Abstract
This work presents the synthesis of a new compound, 1-[aryl-(diphenylphosphono)methyl]-3,4,6-trimethylglycolurils, via the interaction of benzaldehyde and its mononitro- and monohydroxyderivatives with 1,3,4-trimethylglycoluril and triphenylphosphite. By varying the reaction conditions and the catalysts, the obtained product yields ranged from satisfactory to good. The diastereomers formed during the reaction were separated by semipreparative HPLC on the C18 stationary phase. The isolated diastereomers were characterized by 1H, 13C, and 31P NMR, and the structures of the diastereomers were confirmed using a single-crystal X-ray crystal structure analysis and quantum chemical calculations.
1. Introduction
Glycoluril derivatives have attracted the attention of researchers due to their wide range of applications and, above all, due to their biological activities, particularly their nootropic, neurotropic, anxiolytic [1,2], and antibacterial properties [3,4]. Additionally, glycoluril derivatives are widely used as “building blocks” to synthesize supramolecular compounds such as bambus[n]urils [5,6], cucurbit[n]urils [7,8], and molecular clips [9,10]. Special attention has been paid to studying methods for synthesizing new glycoluril derivatives and their structural analogs and precursors as well as to the characterization of their properties [11,12,13,14,15]. Individual phosphorus-containing glycoluril derivatives are described in the literature [16,17], where their catalytic activity in some three-component reactions has been shown. Phosphorylated glycoluril derivatives were investigated as flame-resistant additives for rubber [18] and as moleculars tweezer exhibiting high binding properties to aliphatic amines [19]. However, these reports are sporadic, and to date, only a relatively small number of synthetic approaches have been developed for preparing phosphorus-containing glycoluril derivatives [16,17,18,19], and the main regularities of the formation of stereoisomers of the phosphonate derivatives of glycoluril have not been established either. We previously found [20] that the interaction of 1,3,4-trimethylglycoluril with aliphatic aldehydes and triphenylphosphite in acetonitrile using methanesulfonic acid as a catalyst led to the formation of 1-[1-(diphenylphosphono)alkyl]-3,4,6-trimethylglycoluril.
2. Results and Discussion
2.1. The Effect of Synthesis Conditions on the Yield of Phosphonate Derivatives of 1,3,4-Trimethylglycoluriles
In order to extend the methods for synthesizing phosphorus-containing glycoluril derivatives, we studied the conditions for the synthesis of various new glycoluril phosphonates 4a–f using the Birum–Oleksyszyn approach exemplified by 1,3,4-trimethylglycoluril and benzaldehydes 2a–2f (Scheme 1).
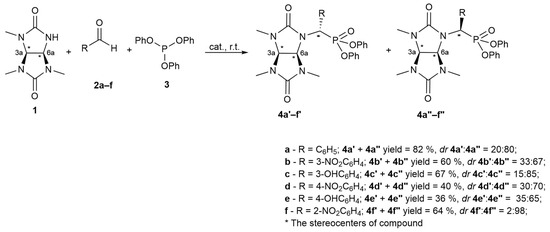
Scheme 1.
Birum–Oleksyszyn synthesis exemplified by 1,3,4-trimethylglycoluril and benzaldehydes 2a–f.
It is noteworthy that the attempt to synthesize phosphonate derivatives of 1,3,4-trimethylglycoluryl 4a was unsuccessful under typical conditions used to prepare diphenyl 1-aminophosphonates [21], namely, with glacial acetic acid as a solvent. Thus, according to the NMR analysis of the reaction mixture, the triphenylphosphite 3 interaction with benzaldehyde 2a in glacial acetic acid with 1,3,4-trimethylglycoluril 1 (80 °C, 2 h) led to the formation of target compound 4a with a low yield of 21%. In addition, the 31P NMR spectra of the reaction mixture contained clear signals within 3.40–−0.57 ppm, indicating both partial and complete hydrolysis of the ester groups for compound 3 as well as the formation of α-hydroxyphosphonate 6a (31P δ = 15.99 ppm) as a by-product (Scheme 2) (Supplementary Materials, Figure S1).
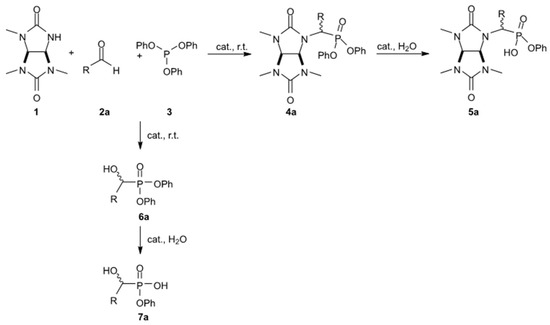
Scheme 2.
The probable formation pathways for by-products 5a, 6a, and 7a in the synthesis of compound 4a.
The authors carried out the selection of catalysts, solvents, temperatures, and reaction times to establish the optimal conditions for synthesizing the target product 4a because the typical conditions for the synthesis of α-aminophosphonates proved to be inefficient in obtaining phosphonate derivatives of 1,3,4-trimethylglycoluryl 1.
A series of model syntheses of compound 4a was carried out to select the optimal reaction conditions considering the fact that Lewis acids and organic Brønsted acids tend to show a catalytic effect in the Birum–Oleksyszyn reaction [22,23]. Therefore, the reaction was carried out in anhydrous acetonitrile; AlCl3, BF3·Et2O, SnCl4, and TiCl4 were used as Lewis acids, whereas trifluoroacetic acid, methanesulfonic acid, and trifluoromethanesulfonic acid were used as Brønsted acids in addition to glacial acetic acid (Table 1). The amount of catalyst equaled 10 mol% of the total mass of the reaction mixture, and the process was carried out at room temperature. The reaction flow was monitored by HPLC.

Table 1.
The effect of a catalyst on the maximum analytical yield of 4a in the reaction mixture at room temperature in acetonitrile.
Table 1 shows that the use of glacial acetic acid as a solvent resulted in a relatively low yield of the target product 4a (23%). The maximum concentration of the latter in the reaction mixture was reached within 60 min according to HPLC (Table 1, entry 10). However, with glacial acetic acid as a catalyst and acetonitrile as a solvent, the content of 4a remained relatively low, although it increased with time (Table 1, entries 1 and 2).
If trifluoroacetic, methanesulfonic, and trifluoromethanesulfonic acids (i.e., organic acids stronger than acetic acid) are applied as catalysts, the content of the target product 4a in the reaction mixture increases by up to nine times (Table 1, entries 3, 4, and 5). When trifluoroacetic acid is applied within a comparable reaction time, the analytical yield of 4a becomes almost six times higher than in the case of synthesis with glacial acetic acid.
For methanesulfonic acid, the maximum yield for trimethylphosphonate glycoluril 4a equaled 91% (82% isolated preparatively). This yield was achieved within 30 min under the selected reaction conditions. An increase in the reaction time when using methanesulfonic acid led to a decrease in the concentration of the target product 4a, which might indicate hydrolysis and the formation of the reaction by-products.
The application of trifluoromethanesulfonic acid also leads to a notable increase in the content of the target substance 4a for a relatively short reaction time (Table 1, entry 5), but the analytical yield of compound 4a is only 38%. We assume that trifluoromethanesulfonic acid not only promotes the main reaction but also catalyzes the side reactions, namely, the hydrolysis of the initial triphenylphosphite 3 and of the target compound 4a. Furthermore, trifluoromethanesulfonic acid might promote the formation of α-hydroxyphosphonate 6a (Scheme 2). To verify this assumption, we conducted an additional experiment and added acetic anhydride to the reaction mixture with trifluoromethanesulfonic acid as a catalyst in order to chemically bind the water formed during the reaction. Thus, we established that the practical yield of the target compound 4a sharply increased by up to 75% for a comparable reaction time, which implicitly confirms the assumption (Table 1, entry 6).
If Lewis acids (AlCl3, BF3·Et2O, SnCl4 and TiCl4) are applied as catalysts in the model reaction to prepare compound 4a (Table 1, entries 7–9), the yields of the target compound reach 15–25%.
In the course of the further selection of the conditions to prepare compound 4a, we studied the effect of solvents on the target product yield under the same conditions (room temperature, methanesulfonic acid as a catalyst, 10 mol%). Table 2 shows the results obtained.
Table 2.
The effect of a solvent on the maximum analytical yield of the target product 4a (methanesulfonic acid as catalyst (10 mol%), room temperature).
Despite a rather low solubility of the initial 1,3,4-trimethylglycoluril 1 in nonpolar solvents at room temperature, the analytical yield of the phosphonate glycoluril 4a is relatively high and varies from 29 to 59% (Table 2, entries 1–3). At the same time, the maximum analytical yield of 91% (82% being a preparative yield) is achieved in an aprotic medium polar solvent, i.e., acetonitrile. The use of a more polar aprotic solvent, DMSO, does not lead to the formation of the product 4a in notable amounts according to HPLC. If methanol is applied as a reaction solvent, the highest analytical yield is achieved in 60 min and amounts to only 8%.
It has been experimentally established that an increase in the reaction temperature within 40 to 70 °C—with acetonitrile as a solvent and methanesulfonic acid as a catalyst—led to a decrease in the yield of compound 4a. Thus, at 40 °C, the maximum analytical yield of product 4a was 64% for 20 min. At 70 °C, the maximum analytical yield of 4a equaled 50% for 20 min.
In sum, the optimal conditions for the three-component Birum–Oleksyszyn reaction in the case of 1,3,4-trimethylglycoluril 1 (Scheme 1) appear to be the use of acetonitrile as a solvent (room temperature) and the use of methanesulfonic acid as a catalyst (10 mol% of the total reaction mixture). A decrease in the amount of catalyst to 5 mol% or its increase to 20 mol% results in decreases in the yield of the desired product 4a to 62% and 54%, respectively.
2.2. Separation and Identification of Diastereomers of Phosphonate Derivatives of 1,3,4-Trimethylglycoluriles 4a′ and 4a″
The individual diastereomers 4a′ and 4a″ were preparatively separated and isolated by HPLC with a C18 stationary phase. The diastereomers were then characterized by NMR and single-crystal X-ray diffraction analysis (SCXRD). Obviously, the order of elution for diastereomers 4a′ and 4a″ depends on the mutual arrangement of the substituents around the asymmetric C7 centers (Scheme 1). The highest retention is characterized by the 4a″ isomer with the most sterically accessible hydrophobic groups.
2.2.1. X-ray Crystal Structure Analysis of the Diastereomers 4a′ and 4a″
Compound 4a′ crystallizes in a monoclinic crystal system, space group C2/c. The asymmetric unit contains one 4a′ molecule and one and a half acetonitrile solvate molecules (Figure 1a). The unit cell contains eight formula units. The crystallographic space group is centrosymmetric; therefore, both enantiomeric forms of compound 4a′ crystallize as a true racemate. The configuration of the stereocenters (3a, 6a, CHPO3) in compound 4a′ is as follows: R,S,R (and enantiomeric S,R,S) (Figure 1).
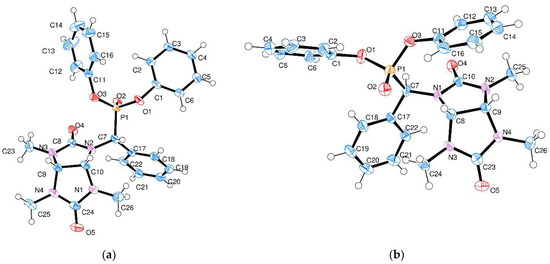
Figure 1.
Molecular X-ray crystal structures for compounds 4a′ (a) and 4a″ (b). Thermal ellipsoids are drawn for 50% probability level. Solvate acetonitrile molecules in panel (a) are omitted for clarity.
The molecules of 4a′ of the same enantiomeric form are joined into homochiral supramolecular chains by short CH···O contacts involving the oxygen atoms of the phosphonate group and the hydrogen atoms of the phenyl (d(O3–H22) = 2.60 Å) or glycoluril fragments (d(O2–H10) = 2.48 Å, Figure 2a). The second type of CH···O contacts involves the carbonyl oxygen atom and aliphatic hydrogen atoms with the distances d(O4–H7) = 2.43 Å and d(O4–H10) = 2.40 Å, respectively. The chains are oriented along the crystallographic axis b. The enantiomeric chains of opposite stereochemistry are joined in the crystal packing by π-π stacking interactions between the phenyl groups of the phosphonate ester moiety with the distance of 3.295 Å between the planes of the rings (Figure 2b).
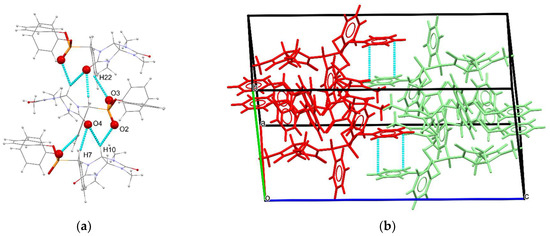
Figure 2.
(a) Homochiral supramolecular chains of 4a′ molecules, viewed along the crystallographic direction (a–c); (b) packing diagram of 4a′, different enantiomers shown in red and green, with blue dashed lines indicating π-π stacking interactions.
Compound 4a″ crystallizes in a monoclinic centrosymmetric space group, P21/c. In contrast to compound 4a′, there are no solvate molecules in the crystal structure, and the asymmetric unit consists of one molecule (Figure 1b). The unit cell contains four formula units. Similarly to compound 4a′, the enantiomeric pair of 4a″ crystallizes as a true racemate. The configuration of the stereocenters (3a, 6a, CHPO3) is S,R,R (and R,S,S). Therefore, compounds 4a′ and 4a″ differ in the absolute configuration of the carbon atom connected to the phosphonate group, in accordance with the two possible directions of the 1,3,4-trimethylglycoluril addition to the carbonyl group of the aldehyde.
Similarly to compound 4a′, the enantiomers of 4a″ of the same configuration are joined into supramolecular chains oriented along the crystallographic axis b (Figure 3a), but the short CH···O contacts of the phosphonate groups involve only the hydrogen atoms of the phenyl rings (d(O2–H6) = 2.53 Å and d(O3–H15) = 2.65 Å). The carbonyl oxygen atoms form only one type of short CH···O contacts with the hydrogen atoms of the glycoluril fragment (d(O4–H9) = 2.65 Å). In the crystal packing, the enantiomers of 4a″ are joined only by C–H···π (d(C19–H25C) = 2.76 Å) contacts, and no other specific supramolecular interactions were found.
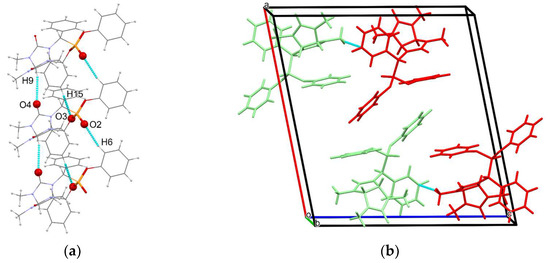
Figure 3.
(a) Homochiral supramolecular chains of 4a″ molecules, viewed along the crystallographic direction (a–c); (b) packing diagram of 4a″, different enantiomers shown in red and green, with blue dashed lines indicating C–H···π stacking interactions.
2.2.2. Stereoisomerization of Nitro- and Hydroxyphosphonate Derivatives of 1,3,4-Trimethylglycoluriles 4b–f
Further studies were focused on establishing the stereochemical features of obtaining phosphonate derivatives of 1,3,4-trimethylglycoluryl 4b–f with the participation of nitro- and hydroxy derivatives of benzaldehyde 2b–f (Scheme 1) using HPLC, NMR spectroscopy, and quantum chemical calculations.
The reactions of nitro- 2b, 2d, and 2f and hydroxybenzaldehydes 2c and 2e with 1,3,4-trimethylglycoluril 1 and triphenylphosphite 3 under the selected conditions (acetonitrile and 10 mol% methanesulfonic acid) proceeded with satisfactory yields of 4b–4f (36–67%). According to HPLC, the compounds with structures 4a″, 4b″, 4c″, 4d″, and 4e″ were formed in a diastereomeric excess (Scheme 1). For compound 4f″, the diastereomeric excess equaled 96%.
The X-ray crystal structures and NMR results for diastereomers 4a′ and 4a″ were compared with the NMR spectroscopy data for compounds 4b–f. Thus, for diastereomer 4′, the signals of the methine protons of 1H CH-CH bond were recorded by two doublets within a narrow region of 5.11–5.37 ppm, while for compound 4″, the signals of these protons were recorded within a broader region of 4.95–5.51 ppm. The 31P spectra for the type 4′ diastereomers are characterized by a signal shift towards the weak field relative to the signal of the type 4″ compounds. The nature of the substituent in the meta- and para-position does not significantly affect the final ratio of the diastereomers of products 4b–e. However, we observed a notable decrease in the yield of reaction products with a para-substituent (the yields for 4d and 4e are 40% and 36%, respectively) compared to the reaction products with a meta-substituent (yields of 4b and 4c are 60% and 67%, respectively). In the case of compound 4f, only one diastereomer was isolated from the reaction mixture and assigned to the 4f″ structure. According to HPLC-MS, the application of 2-hydroxybenzaldehyde under the conditions of interest did not allow for the detection of any hydrolysis products of compound 4f′ or any ions with the mass that could be assigned to this compound.
Comparing the positions of diastereomeric structures for the phosphonates of 1,3,4-trimethylglycoluriles (4a–f) on the energy profile (Table S1 Supplementary Materials) indicates a larger thermodynamic stability of the 4″-type structures regardless of the nature and position of the substituent in the aromatic ring. This correlates with the experimental data on the ratio of 4′:4″ stereoisomers (Scheme 1). The changes in the electron energy and enthalpy of the isomeric transition of 4″-type structures to 4′-type structures range from 2.85 to 3.58 kcal/mol and from 2.92 to 3.93 kcal/mol, respectively (Table S2 Supplementary Materials).
2.2.3. The Diastereomers of the Phosphonate Derivatives of 1,3,4-Trimethylglycolyrils (4a–f): General Regularities in the NMR Spectra
NMR spectroscopy was applied to reliably identify the diastereomers of phosphonate derivatives of 1,3,4-trimethylglycolyrils 4a–f. Table 3 presents the results of comparing the experimental and pre-calculated values for the chemical shifts of the hydrogen atoms in bridging the methine groups of the glycoluril fragment (3a and 6a) and the hydrogen atom bonded to the carbon atom in the CHPO3 group. The correlations between the experimental and pre-calculated δ values for all of these atoms can be found in Supplementary Materials (Figure S52).
Table 3.
The comparison of experimental and pre-calculated chemical shifts for hydrogen atoms of the 3a, 6a, and CHPO3 carbon atom of the compounds 4a–f.
Table 3 shows that the δ for the hydrogen atom bonded to the carbon atom in the CHPO3 group, as well as the methine protons of the bridging CH-CH (3a, 6a) group, can indicate the configuration of the phosphonate substituent in structure 4. Thus, the signals of the protons in the carbon atom with a phosphonate substituent (CHPO3) of type 4′ (RSR, SRS) are shifted towards lower values compared to the signals of the enantiomers of type 4″ (SRR, RSS). For example, the experimental and pre-calculated chemical shifts equal 5.77 and 5.33 ppm, respectively, for the hydrogen atoms bonded to the carbon atom of the phosphonate group (CHPO3) in the RSR and SRS enantiomers of compound 4a′. However, these shifts equal 5.85 and 6.57 ppm, respectively, for the hydrogen atoms of the SRR and RSS enantiomers of compound 4a″. This pattern persists for the unsubstituted and OH-substituted synthesized glycoluril phosphonates 4a, 4c, and 4e. The experimental signals for similar hydrogen atoms in the indicated pairs of diastereomers of the nitro derivatives (4b, 4d, and 4f) have identical chemical shifts, while the pre-calculated chemical shifts sustain the trend. At the same time, for δ 31P, we observed a significant discrepancy between the pre-calculated values and the experimental ones. This latter fact might be related to the calculation methodology.
A comparison of the experimental and pre-calculated δ values for the hydrogen atoms bonded to the carbon atom in the phosphonate substituent (CHPO3) and for the bridging CH-CH (3a, 6a) group demonstrates the significant discrepancy in these values for the enantiomeric pairs of the SRR and RSS (4″) types and indicates the values’ precision for the RSR and SRS (4′) pairs. For example, the experimental δ of the carbon proton (CHPO3) and CH-CH protons (3a, 6a) in the compound of group 4a″ equal 5.85, 5.50, and 5.00 ppm, whereas the pre-calculated ones equal 6.57, 5.20, and 2.74 ppm. This discrepancy might be caused by the fact that the most stable pre-calculated conformations of structure 4a″ (SRR and RSS) contain the phenyl rings located relative to the glycoluril fragment, thus creating anisotropy cones and shielding the farthest methine proton in the bridging group (CH-CH). At the same time, the hydrogen atom in the methine group of the phosphonate substituent remains in the de-shielding region (Figure 4). This effect is not observed in the experimental 1H NMR spectra, which might be linked to the impossibility of tracking the solvating effect of the solvent using ab initio methods and to the respective change in the structure conformations in the actual solution. The observed effect of phenyl rings on the SRR- and RSS-type enantiomers is retained for the OH and NO2 derivatives (4b″–4f″).
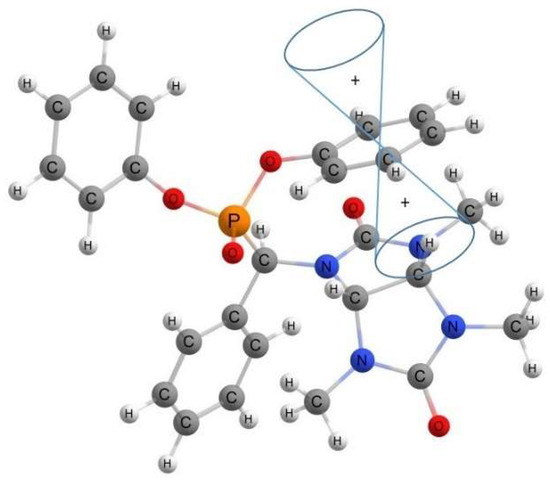
Figure 4.
Location of anisotropy cone of phenyl rings in the structure of SRR diastereomer of 4a″.
The 2D correlation experiment (NOESY) was performed for compounds 4a′ and 4a″. The interactions between the protons of the 3a, 6a, and CHPO3 sites were identified for compound 4a′, while these interactions were not detected for compound 4a″ (Supplementary Materials, Figures S35 and S36).
3. Materials and Methods
All reagents (Acros Organics, Merck, Rahway, NJ, USA) were used as purchased, unless indicated otherwise. Solvents for HPLC (PanReac AppliChem, Darmstadt, Germany) were used for the reactions, benzene was dried over molecular sieves 3A for 48 h. 1,3,4-trimethylglycoluril was prepared according to a known procedure [24] and used as a racemate. 1H NMR (400 MHz, Chloroform-d): δ 7.46 (s, 1H), 5.12 (dd, J = 1.7, 8.3 Hz, 1H), 4.97 (d, J = 8.3 Hz, 1H), 2.92 (s, 3H), 2.88 (s, 3H), 2.77 (d, J = 1.6 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 160.61, 158.41, 73.83, 65.94, 30.47, 29.55, 27.96.
3.1. General Procedure for the Synthesis of Compounds 4a–4f
Aldehyde 2 (2 mmol), triphenyl phosphite 3 (2 mmol), and methanesulfonic acid (10 mol%) were added to the suspension of 1,3,4-trimethylglycoluril 1 (2 mmol) in dry acetonitrile (4 mL) in an argon atmosphere. The mixture was stirred for 1 h at room temperature and then distilled on a rotary evaporator. The residue was dissolved in 10 mL of toluene and washed with 5% aqueous Na2CO3 solution (3 × 10 mL). The toluene solution was then washed with water (3 × 10 mL). Upon washing, the organic layer was dried over Na2SO4 and concentrated on a rotary evaporator. Concentrated viscous oil was purified by preparative HPLC with C18 stationary phase and water–acetonitrile (60:40) mobile phase.
3.2. NMR Spectroscopy
1H, 13C, and 31P NMR spectra were recorded using the Bruker AVANCE III HD 400 MHz NMR spectrometer (Brucker BioSpin GmbH, Ettlingen, Germany) with the PA BBO 400S1 BBF-H-D-05 Z SP probe head and the BCU temperature control unit, PLC on TTY1 of ELCB 1 autosampler, and TopSpin 3.5 pl5 interface. The 1H and 13C NMR signals of the samples were assigned to the 1H and 13C NMR signals of tetramethylsilane (TMS) (0.0 ppm), and the 31P NMR signals were assigned to the 31P NMR signals of H3PO4 (0.0 ppm). Figures S2–S34 (Supplementary Materials) show the 1H, 13C, and 31P NMR spectra of the compounds.
Diphenyl ((1S)-phenyl(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4a′), white solid. 1H NMR (400 MHz, DMSO-d6): δ 7.75–7.70 (m, 2H), 7.54–7.45 (m, 4H), 7.42–7.33 (m, 4H), 7.28–7.19 (m, 2H), 7.12–7.05 (m, 2H), 6.97 (dt, J = 8.5, 1.2 Hz, 2H), 5.77 (d, J = 27.4 Hz, 1H), 5.28 (dd, J = 8.5, 1.3 Hz, 1H), 5.24 (d, J = 8.5 Hz, 1H), 2.94 (s, 3H), 2.88 (s, 3H), 2.57 (s, 3H). 13C NMR (101 MHz, DMSO): 159.06, 158.37 (d, J = 3.7 Hz), 150.59 (d, J = 9.9 Hz), 134.76, 130.17 (d, J = 18.7 Hz), 129.45 (d, J = 6.6 Hz), 129.12, 125.55 (d, J = 15.0 Hz), 120.90 (dd, J = 9.1, 4.1 Hz), 72.40, 72.14 (d, J = 5.0 Hz), 57.37 (d, J = 155.9 Hz), 30.70, 30.27, 30.13; 31P NMR (162 MHz, DMSO-d6): δ 14.06 (d, J = 27.1 Hz). MS (HRMS-ESI): Calcd. For C26H27N4O5P, [M + H]+: 507.1792, found: m/z 507.1823.
Diphenyl ((1R)-phenyl(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4a″) white solid. 1H NMR (400 MHz, DMSO-d6): δ 7.65 (d, J = 7.5 Hz, 2H), 7.50–7.35 (m, 8H), 7.30–7.21 (m, 2H), 7.17 (d, J = 8.1 Hz, 2H), 7.13–7.07 (m, 2H), 5.85 (d, J = 25.8 Hz, 1H), 5.50 (d, J = 8.5 Hz, 1H), 5.00 (d, J = 8.5 Hz, 1H), 2.88 (s, 3H), 2.82 (s, 3H), 2.16 (s, 3H). 13C NMR (101 MHz, DMSO): δ 159.41, 158.11 (d, J = 2.9 Hz), 150.02 (dd, J = 9.7, 3.3 Hz), 130.57 (d, J = 7.9 Hz), 129.28, 129.03, 129.00, 128.95, 126.15, 120.73 (dd, J = 20.8, 4.1 Hz), 72.32, 70.78, 53.84 (d, J = 155.3 Hz), 30.85, 30.75, 30.26; 31P NMR (162 MHz, DMSO-d6): δ 13.35 (d, J = 25.9 Hz). MS (HRMS-ESI): Calcd. for C26H27N4O5P, [M + H]+: 507.1792, found: m/z 507.1825.
Diphenyl ((1S)-(3-nitrophenyl)(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4b′) yellow viscous oil. 1H NMR (400 MHz, DMSO-d6): δ 8.57 (s, 1H), 8.28–8.24 (m, 1H), 8.08 (d, J = 5.7 Hz, 1H), 7.77 (t, J = 8.0 Hz, 1H), 7.46–7.33 (m, 4H), 7.32–7.24 (m, 2H), 7.24–7.16 (m, 2H), 7.14–7.07 (m, 2H), 5.98 (d, J = 27.4 Hz, 1H), 5.36 (d, J = 8.4 Hz, 1H), 5.25 (d, J = 8.5 Hz, 1H), 2.89 (s, 3H), 2.85 (s, 3H), 2.49 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 158.79, 158.42 (d, J = 3.1 Hz), 150.59 (d, J = 10.2 Hz), 150.45 (d, J = 10.0 Hz), 148.22, 137.96 (d, J = 2.3 Hz), 135.77 (d, J = 5.9 Hz), 130.72, 130.39, 130.08, 125.79, 125.50, 124.10, 124.04, 123.72, 120.82, 120.78, 120.72, 120.68, 72.69 (d, J = 4.9 Hz), 72.41, 57.44 (d, J = 152.4 Hz), 30.75, 30.16, 29.86. 31P NMR (162 MHz, DMSO-d6): δ 12.77 (d, J = 27.8 Hz); MS (HRMS-ESI): Calcd. for C26H26N5O7P, [M + H]+: 552.1643, found: m/z 552.1641.
Diphenyl ((1R)-(3-nitrophenyl)(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4b″) yellow viscous oil. 1H NMR (400 MHz, DMSO-d6): δ 8.57 (s, 1H), 8.28 (dd, J = 2.3, 8.1 Hz, 1H), 8.10 (d, J = 7.7 Hz, 1H), 7.78 (t, J = 8.1 Hz, 1H), 7.45–7.36 (m, 5H), 7.31–7.24 (m, 2H), 7.20–7.16 (m, 2H), 7.13–7.09 (m, 2H), 5.98 (d, J = 27.0 Hz, 1H), 5.51 (d, J = 8.5 Hz, 1H), 5.10 (d, J = 8.5 Hz, 1H), 2.90 (s, 3H), 2.85 (s, 3H), 2.40 (s, 3H).13C NMR (101 MHz, DMSO-d6): δ 159.47, 157.94 (d, J = 2.9 Hz), 150.00 (d, J = 5.5 Hz), 149.90 (d, J = 5.4 Hz), 148.17, 136.84 (d, J = 6.4 Hz), 135.68 (d, J = 7.9 Hz), 130.89, 130.65, 130.52, 126.30, 126.18, 123.95, 123.71, 123.63, 120.83, 120.78, 120.72, 120.68, 72.46, 71.00, 54.02 (d, J = 157.3 Hz), 31.08, 30.86, 30.29. 31P NMR (162 MHz, DMSO-d6): δ 12.32 (d, J = 26.8 Hz); MS (HRMS-ESI): Calcd. for C26H26N5O7P, [M + H]+: 552.1643, found: m/z 552.1646.
Diphenyl ((1S)-(3-hydroxyphenyl)(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4c′) white viscous oil. 1H(400 MHz, DMSO), δ, ppm: 9.63 (s, 1H), 7.38–7.29 (m, 4H), 7.26–7.12 (m, 6H), 7.04 (d, J = 8.1 Hz, 2H), 6.99–6.91 (m, 2H), 6.78 (d, J = 8.2 Hz, 1H), 5.64 (d, J = 27.2 Hz, 1H), 5.15 (s, 2H), 2.88 (s, 3H), 2.82 (s, 3H), 2.56 (s, 3H). 13C NMR (101 MHz, DMSO), δ, ppm: 157.04, 156.22 (d, J = 3.7 Hz), 155.82, 128.03 (d, J = 13.3 Hz), 123.42 (d, J = 7.3 Hz), 118.81 (dd, J = 16.8, 4.1 Hz), 118.48, 117.94, 114.21, 113.79, 70.30, 69.79, 54.92 (d, J = 158.1 Hz), 28.57, 28.17, 28.14; 31P (162 MHz, DMSO), δ, ppm: 14.16 (d, J = 27.1 Hz). MS (HRMS-ESI): Calcd. for C26H27N4O6P, [M + H]+: 523.1741, found: m/z 523.1777.
Diphenyl ((1R)-(3-hydroxyphenyl)(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4c″) white viscous oil. 1H(400 MHz, DMSO), δ, ppm: δ 9.67 (s, 1H), 7.54–7.34 (m, 4H), 7.30–7.20 (m, 3H), 7.20–7.15 (m, 2H), 7.15–7.04 (m, 4H), 6.78 (dd, J = 2.4, 8.1 Hz, 1H), 5.76 (d, J = 25.9 Hz, 1H), 5.49 (d, J = 8.5 Hz, 1H), 4.97 (d, J = 8.6 Hz, 1H), 2.88 (s, 3H), 2.82 (s, 3H), 2.19 (s, 3H). 13C NMR (101 MHz, DMSO), δ, ppm: 157.27, 155.99 (d, J = 3.1 Hz), 155.90, 147.92 (dd, J = 9.7, 6.4 Hz), 133.51 (d, J = 7.3 Hz), 128.43 (d, J = 6.5 Hz), 124.00, 118.59 (dd, J = 25.6, 4.1 Hz), 117.32 (d, J = 8.1 Hz), 113.86, 113.73 (d, J = 8.8 Hz), 70.16, 68.62, 51.47 (d, J = 155.0 Hz), 28.62, 28.12; 31P (162 MHz, DMSO), δ, ppm: 13.35 (d, J = 25.7 Hz); MS (HRMS-ESI): Calcd. for C26H27N4O6P, [M + H]+: 523.1741, found: m/z 523.1780.
Diphenyl ((1S)-(4-nitrophenyl)(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4d′) yellow viscous oil. 1H (400 MHz, Chloroform-d), δ, ppm: 8.19 (2H, d, J = 8.1 Hz), 7.84 (2H, d, J = 8.2 Hz), 7.35–7.20 (4H, m), 7.21–7.02 (6H, m), 5.26 (1H, d, J = 26.4 Hz), 4.93 (2H, d, J = 5.1 Hz), 2.95 (3H, s), 2.88 (3H, s), 2.83 (3H, s); 13C NMR (101 MHz, Chloroform-d), δ, ppm: 158.42, 158.22 (d, J = 4.2 Hz), 150.15 (d, J = 9.7 Hz), 147.94 (d, J = 3.1 Hz), 140.66, 130.19, 129.96 (d, J = 6.0 Hz), 129.69 (d, J = 8.7 Hz), 125.97 (d, J = 28.7 Hz), 125.55 (d, J = 24.6 Hz), 123.97 (d, J = 14.4 Hz), 120.42, 120.26 (d, J = 4.3 Hz), 119.99 (d, J = 4.0 Hz), 72.77, 72.26, 57.88 (d, J = 156.2 Hz), 30.65, 30.49, 30.04; 31P (162 MHz, Chloroform-d), δ, ppm: 11.29 (d, J = 26.4 Hz); MS (HRMS-ESI): Calcd. for C26H26N5O7P, [M + H]+: 552.1643, found: m/z 552.1674.
Diphenyl ((1R)-(4-nitrophenyl)(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4d″) yellow viscous oil. 1H (400 MHz, Chloroform-d), δ, ppm: 8.15 (2H, d, J = 8.2 Hz), 7.93 (2H, d, J = 7.9 Hz), 7.34–7.24 (4H, m), 7.20–7.10 (6H, m), 6.18 (1H, d, J = 26.2 Hz), 5.43 (1H, d, J = 8.6 Hz), 4.29 (1H, dd, J = 8.6, 1.4 Hz), 2.90 (2H, s), 2.82 (2H, s), 2.09 (2H, s); 13C NMR (101 MHz, Chloroform-d), δ, ppm: 157.03, 155.93 (d, J = 4.1 Hz), 148.27 (d, J = 9.0 Hz), 147.43 (d, J = 10.6 Hz), 146.30, 134.72 (d, J = 8.8 Hz), 132.98 (d, J = 9.2 Hz), 128.22, 128.01, 124.17, 123.84, 121.68 (d, J = 7.1 Hz), 118.46 (d, J = 4.2 Hz), 118.06 (d, J = 4.4 Hz), 70.31, 68.62, 50.53 (d, J = 158.2 Hz), 29.22, 28.69, 28.57; 31P (162 MHz, Chloroform-d), δ, ppm: 10.48 (d, J = 26.2 Hz); MS (HRMS-ESI): Calcd. for C26H26N5O7P, [M + H]+: 552.1643, found: m/z 552.1670.
Diphenyl ((1S)-(4-hydroxyphenyl)(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4e′) white viscous oil. 1H(400 MHz, DMSO), δ, ppm: 9.72 (s, 1H), 7.50 (d, J = 7.8 Hz, 2H), 7.39–7.27 (m, 5H), 7.20–7.14 (m, 3H), 7.10–7.04 (m, 2H), 6.98–6.92 (m, 2H), 6.84 (d, J = 7.6 Hz, 2H), 5.63 (d, J = 26.5 Hz, 1H), 5.11 (s, 2H), 2.88 (s, 3H), 2.81 (s, 3H), 2.63 (s, 3H). 13C NMR (101 MHz, DMSO), δ, ppm: 157.19, 156.37 (d, J = 4.3 Hz), 155.69, 148.53 (t, J = 10.0 Hz), 129.17 (d, J = 7.3 Hz), 128.05 (d, J = 11.1 Hz), 123.42 (d, J = 5.5 Hz), 118.89 (d, J = 4.0 Hz), 118.74 (d, J = 4.0 Hz), 113.88, 70.40, 69.61, 54.34 (d, J = 160.3 Hz), 29.03, 28.55, 28.20; 31P (162 MHz, DMSO), δ, ppm: 14.56 (d, J = 26.3 Hz); MS (HRMS-ESI): Calcd. for C26H27N4O6P, [M + H]+: 523.1741, found: m/z 523.1767.
Diphenyl ((1R)-(4-hydroxyphenyl)(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4e″) white viscous oil. 1H(400 MHz, DMSO), 9.70 (1H, s), 7.50 (2H, d, J = 7.8 Hz) 7.47–7.34 (5H, m), 7.24 (3H, dt, J = 14.5, 7.4 Hz), 7.16 (2H, d, J = 8.0 Hz), 7.08 (2H, d, J = 8.0 Hz), 6.81 (2H, d, J = 8.5 Hz), 5.71 (1H, d, J = 25.2 Hz), 5.48 (1H, d, J = 8.4 Hz), 4.95 (1H, d, J = 8.4 Hz), 2.85 (3H, s), 2.81 (3H, s), 2.16 (3H, s); 13C NMR (101 MHz, DMSO), δ, ppm: 159.34, 158.13 (d, J = 2.3 Hz), 158.02, 150.10 (dd, J = 9.9, 4.6 Hz), 130.71 (d, J = 8.6 Hz), 130.53 (d, J = 8.4 Hz), 126.06, 120.86 (d, J = 4.0 Hz), 120.60 (d, J = 4.1 Hz), 115.99, 72.27, 70.64, 53.36 (d, J = 155.0 Hz), 30.82, 30.64, 30.19; 31P (162 MHz, DMSO), δ, ppm:13.87 (d, J = 25.4 Hz); MS (HRMS-ESI): Calcd. for C26H27N4O6P, [M + H]+: 523.1741, found: m/z 523.1776.
Diphenyl ((1S)-(2-nitrophenyl)(3,4,6-trimethyl-2,5-dioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl)methyl)phosphonate (4f″) yellow viscous oil. 1H(400 MHz, DMSO), δ, ppm: 8.24 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 8.1 Hz, 1H), 7.86 (td, J = 1.4, 7.8 Hz, 1H), 7.73 (t, J = 7.7 Hz, 1H), 7.44–7.17 (m, 7H), 7.17–7.03 (m, 2H), 7.01–6.94 (m, 2H), 6.35 (d, J = 26.2 Hz, 1H), 5.49 (d, J = 8.4 Hz, 1H), 5.11 (d, J = 8.4 Hz, 1H), 2.84 (d, J = 5.5 Hz, 6H), 2.39 (s, 3H). 13C NMR (101 MHz, DMSO), δ, ppm: 159.46, 157.74, 150.21–149.95 (m), 133.92, 132.08 (d, J = 4.3 Hz), 131.19, 130.47 (d, J = 17.6 Hz), 127.62 (d, J = 7.0 Hz), 126.06 (d, J = 9.2 Hz), 125.74, 120.56 (dd, J = 21.0, 4.2 Hz), 72.48, 70.83 (d, J = 3.2 Hz), 50.04 (d, J = 158.9 Hz), 30.90, 30.76, 30.29; 31P (162 MHz, DMSO), δ, ppm: 11.67 (d, J = 26.4 Hz); MS (HRMS-ESI): Calcd. for C26H26N5O7P, [M + H]+: 552.1643, found: m/z 552.1692.
3.3. X-ray Crystallography
The X-ray diffraction data were collected at 150 °K with the Bruker D8 Venture diffractometer (Bruker Optik GmbH, Ettlingen, Germany) (0.5° ω- and φ-scans, fixed-χ three-circle goniometer, CMOS PHOTON III detector, Mo-IμS 3.0 microfocus source, focusing Montel mirrors, λ = 0.71073 Å MoKα radiation, N2-flow thermostat). Data reduction was performed via the APEX 3 suite. The crystal structure was solved using the ShelXT [25] and was refined using the ShelXL [26] programs assisted by the Olex2 GUI [27]. The atomic displacements for the non-hydrogen atoms were refined with harmonic anisotropic approximation. Hydrogen atoms were located geometrically and refined in the riding model. Table 4 represents the crystallographic parameters for compounds 4a′ and 4a″.
Table 4.
Crystallography data for compounds 4a’ and 4a’’.
3.4. HPLC Analysis
The analysis was performed with the Agilent 1200 chromatograph (Agilent Technologies, Santa Clara, CA, USA). Spectrophotometric detection was carried out using the Zorbax Eclipse Plus C18 4.6 × 100 mm 3.5 μm chromatographic column (Agilent Technologies, Santa Clara, CA, USA) as a stationary phase. The chromatographic mode was gradient, mobile phase A was a buffer solution (25 mM formic acid (Honeywell Fluka, Charlotte, NC, USA) and 5 mM ammonia (Honeywell Fluka, Charlotte, NC, USA)), and mobile phase B was acetonitrile (PanReac AppliChem, Darmstadt, Germany). Gradient mode: 0.0 min–50% A, 0.5 min–50% A, 7.5 min–20% A, 9.0–20% A, 9.01–50% A, 11.0–50% A. Flow rate was 1.5 mL/min, and thermostat temperature was 25 °C.
3.5. Preparative HPLC
The preparative purification and separation of diastereomers 4a′–e′ and 4a″–f″ were performed using the Shimadzu LC 20 Prominence chromatograph (Shimadzu, Kyoto, Japan) with the Kromasil C18 column 20 × 250 mm, 5 µm particle size (Nouryon, Bohus, Sweden). The column temperature was set to 25 °C (±1 °C). The mobile phase consisted of a water–acetonitrile mixture (60:40). The flow rate was 5 mL/min in the isocratic mode. The detection wavelength for the UV detector equaled 250 nm. The samples were preliminarily dissolved in acetonitrile (1:10, v/v). Figures S48–S53 (Supplementary Materials) show the HRMS of the compounds 4a–4f.
3.6. HRMS
The analysis was performed using the HPLC-MS method in the electro-spray ionization mode using the Agilent QTOF-6550 (Agilent Technologies, Santa Clara, CA, USA) connected with Agilent 1260 Infinity II (Agilent Technologies, Santa Clara, CA, USA). The samples were analyzed with the Poroshell 120 EC-C18 column 2.1 × 100 mm, 2.7 µm particle size. The column temperature was set as 30 °C (±0.4 °C). The chromatographic analysis was performed in isocratic mode. The 0.05% formic acid (Honeywell Fluka, Charlotte, NC, USA) aqueous solution–acetonitrile mixture (60:40) was applied as a mobile phase. The flow rate was 0.5 mL/min. The samples were analyzed in a positive ionization mode with a scan range of 300–900 Da. Capillary and nozzle voltages were set to 2000 and 100 V, respectively. Drying gas temperature was 200 °C, and sheath gas temperature was 250 °C. The drying gas flow was 13 L/min, and the sheath gas flow was 11 L/min. The samples were preliminarily dissolved in acetonitrile (1:1000, v/v). Figures S37–S47 (Supplementary Materials) show the preparative elution profiles of the compounds 4a–4f.
3.7. Computational Details
All model calculations were performed using the Gaussian’09 program package [28] installed on the SKIF “Cyberia” supercomputer of Tomsk State University. The geometries of the structures under analysis were fully optimized at the meta-hybrid functional M062X [29] with the split-valence basis set 6-311+G(2d,p) in DMSO (ε = 46.826) as a solvent using PCM. The PCM was applied using a scaled van der Waals surface cavity with an alpha value of 1.1, and atomic radii modelling using a universal forcefield. The geometry optimization was performed on the basis of the experimentally determined X-ray structures of 4a’ and 4a″ without conformational analysis. Frequency calculations were also performed at the M062X/6-311+G(2d,p) level of theory. The freely available GoodVibes script developed by Paton and Funes-Ardoiz [30] was used to recalculate the thermochemical data with the low-frequency corrections and correction from the gas with the pressure of 1 atm to solution with the concentration of 1 mol/L.
Tables S3–S6 (Supplementary Materials) show the correlation between the geometric parameters for the optimized 4a molecular structures and the experimental data. Table S7 (Supplementary Materials) presents the XYZ coordinates for all of the compounds under analysis.
Magnetic shielding tensors were calculated with the gauge including atomic orbitals (GIAO) DFT method using the Gaussian’09 at the PBE0/6-311+G(2d,p) level theory.
The chemical shifts (δ 31Pcalc) were detected as δ 31Pcalc = σ H3PO4–σ calc, where σ H3PO4 is the shielding constant of 31P in phosphoric acid (H3PO4). The σ H3PO4 values of 31P of the H3PO4 were computed using the same strategy as for structure 4. Figure S54 (Supplementary Materials) shows the correlations between experimental and calculation values δ 1H, 13C atoms of 3a, 6a, CHPO3 and δ 31P of the compounds 4a–4f.
4. Conclusions
The results demonstrate an approach to the synthesis of new phosphonate derivatives of 1,3,4-trimethylglycoluril via the Birum–Oleksyszyn reaction. This approach allowed for the obtainment of a series of diastereomers that were formed in the reaction medium with the predominance of structure 4″. The obtained diastereomers can be separated by the preparative HPLC with the C18 stationary phase. The diastereomers’ structures were confirmed via NMR spectroscopy and HRMS; for compound 4a, the structure was confirmed with a single-crystal X-ray diffraction analysis. The results of the quantum chemical calculations were consistent with the experimental data. Additionally, the authors revealed a correlation between the spatial arrangement of substituents in the structure of 1-[aryl-(diphenylphosphono)methyl]-3,4,6-trimethylglycolurils and the δ values of the proton on the carbon atom in the phosphonate group (CHPO3) as well as the proton bridging CH-CH group. For the compounds with structure 4′, the signals of the methine protons in the CH-CH bond were recorded in the 1H NMR spectrum as two doublets within the narrow region (5.11–5.37 ppm), while for the compounds with structure 4″, the signals of these protons were recorded within a wider range (4.95–5.51 ppm). At the same time, the signal of the proton bonded to the carbon atom in the phosphonate group (CHPO3) was shifted to a higher field for the compounds with structure 4′ compared to the similar proton in structure 4″.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/ijms242317082/s1. The CIF and CheckCIF files of the 4a′ and 4a″structures files were attached.
Author Contributions
Conceptualization, S.I.G. and V.P.T.; methodology, A.A.B., V.S.M. and A.S.K.; validation, S.I.G., V.P.T., A.V.K., D.A.K. and M.V.M.; investigation, S.I.G., V.P.T. and M.V.M.; resource, A.S.K. and V.S.M.; data curation S.I.G., V.P.T., A.V.K., G.O.S., D.I.P. and M.V.M.; writing—original draft, S.I.G., V.P.T., D.I.P. and A.V.K.; writing—review and editing, A.A.B., V.S.M. and A.S.P.; visualization, G.O.S., A.V.K. and D.I.P.; supervision, A.A.B. and A.S.K.; project administration, S.I.G. and V.P.T.; funding acquisition, V.P.T., S.I.G., A.A.B. and V.S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Tomsk State University Development Programme (Priority 2030).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available within the article and the Supplementary Materials. Further data are available from the corresponding author upon reasonable request. Crystallographic Data Centre, CCDC no. 2294213 provided compound 4a’, and CCDC no. 2294214 provided compound 4a″. Copies of the data can be obtained free of charge from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-1223-336-033; e-mail: deposit@ccdc.cam.ac.uk).
Acknowledgments
A.S.P. and D.I.P. acknowledge the Ministry of Science and Higher Education of the Russian Federation (121031700321-3). The authors thank Mikhail Salaev (Tomsk State University) for conducting a language review. The authors also thank Ksenia Kazantseva and Natalia Selikhova (Tomsk State University) for recording MS. The authors thank Alexander Fateev (Tomsk State Pedagogical University) for helping with the quantum chemical calculations and Oleg Kotelnikov (Tomsk State University) for recording the NMR spectra.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ryzhkina, I.S.; Kiseleva, Y.V.; Mishina, O.A.; Timosheva, A.P.; Sergeeva, S.Y.; Kravchenko, A.N.; Konovalov, A.I. Correlations between the self-organization, physicochemical properties and biological activity of Mebicar in dilute aqueous solutions. Mendeleev Commun. 2013, 23, 262–264. [Google Scholar] [CrossRef]
- Kravchenko, A.N.; Baranov, V.V.; Anikina, L.V.; Vikharev, Y.B.; Bushmarinov, I.S.; Nelyubina, Y.V. Neuroprotective Activity of (+)-(S)-2-[(1S,5R)-(3,7-Dioxo-2,4,6,8-Tetraazabicyclo[3.3.0]oct-2-yl)]-4-methylthiobutanoic acid. Russ. J. Bioorg. Chem. 2012, 38, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, K.; Beilfus, W.; Krull, I.; Gradtke, R. Verwendung von Formaldehyd und Formaldehyd Freisetzenden Verbindungen in einer Zusammensetzung zur Bekämpfung von Mykobakterien. DE Patent 102004059041, 8 June 2006. [Google Scholar]
- Mcbride, R.; Elsmore, R.; Houghton, M.P. Bactericide Combinations in Detergents. GB Patent 2354771, 4 April 2001. [Google Scholar]
- Lizal, T.; Sindelar, V. Bambusuril Anion Receptors. Isr. J. Chem. 2018, 58, 326–333. [Google Scholar] [CrossRef]
- Lafosse, M.; Liang, Y.; Schneider, J.P.; Cartier, E.; Bodlenner, A.; Compain, P.; Heck, M.-P. Bambus[4,6]urils as Dual Scaffolds for Multivalent Iminosugar Presentation and Ion Transport: Access to Unprecedented Glycosidase-Directed Anion Caging Agents. Molecules 2022, 27, 4772. [Google Scholar] [CrossRef] [PubMed]
- Cong, H.; Ni, X.L.; Xiao, X.; Huang, Y.; Zhu, Q.-J.; Xue, S.-F.; Tao, Z.; Leonard, F. Lindoy and Gang Wei. Synthesis and separation of cucurbit[n]urils and their derivatives. Org. Biomol. Chem. 2016, 14, 4335–4364. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Zhao, J.; Xu, J.-F.; Zhang, X. Cucurbit[n]urils for Supramolecular Catalysis. Chem. Eur. J. 2020, 26, 15446. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, M.; Gosecki, M.; Gostynskia, B.; Gosecka, M. Synthesis of a monofunctional glycoluril molecular clip via cyclic imide formation on the convex site. New J. Chem. 2020, 44, 596–604. [Google Scholar] [CrossRef]
- Sokolov, J.; Šindelář, V. Synthesis of Glycoluril Dimers with the Ability to Form Polymeric Self-Associates in Water. Chemistry 2022, 4, 53. [Google Scholar] [CrossRef]
- Amita, N.; Sushma, J.; Singh, B.; Mane, M.V.; Kumbhar, A.S. Heteroleptic Copper(I) complexes of bipyridine glycoluril and phosphine ligands: Photophysical and computational studies. Inorganica Chim. Acta 2022, 538, 120934. [Google Scholar] [CrossRef]
- Berdimurodov, E.; Kholikov, A.; Akbarov, K.; Guo, L.; Kaya, S.; Verma, D.K.; Rbaa, M.; Dagdag, O. Novel glycoluril pharmaceutically active compound as a green corrosion inhibitor for the oil and gas industry. J. Electroanal. Chem. 2022, 907, 116055. [Google Scholar] [CrossRef]
- Baranov, V.V.; Vol’khina, T.N.; Kolotyrkina, N.G.; Kravchenko, A.N. Synthesis and crystal structures of novel glycoluril carboxylic acids conglomerates. Mendeleev Commun. 2022, 32, 537–539. [Google Scholar] [CrossRef]
- Kalichkina, L.E.; Fateev, A.V.; Krivolapenko, P.K.; Isakova, K.A.; Knyazev, A.S.; Malkov, V.S.; Bakibaev, A.A.; Tuguldurova, V.P. The Study of Structural Features of N- and O-Derivatives of 4,5-Dihydroxyimidazolidine-2-Thione by NMR Spectroscopy and Quantum Chemical Calculations. Magnetochemistry 2023, 9, 15. [Google Scholar] [CrossRef]
- Sinitsyna, A.A.; Il’yasov, S.G.; Chikina, M.V.; Eltsov, I.V.; Nefedov, A.A. A search for synthetic routes to tetrabenzylglycoluril. Chem. Pap. 2020, 74, 1019–1025. [Google Scholar] [CrossRef]
- Moradi, S.; Zolfigol, M.A.; Zarei, M.; Alonso, D.A.A. Khoshnood Synthesis of a Biological-Based Glycoluril with Phosphorous Acid Tags as a New Nanostructured Catalyst: Application for the Synthesis of Novel Natural Henna-Based Compounds. Chem. Sel. 2018, 3, 3042. [Google Scholar] [CrossRef]
- Danishyar, B.; Sepehrmansourit, H.; Zarei, M.; Zolfigol, M.A.; As’Habi, M.A.; Gu, Y. Synthesis and Application of Novel Magnetic Glycoluril Tetrakis(Methylene Phosphorous Acid) as a Nano Biological Catalyst for the Preparation of Nicotinonitriles via a Cooperative Vinylogous Anomeric-Based Oxidation. Polycycl. Aromat. Compd. 2022, 43, 6837–6857. [Google Scholar] [CrossRef]
- Sal’keeva, L.K.; Bakibaev, A.A.; Khasenova, G.T.; Taishibekova, Y.K.; Sugralina, L.M.; Minaeva, Y.V.; Sal’keeva, A.K. Effect of glycoluril and its derivatives on the flame resistance and physico-mechanical properties of rubber. Russ. J. Appl. Chem. 2016, 89, 132–139. [Google Scholar] [CrossRef]
- Heilmann, M.; Tiefenbacher, K. A Modular Phosphorylated Glycoluril-Derived Molecular Tweezer for Potent Binding of Aliphatic Diamines. Chem. Eur. J. 2019, 25, 12900–12904. [Google Scholar] [CrossRef]
- Gorbin, S.I.; Bakibaev, A.A.; Kurgachev, D.A.; Malkov, V.S.; Novolokov KYu Sysoev, G.O. Synthesis of 1-[1-(diphenoxyphosphoryl)alkyl]-3,4,6-trimethylglycolurils. Mendeleev Commun. 2023, 33, 638–639. [Google Scholar] [CrossRef]
- Oleksyszyn, J.; Subotkowska, L.; Mastalerz, P. Diphenyl 1-Aminoalkanephosphonates. Synthesis 1979, 12, 985–986. [Google Scholar] [CrossRef]
- Van der Veken, P.; El Sayed, I.; Joossens, J.; Stevens, C.V.; Augustyns, K. Achiel Haemers Lewis Acid Catalyzed Synthesis of N-Protected Diphenyl 1-Aminoalkylphosphonates. Synthesis 2005, 4, 634–638. [Google Scholar] [CrossRef]
- Birum, G.H. Urylenediphosphonates. General method for the synthesis of α-ureidophosphonates and related structures. J. Org. Chem. 1974, 39, 209–213. [Google Scholar] [CrossRef]
- Kravchenko, A.N.; Sigachev, A.S.; Maksareva, E.Y.; Gazieva, G.A.; Trunova, N.S.; Lozhkin, B.V.; Pivina, T.S.; Il’in, M.M.; Lyssenko, K.A.; Nelyubina, Y.V.; et al. Synthesis of new chiral mono-, di-, tri-, and tetraalkylglycolurils. Russ. Chem. Bull. 2005, 54, 691–704. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A complete structure solution, refinement and analysis program. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functional. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Funes-Ardoiz, I.; Paton, R. GoodVibes Version 3.2. Available online: https://github.com/bobbypaton/GoodVibes (accessed on 12 August 2022).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).