Huber, S.C. Sucrose phosphate synthase and other sucrose metabolizing enzymes in fruits of various species. Physiol. Plant 2006, 82, 191–196. [Google Scholar] [CrossRef]
- Byrne, D.; Nikolic, A.; Burns, E. Variability in sugars, acids, firmness, and color characteristics of 12 peach genotypes. J. Am. Soc. Hortic. Sci. 1991, 116, 1004–1006. [Google Scholar] [CrossRef]
- Takaya, M.; Yuri, I.; Tetsuro, S. Differences in sugar composition in prunus persica fruit and the classification by the principal component analysis. J. Jpn. Soc. Hortic. Sci. 1990, 59, 307–312. [Google Scholar] [CrossRef]
- Jiang, L. Study on HXK Molecular Mechanism in the Regulation of Loquat Sugar Accumulation. Master’s Thesis, Zhejiang A&F University, Lin’an, China, June 2013. [Google Scholar]
- Dondini, L.; Domenichini, C.; Dong, Y.; Gennari, F.; Bassi, D.; Foschi, S.; Lama, M.; Adami, M.; Franceschi, P.; Cervellati, C.; et al. Quantitative trait loci mapping and identification of candidate genes linked to fruit acidity in apricot (Prunus armeniaca L.). Front. Plant Sci. 2022, 13, 838370. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, K.; Cheng, L.; Li, Y.; Guo, J.; Yang, X.; Wang, J.; Khan, I.; Zhu, G.; Fang, W.; et al. Multi-omics approaches identify a key gene, PpTST1, for organic acid accumulation in peach. Hortic. Res. 2022, 9, uhac026. [Google Scholar] [CrossRef]
- Lombardo, V.; Osorio, S.; Borsani, J.; Lauxmann, M.; Bustamante, C.; Budde, C.; Andreo, C.; Lara, M.; Fernie, A.; Drincovich, M.F. Metabolie profiling during peach fruit development and ripening reveals the metabolic networks that underpin each developmental stage. Plant Physiol. 2011, 157, 1696–1710. [Google Scholar] [CrossRef]
- Wang, F.; Sha, J.; Chen, Q.; Xu, X.; Zhu, Z.; Ge, S.; Jiang, Y. Exogenous abscisic acid regulates distribution of 13C and 15N and anthocyanin synthesis in ‘Red Fuji’ apple fruit under high nitrogen supply. Front. Plant Sci. 2019, 10, 1738. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Y.; Xu, J.; Korban, S.; Fei, Z.; Tao, S.; Ming, R.; Tai, S.; Khan, A.; Postman, J.; et al. Diversification and independent domestication of Asian and European pears. Genome Biol. 2018, 19, 77. [Google Scholar] [CrossRef]
- Müller, M.; Irkens-kiesecker, U.; Rubinstein, B.; Taiz, L. On the mechanism of hyperacidification in lemon. Comparison of the vacuolar H(+)-ATPase activities of fruits and epicotyls. J. Biol. Chem. 1996, 271, 1916–1924. [Google Scholar] [CrossRef]
- Luo, A.; Yang, X.; Deng, Y.; Li, C.; Xiang, K.; Li, D. Organic acid concentrations and the relative enzymatic changes during the development of citrus fruits. Sci. Agric. Sin. 2003, 2, 653–657. [Google Scholar]
- Moing, A.; Rothan, C.; Svanella, L.; Just, D.; Diakou, P.; Raymond, P.; Gaudillère, J.; Monet, R. Role of phosphoenolpyruvate carboxylase in organic acid accumulation during peach fruit development. Physiol. Plant 2000, 108, 1–10. [Google Scholar] [CrossRef]
- Pangborn, R.M. Relative taste intensities of selected sugars and organic acids. J. Food Sci. 1963, 28, 726–733. [Google Scholar] [CrossRef]
- Bassi, D.; Selli, R. Evaluation of fruit quality in peach and apricot. Adv. Hortic. Sci. 1990, 2, 107–111. [Google Scholar]
- Nguyen-Quoc, B.; Foyer, C. A role for ‘futile cycles’ involving invertase and sucrose synthase in sucrose metabolism of tomato fruit. J. Exp. Bot. 2001, 52, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Feng, F.; Cheng, L. Expression patterns of genes involved in sugar metabolism and accumulation during apple fruit development. PLoS ONE 2012, 7, e33055. [Google Scholar] [CrossRef]
- Granot, D.; David-Schwartz, R.; Kelly, G. Hexose kinases and their role in sugar-sensing and plant development. Front. Plant Sci. 2013, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Miron, D.; Schafferm, A. Sucrose phosphate synthase, sucrose synthase, and invertase activities in developing fruit of lycopersicon esculentum mill. and the sucrose accumulating lycopersicon hirsutum humb. and bonpl. Plant Physiol. 1991, 95, 623–627. [Google Scholar] [CrossRef]
- Chen, L.; Qu, X.; Hou, B.; Sosso, D.; Osorio, S.; Fernie, A.; Frommer, W. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 2012, 335, 207–211. [Google Scholar] [CrossRef]
- Chen, L.; Cheung, L.; Feng, L.; Tanner, W.; Frommer, W. Transport of sugars. Annu. Rev. Biochem. 2015, 84, 865–894. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Zhang, J.; Ren, Y.; Li, M.; Tian, S.; Yu, Y.; Zuo, Y.; Gong, G.; Zhang, H.; et al. The NAC transcription factor ClNAC68 positively regulates sugar content and seed development in watermelon by repressing ClINV and ClGH3.6. Hortic. Res. 2021, 8, 214. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, Y.; Ramakrishnan, M.; Chen, C.; Xie, F.; Hua, Q.; Chen, J.; Zhang, R.; Zhao, J.; Hu, G.; et al. Transcriptomics-based identification and characterization of genes related to sugar metabolism in ‘Hongshuijing’ pitaya. Hortic. Plant J. 2021, 8, 450–460. [Google Scholar] [CrossRef]
- Diakou, P.; Svanella, L.; Raymond, P.; Gaudillère, J.; Moing, A. Phosphoenolpyruvate carboxylase during grape berry development: Protein level, enzyme activity and regulation. Funct. Plant Biol. 2000, 27, 221–229. [Google Scholar] [CrossRef]
- Wang, L.; Bao, W.; Xu, W.; Huang, M.; Zhu, G.; Zhao, H.; Chen, C.; Bai, H.; Chen, J.; Liu, H.; et al. Transcriptome analysis reveals genes associated with kernel size in apricots cultivated for kernel consumption (Prunus Armeniaca × Prunus Sibirica). Sci. Hortic. 2022, 302, 11141. [Google Scholar] [CrossRef]
- Garg, R.; Singh, V.; Rajkumar, M.; Kumar, V.; Jain, M. Global transcriptome and coexpression network analyses reveal cultivar-specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J. 2017, 91, 1088–1107. [Google Scholar] [CrossRef] [PubMed]
- Panni, S.; Lovering, R.; Porras, P.; Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194417. [Google Scholar] [CrossRef] [PubMed]
- Vance, K.; Ponting, C. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014, 30, 348–355. [Google Scholar] [CrossRef]
- Yu, J.; Bennett, D.; Dardick, C.; Zhebentyayeva, T.; Abbott, A.; Liu, Z.; Staton, M. Genome-wide changes of regulatory non-coding RNAs reveal pollen development initiated at ecodormancy in Peach. Front. Mol. Biosci. 2021, 8, 612881. [Google Scholar] [CrossRef]
- Ghorbani, F.; Abolghasemi, R.; Haghighi, M.; Etemadi, N.; Wang, S.; Karimi, M.; Soorni, A. Global identification of long non-coding RNAs involved in the induction of spinach flowering. BMC Genom. 2021, 22, 704. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, J.; Liang, Q.; Zhang, Y.; Li, Y. Genome-wide analysis of long noncoding RNAs affecting floral bud dormancy in pears in response to cold stress. Tree Physiol. 2020, 41, 771–790. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Chen, Q.; Jiang, L.; Lin, Y.; Ye, Y.; Liu, P.; Wang, X.; Tang, H. Comparative transcriptome analysis uncovers the regulatory functions of long noncoding RNAs in fruit development and color changes of Fragaria pentaphylla. Hortic. Res. 2019, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Fu, D.; Zhu, B.; Luo, Y.; Zhu, H. CRISPR/Cas9-mediated mutagenesis of lncRNA1459 alters tomato fruit ripening. Plant J. 2018, 94, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yang, H.; Qu, D.; Zhu, Z.; Yang, Y.; Zhao, Z. The MdBBX22–miR858–MdMYB9/11/12 module regulates proanthocyanidin biosynthesis in apple peel. Plant Biotechnol. J. 2022, 20, 1683–1700. [Google Scholar] [CrossRef]
- Ma, H.; Yang, T.; Li, Y.; Zhang, J.; Wu, T.; Song, T.; Yao, Y.; Tian, J. The long noncoding RNA MdLNC499 bridges MdWRKY1 and MdERF109 function to regulate early-stage light-induced anthocyanin accumulation in apple fruit. Plant Cell 2021, 33, 3309–3330. [Google Scholar] [CrossRef]
- Mo, R.; Wang, F.; Ni, Z.; Tang, F.; Shao, S.; Deng, J. Simultaneous Determination of Sugar, Organic Acids, and Vitamin C in Agricultural Products by Ion-Exclusion Chromatography with Refractive Index and Diode Array Detectors. Mod. Food Sci. Technol. 2016, 4, 044. [Google Scholar] [CrossRef]
- Cao, J.; Jiang, W.; Zhao, Y. Experiment Guidance of Postharvest Physiology and Biochemistry of Fruits and Vegetables, 1st ed.; China Light Industry Press Ltd.: Beijing, China, 2007; pp. 76–84. [Google Scholar]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.; Antonescu, C.; Chang, T.; Mendell, J.; Salzberg, S. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.; Pertea, G.; Mortazavi, A.; Kwan, G.; Baren, M.; Salzberg, S.; Wold, B.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 25, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.; Huttner, M.; Dueck, A.; Meister, G.; Engelmann, J. miRA: Adaptable novel miRNA identification in plants using small RNA sequencing data. BMC Bioinform. 2015, 16, 370. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Abdi, H. The Bonferonni and Šidák Corrections for Multiple Comparisons. Comput. Sci. 2007, 3, 2007. [Google Scholar]
- Tafer, H.; Hofacker, I. RNAplex: A fast tool for RNA-RNA interaction search. Bioinformatics 2008, 24, 2657–2663. [Google Scholar] [CrossRef]
- Wu, H.; Ma, Y.; Chen, T.; Wang, M.; Wang, X. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids. Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Bonnet, E.; He, Y.; Billiau, K.; Peer, Y. TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 2010, 26, 1566–1568. [Google Scholar] [CrossRef]
- Fahlgren, N.; Carrington, J. miRNA Target Prediction in Plants. Methods Mol. Biol. 2010, 592, 51–57. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Tong, Z.; Gao, Z.; Wang, F.; Zhou, J.; Zhang, Z. Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol. Biol. 2009, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gou, N.; Chen, C.; Huang, M.; Zhang, Y.; Bai, H.; Li, H.; Wang, L.; Wuyun, T. Transcriptome and Metabolome Analyses Reveal Sugar and Acid Accumulation during Apricot Fruit Development. Int. J. Mol. Sci. 2023, 24, 16992. https://doi.org/10.3390/ijms242316992
Gou N, Chen C, Huang M, Zhang Y, Bai H, Li H, Wang L, Wuyun T. Transcriptome and Metabolome Analyses Reveal Sugar and Acid Accumulation during Apricot Fruit Development. International Journal of Molecular Sciences. 2023; 24(23):16992. https://doi.org/10.3390/ijms242316992
Chicago/Turabian StyleGou, Ningning, Chen Chen, Mengzhen Huang, Yujing Zhang, Haikun Bai, Hui Li, Lin Wang, and Tana Wuyun. 2023. "Transcriptome and Metabolome Analyses Reveal Sugar and Acid Accumulation during Apricot Fruit Development" International Journal of Molecular Sciences 24, no. 23: 16992. https://doi.org/10.3390/ijms242316992