
Does Background Matter? A Comparative Characterization of Mouse Models of Autosomal Retinitis Pigmentosa rd1 and Pde6b-KO

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Development and Biochemical Characterization of the Pde6b Knockout Mouse Model in Comparison with the rd1 Mouse Model
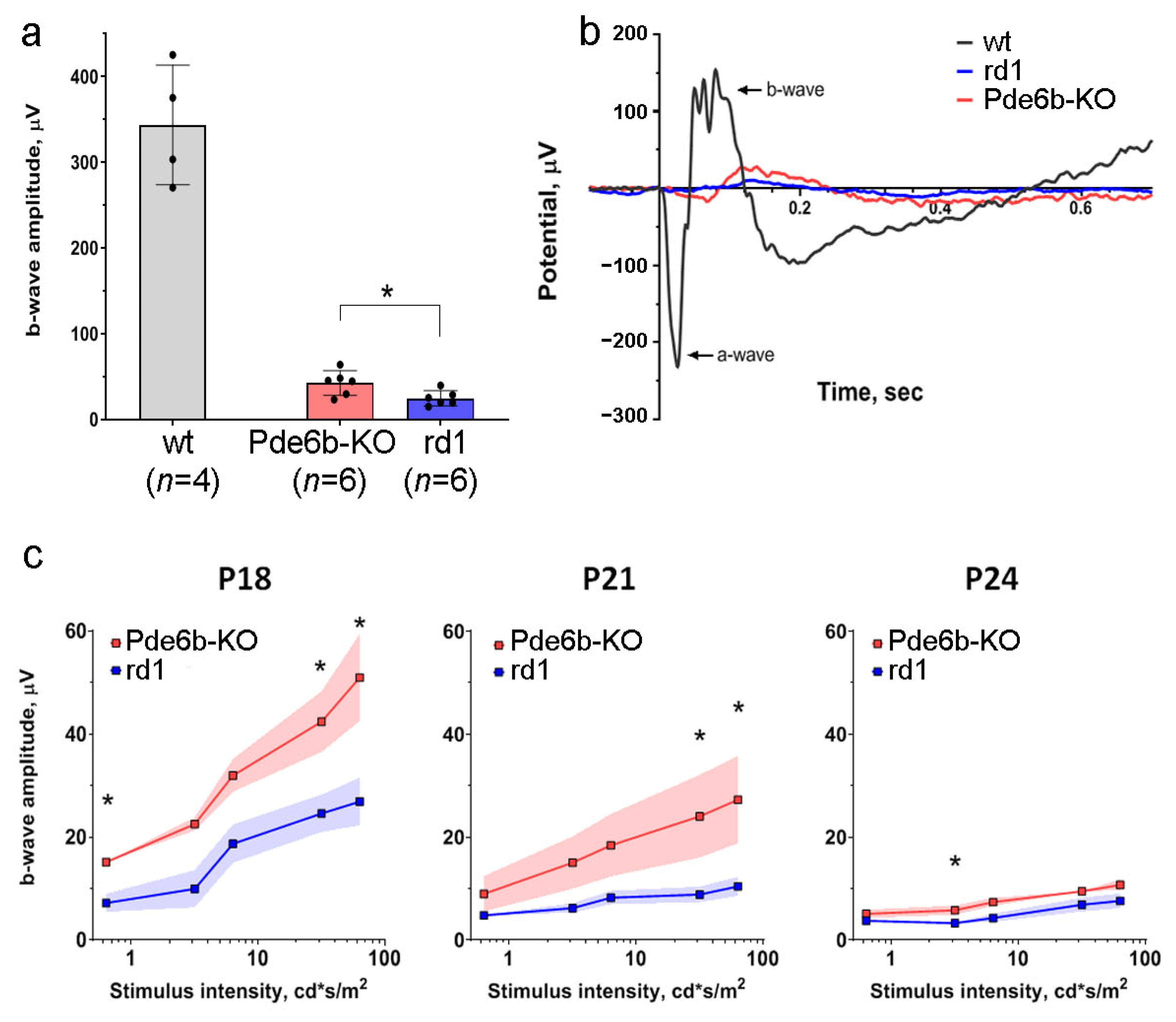
2.2. Electroretinography of Pde6b Knockout Mice in Comparison with rd1 Mice
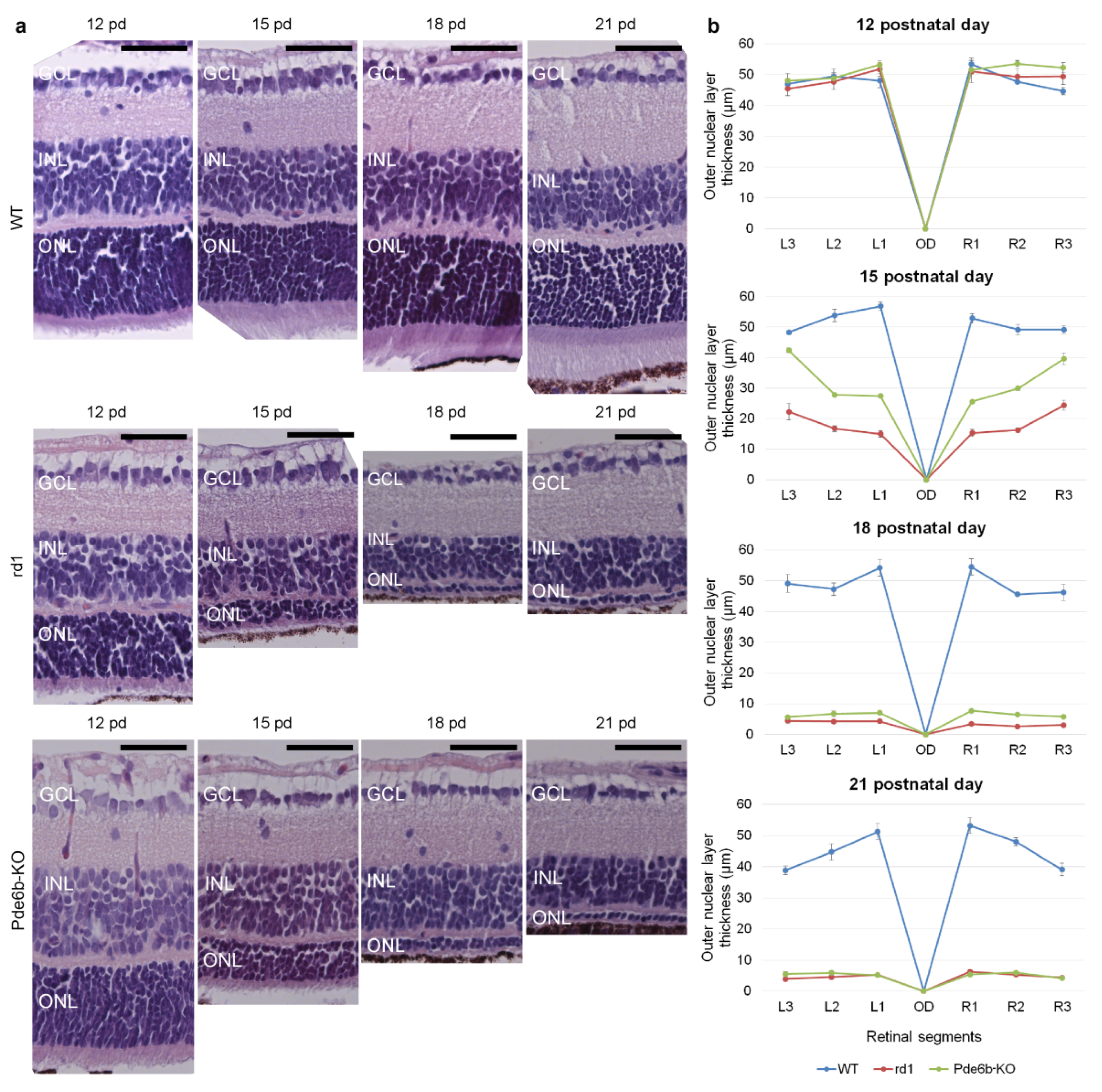
2.3. Retinal Morphology of Pde6b Knockout Mice in Comparison with rd1 Mice
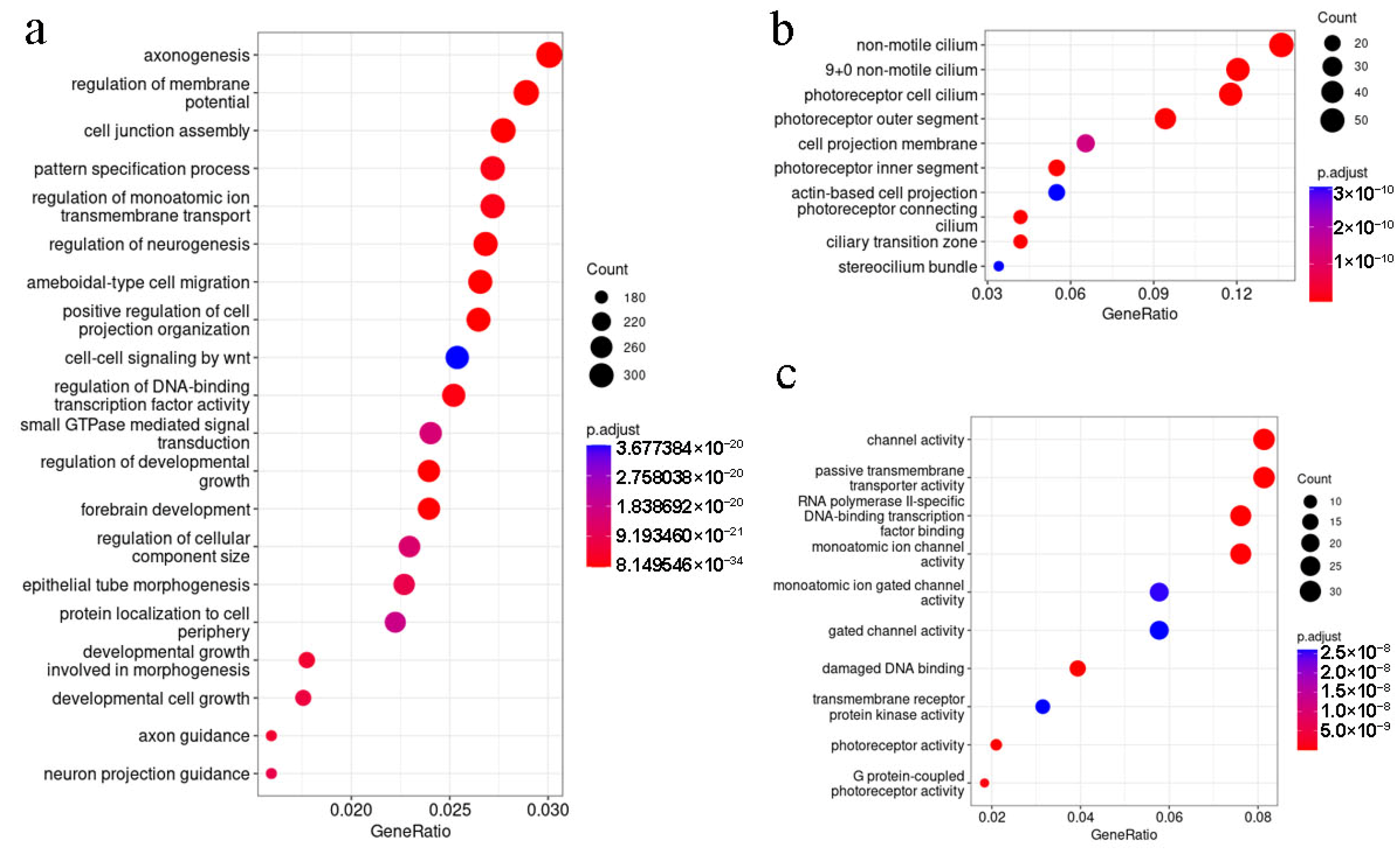
2.4. Functional Analysis of Genes
3. Discussion
4. Materials and Methods
4.1. Production of Pde6b KO Mouse Model
4.2. Electroretinography
4.3. Histology
4.4. Total RNA Extraction and RT-PCR
4.5. Protein Extraction and Western Blot Hybridization
4.6. The Identification and Description of Genetic Polymorphisms
4.7. Function and Enrichment Analysis
4.8. Functional Analysis of Genes Harboring Nucleotide Polymorphisms in C3H
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and Mutations Causing Retinitis Pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.; Ullah, M.; Rashid, A.; Hussain, Z.; Shah, K.; Awan, A.; Khan, M.; Ullah, I.; Rehman, A.U. A Novel Homozygous Missense Substitution p.Thr313Ile in the PDE6B Gene Underlies Autosomal Recessive Retinitis Pigmentosa in a Consanguineous Pakistani Family. BMC Ophthalmol. 2023, 23, 116. [Google Scholar] [CrossRef]
- Yeo, J.H.; Jung, B.K.; Lee, H.; Baek, I.-J.; Sung, Y.H.; Shin, H.-S.; Kim, H.K.; Seo, K.Y.; Lee, J.Y. Development of a Pde6b Gene Knockout Rat Model for Studies of Degenerative Retinal Diseases. Invest. Ophthalmol. Vis. Sci. 2019, 60, 1519. [Google Scholar] [CrossRef]
- Wu, W.-H.; Tsai, Y.-T.; Justus, S.; Lee, T.-T.; Zhang, L.; Lin, C.-S.; Bassuk, A.G.; Mahajan, V.B.; Tsang, S.H. CRISPR Repair Reveals Causative Mutation in a Preclinical Model of Retinitis Pigmentosa. Mol. Ther. 2016, 24, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Collin, G.B.; Gogna, N.; Chang, B.; Damkham, N.; Pinkney, J.; Hyde, L.F.; Stone, L.; Naggert, J.K.; Nishina, P.M.; Krebs, M.P. Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss. Cells 2020, 9, 931. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Dinculescu, A.; Dai, X.; Du, W.; Smith, W.C.; Pang, J. Review: The History and Role of Naturally Occurring Mouse Models with Pde6b Mutations. Mol. Vis. 2013, 19, 2579–2589. [Google Scholar] [PubMed]
- Wang, T.; Reingruber, J.; Woodruff, M.L.; Majumder, A.; Camarena, A.; Artemyev, N.O.; Fain, G.L.; Chen, J. The PDE6 Mutation in the Rd10 Retinal Degeneration Mouse Model Causes Protein Mislocalization and Instability and Promotes Cell Death through Increased Ion Influx. J. Biol. Chem. 2018, 293, 15332–15346. [Google Scholar] [CrossRef]
- van Wyk, M.; Schneider, S.; Kleinlogel, S. Variable Phenotypic Expressivity in Inbred Retinal Degeneration Mouse Lines: A Comparative Study of C3H/HeOu and FVB/N Rd1 Mice. Mol. Vis. 2015, 21, 811–827. [Google Scholar]
- Leinonen, H.; Tanila, H. Vision in Laboratory Rodents—Tools to Measure It and Implications for Behavioral Research. Behav. Brain Res. 2018, 352, 172–182. [Google Scholar] [CrossRef]
- Vollrath, D.; Yasumura, D.; Benchorin, G.; Matthes, M.T.; Feng, W.; Nguyen, N.M.; Sedano, C.D.; Calton, M.A.; LaVail, M.M. Tyro3 Modulates Mertk-Associated Retinal Degeneration. PLoS Genet. 2015, 11, e1005723. [Google Scholar] [CrossRef]
- Benchorin, G.; Calton, M.; Beaulieu, M.; Vollrath, D. Assessment of Murine Retinal Function by Electroretinography. Bio-Protocol 2017, 7, e2218. [Google Scholar] [CrossRef]
- Lowe, D.G. Object Recognition from Local Scale-Invariant Features. In Proceedings of the Seventh IEEE International Conference on Computer Vision, Kerkyra, Greece, 20–27 September 1999; Volume 2, pp. 1150–1157. [Google Scholar]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P. PhD-SNPg: A Webserver and Lightweight Tool for Scoring Single Nucleotide Variants. Nucleic Acids Res. 2017, 45, W247–W252. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Patch, A.-M.; Ellard, S. Using SIFT and PolyPhen to Predict Loss-of-Function and Gain-of-Function Mutations. Genet. Test. Mol. Biomark. 2010, 14, 533–537. [Google Scholar] [CrossRef]
- Masica, D.L.; Sosnay, P.R.; Cutting, G.R.; Karchin, R. Phenotype-Optimized Sequence Ensembles Substantially Improve Prediction of Disease-Causing Mutation in Cystic Fibrosis. Hum. Mutat. 2012, 33, 1267–1274. [Google Scholar] [CrossRef]
- Sancho-Pelluz, J.; Arango-Gonzalez, B.; Kustermann, S.; Romero, F.J.; Van Veen, T.; Zrenner, E.; Ekström, P.; Paquet-Durand, F. Photoreceptor Cell Death Mechanisms in Inherited Retinal Degeneration. Mol. Neurobiol. 2008, 38, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Doe, B.; Brown, E.; Boroviak, K. Generating CRISPR/Cas9-Derived Mutant Mice by Zygote Cytoplasmic Injection Using an Automatic Microinjector. Methods Protoc. 2018, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Goriachenkov, A.A.; Rotov, A.Y.; Firsov, M.L. Developmental Dynamics of the Functional State of the Retina in Mice with Inherited Photoreceptor Degeneration. Neurosci. Behav. Physiol. 2021, 51, 807–815. [Google Scholar] [CrossRef]
- Li, H. New Strategies to Improve Minimap2 Alignment Accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chirinskaite, A.V.; Rotov, A.Y.; Ermolaeva, M.E.; Tkachenko, L.A.; Vaganova, A.N.; Danilov, L.G.; Fedoseeva, K.N.; Kostin, N.A.; Sopova, J.V.; Firsov, M.L.; et al. Does Background Matter? A Comparative Characterization of Mouse Models of Autosomal Retinitis Pigmentosa rd1 and Pde6b-KO. Int. J. Mol. Sci. 2023, 24, 17180. https://doi.org/10.3390/ijms242417180
Chirinskaite AV, Rotov AY, Ermolaeva ME, Tkachenko LA, Vaganova AN, Danilov LG, Fedoseeva KN, Kostin NA, Sopova JV, Firsov ML, et al. Does Background Matter? A Comparative Characterization of Mouse Models of Autosomal Retinitis Pigmentosa rd1 and Pde6b-KO. International Journal of Molecular Sciences. 2023; 24(24):17180. https://doi.org/10.3390/ijms242417180
Chicago/Turabian StyleChirinskaite, Angelina V., Alexander Yu. Rotov, Mariia E. Ermolaeva, Lyubov A. Tkachenko, Anastasia N. Vaganova, Lavrentii G. Danilov, Ksenia N. Fedoseeva, Nicolay A. Kostin, Julia V. Sopova, Michael L. Firsov, and et al. 2023. "Does Background Matter? A Comparative Characterization of Mouse Models of Autosomal Retinitis Pigmentosa rd1 and Pde6b-KO" International Journal of Molecular Sciences 24, no. 24: 17180. https://doi.org/10.3390/ijms242417180
APA StyleChirinskaite, A. V., Rotov, A. Y., Ermolaeva, M. E., Tkachenko, L. A., Vaganova, A. N., Danilov, L. G., Fedoseeva, K. N., Kostin, N. A., Sopova, J. V., Firsov, M. L., & Leonova, E. I. (2023). Does Background Matter? A Comparative Characterization of Mouse Models of Autosomal Retinitis Pigmentosa rd1 and Pde6b-KO. International Journal of Molecular Sciences, 24(24), 17180. https://doi.org/10.3390/ijms242417180