
Transcriptomic Changes in Response to Form of Selenium on the Interferon-Tau Signaling Mechanism in the Caruncular Tissue of Beef Heifers at Maternal Recognition of Pregnancy


Abstract
:1. Introduction
2. Results
2.1. Real-Time PCR Analysis of Selenoproteins and Selenoprotein P Receptors mRNA
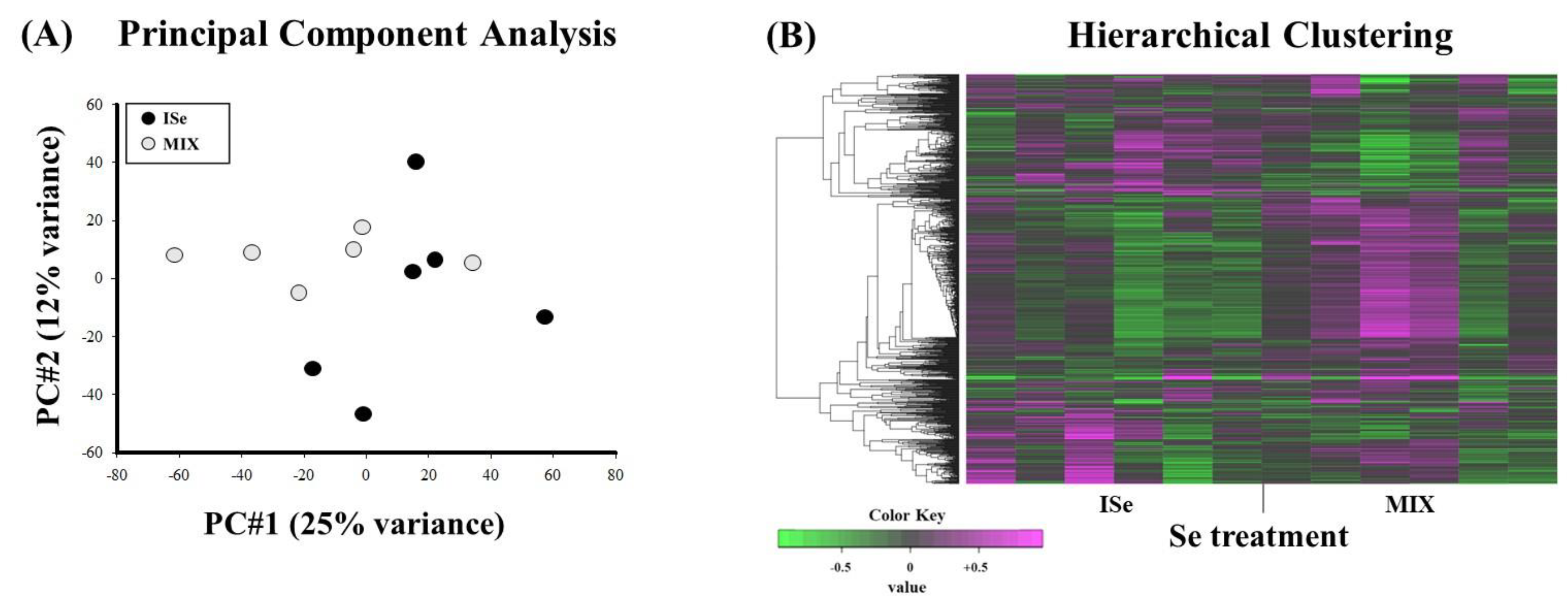
2.2. Cluster Analyses
2.3. Differentially Expressed Genes
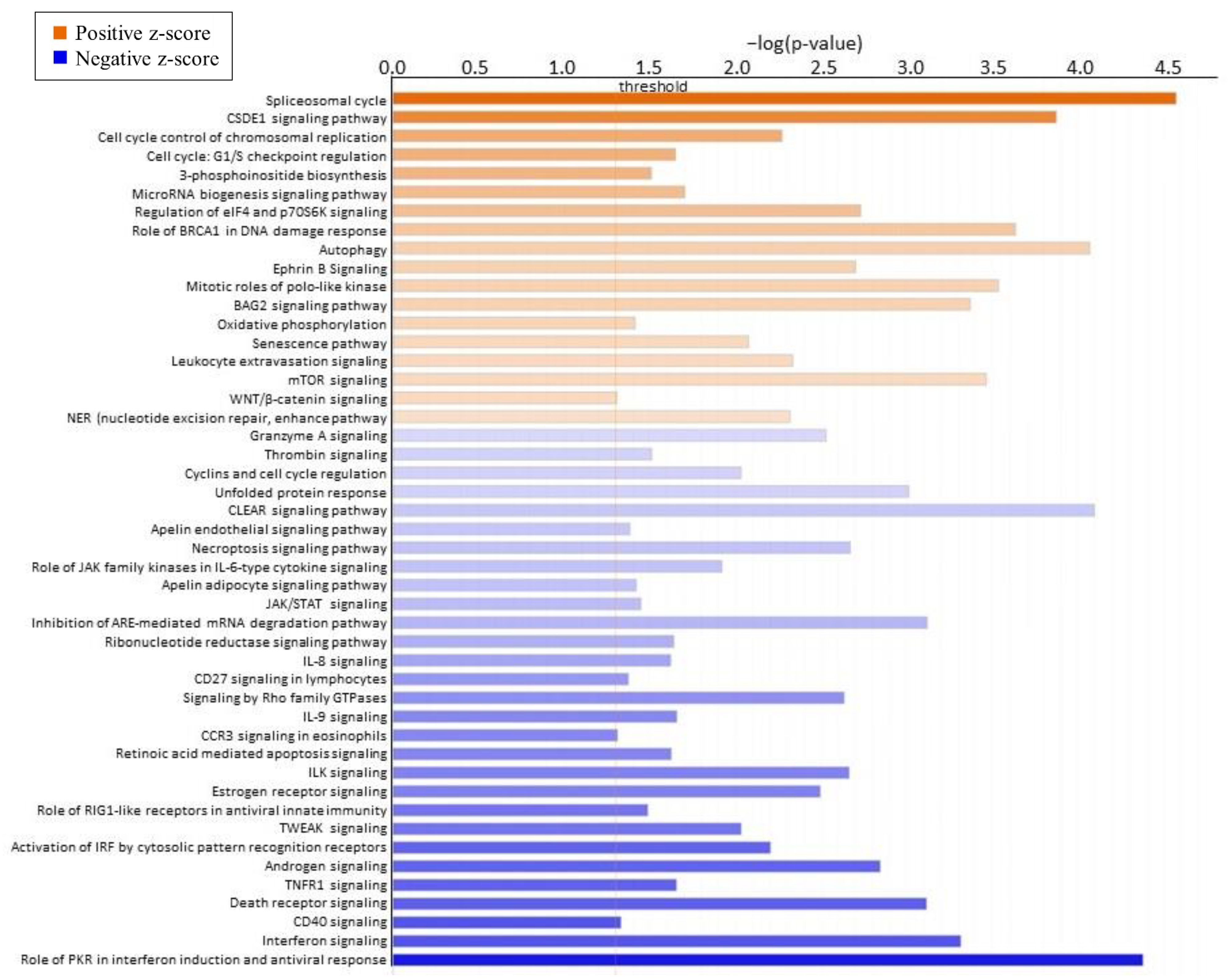
2.4. Pathway and Gene Network Analysis
2.5. Real-Time PCR Analysis of Select mRNA Transcripts
3. Discussion
3.1. Selenoproteins
3.2. Caruncular Transcriptomics
4. Materials and Methods
4.1. Animals and Experimental Procedure
4.2. Experimental Regimen and Serum Collection
4.3. RNA Extraction
4.4. RNA-Sequencing
4.5. Real-Time PCR Analysis
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Name | Accession Number 1 | Oligonucleotide Primer Design (5′ to 3′) Direction | Amplicon Length (bp) | Product Identity (%) 2 |
|---|---|---|---|---|---|
| Enzymatic transcripts | |||||
| Dio1 | Iodothyronine deiodinase 1 | NM_001122593.2 | F: TCCTGTAGTCCGCCTGTCA R: TCCGGTGATTCTTGATGTCCA | 242 | 99 |
| Dio2 | Iodothyronine deiodinase 2 | NM_001010992.4 | F: GATGGGCATCCTCAGCGTAG R: TTCTCCTGGGCACCATTTCC | 315 | 100 |
| Dio3 | Iodothyronine deiodinase 3 | NM_001010993.3 | F: AAGTGGAGCTCAACAGCGAT R: AGTCGAGGATGTGCTGGTTC | 213 | 100 |
| Glutathione peroxidases | |||||
| Gpx1 | Glutathione peroxidase 1 | NM_174076.3 | F: GCAACCAGTTTGGGCATCAG R: TAGGGTCGGTCATGAGAGCA | 210 | 100 |
| Gpx2 | Glutathione peroxidase 2 | NM_001163139.2 | F: AACAGCCTCAAGTACGTCCG R: TCGGTCATGAGGGAAAACGG | 158 | 100 |
| Gpx3 | Glutathione peroxidase 3 | NM_174077.5 | F: GCACCATCTATGAGTACGGGG R: CCCCATTCACATCGCCTTTC | 315 | 100 |
| Gpx4 | Glutathione peroxidase 4 | NM_174770.3 | F: GATCAAAGAGTTCGCCGCTG R: CCATACCGCTTCACCACACA | 198 | 100 |
| Gpx6 | Glutathione peroxidase 6 | NM_001163142.1 | F: CACTGTTCCTGGTCGGCTTA R: CCCAGCACAACTACACCGAA | 259 | 100 |
| Thioredoxin reductases | |||||
| Txnrd1 | Thioredoxin reductase 1 | NM_174625.5 | F: AAGGCCGCGTTATTTGGGTA R: CCTGGTGTCCCTGCTTCAAT | 306 | 100 |
| Txnrd2 | Thioredoxin reductase 2 | NM_174626.2 | F: CAAATGGCTTCGCTGGTCAC R: TTCGTATGCACACCAGCCTT | 230 | 100 |
| Txnrd3 | Thioredoxin reductase 3 | XM_015468824.1 | F: CGGCGTATGACTACGACCTC R: GACTGTACTCCCAGCCGAAC | 249 | 100 |
| Other selenoproteins | |||||
| Selenof | Selenoprotein F | NM_001034759.2 | F: GCAGCTCCTGTGATTTGCTT R: TTTAGCACAGGGTCTGAACCG | 241 | 100 |
| Selenoh | Selenoprotein H | NM_001321327.1 | F: CACGAGCTGACGAGTCTACG R: CTTCTTCAGCTCCTCCAGCA | 235 | 100 |
| Selenoi | Selenoprotein I | NM_001075257.2 | F: TCTGGCTTTCTGCTGGTTGT R: TGGTCAAAAAGCTCCCCCAG | 212 | 100 |
| Selenok | Selenoprotein K | NM_001037489.3 | F: CCGTTTTGTCGATTCACGGC R: CAGATGAGCTTCCGTAGCCT | 278 | 100 |
| Selenom | Selenoprotein M | NM_001163171.2 | F: CCCACTCTACCACAACCTGG R: ACCTAAAGGTCTGCGTGGTC | 249 | 100 |
| Selenon | Selenoprotein N | NM_001114976.2 | F: GTGGCCATGTACCCCTTCAA R: GGGATGGGTTCTCCTGGTTG | 265 | 100 |
| Selenoo | Selenoprotein O | NM_001163193.2 | F: TGGACAGGTATGACCCCGAT R: ATCTTCTGCAGGTAGTGCCG | 202 | 100 |
| Selenop | Selenoprotein P | NM_174459.3 | F: TCAGGTCTTCATCACCACCA R: GTGGCAACAGCAGCTACTCA | 201 | 100 |
| Selenor | Selenoprotein R, Methionine sulfoxide reductase B1 (MSRB1) | NM_001034810.2 | F: GAACCACTTTGAGCCGGGTA R: GGCCATCGTTCAGGAACTCA | 221 | 100 |
| Selenos | Selenoprotein S | NM_001046114.3 | F: CCCACCCTCGAGACCGA R: GCCCAGGACTGTCTTCTTCC | 394 | 100 |
| Selenot | Selenoprotein T | NM_001103103.2 | F: TGGTCACCTTCCATCCATGC R: AAGAGGTACAACGAGCCTGC | 240 | 100 |
| Selenov | Selenoprotein V | NM_001163244.2 | F: ACTCCATTGGCCACCGATTT R: AGGCCACAGTAAACCACTCG | 224 | 100 |
| Selenow | Selenoprotein W | NM_001163225.1 | F: AGTGTTCGTAGCGGGAAAGC R: CGCGAGAACATCAGGGAAGG | 233 | 98 |
| Selenophosphate synthetase | |||||
| Sephs2 | Selenophosphate synthetase 2 | NM_001114732.2 | F: GATCCCTACATGATGGGGCG R: GTTTACCACCGTTTGCCCAC | 219 | 100 |
| Selenoprotein P receptors | |||||
| Lrp2 | LDL receptor related protein 2 | XM_024983502.1 | F: GTGGTTTGGGTTACCGTTGC R: GGCACCCTGTTAGCTGTGAT | 304 | 99 |
| Lrp8 | LDL receptor related protein 8 | NM_001097565.1 | F: AGCCACCCTTTTGGGATAGC R: AAGGCACAGGTACTCACAGC | 231 | 100 |
| Tfrc | Transferrin receptor | NM_001206577.1 | F: CCAGGTTTAGTCTGGCTCGG R: GGTCTGCCCAGAATATGCGA | 339 | 99 |
| Gene | Gene Name | Accession Number 1 | Primer Design (5′ to 3′) Direction | Amplicon Length (bp) | Product Identity (%) 2 |
|---|---|---|---|---|---|
| Ifit2 | Interferon induced protein with tetratricopeptide repeats 2 | XM_002698356.5 | F: CAGATGTGATTCGAGGGGCA R: CATGGAGGCAGGCGAGATAG | 282 | 100 |
| Ifit3 * | Interferon induced protein with tetratricopeptide repeats 3 | NM_001075414.1 | F: ATTCTGAAGCAGGCCGTTGA R: TCCAGTGCCCTTAGCAACAG | 224 | 100 |
| Irf1 * | Interferon regulatory factor 1 | NM_001191261.2 | F: ACAGCCCCGATACCTTCTCT R: CTTCCCATCCACGCTTGTCT | 338 | 100 |
| Irf9 | Interferon regulatory factor 9 | NM_001024506.1 | F: GCGCTGTGCTCTCAACAAAA R: AAGTCTAAACGGCCAGCTCC | 285 | 100 |
| Isg15 * | ISG15 ubiquitin like modifier | NM_174366.1 | F: CCATCCTGGTGAGGAACGAC R: GAACACGGTGCACCCCTTCA | 200 | 99 |
| Isg20 | Interferon stimulated exonuclease gene 20 | XM_002696514.5 | F: CTTGTGGACTACCACGGCTC R: GATGGCGTAGTCGCTCATGT | 234 | 98 |
| Oas2 * | 2′-5′-oligoadenylate synthetase 2 | NM_001024557.1 | F: ACTGGTTTCAAAAGTGCCAGG R: CAGCCAGCAGGTGTTATCCA | 314 | 98 |
| Rsad2 * | Radical S-adenosyl methionine domain containing 2 | NM_001045941.1 | F: GTGGTTCCAGAAGTACGGTGA R: AACCGTTCCGCTTCTCTCAG | 315 | 100 |
| Scara5 | Scavenger receptor class A member 5 | NM_001102499.1 | F: AGGACCTACGCCTCAAGGAT R: GGGCCTCGATCACCTTTGAA | 256 | 100 |
| Scarb1 | Scavenger receptor class B member 1 | NM_174597.2 | F: GCAGACATGGGCAACCTCT R: GCCTTGGATGATCCCCTCAG | 249 | 100 |
| Timp2 ⱡ | TIMP metallopeptidase inhibitor 2 | NM_174472.4 | F: GGGTCTCGCTGGACATTG R: TTGATGTTCTTCTCCGTGACC | 256 | 100 |
| Timp3 | Bos taurus TIMP metallopeptidase inhibitor 3 (TIMP3), mRNA | NM_174473.4 | F: GGATTCACCAAGATGCCCCA R: GCAGTTACAGCCCAGGTGAT | 222 | 99 |
| Trim56 | Tripartite motif containing 56 | NM_001206574.1 | F: TTCAGACCCCAAATCAGGAC R: TCTGGGCTCTGCTCTCTTTC | 126 | 99 |
| Housekeeping Transcripts | |||||
| Gapdh | Glyceraldehyde 3-phosphate dehydrogenase | NM_001034034.2 | F: ACATCAAGTGGGGTGATGCT R: GGCATTGCTGACAATCTTGA | 200 | 99 |
| Hprt1 | Hypoxanthine phosphoribosyltransferase 1 | NM_001034035.2 | F: GCCAGCCGGCTACGTTAT R: ATCCAACAGGTCGGCAAAGA | 256 | 100 |
| Rps11 | Ribosomal protein S11 | NM_001024568.2 | F: AAGATGGCGGACATTCAGAC R: GCCCTCGAATGGAGACATTA | 214 | 99 |
| Sdha | Succinate dehydrogenase complex flavoprotein subunit A | NM_174178.2 | F: GCAGAACCTGATGCTTTGTG R: CGTAGGAGAGCGTGTGCTT | 185 | 99 |
References
- Franke, K.W. A new toxicant occurring naturally in certain samples of plant foodstuffs. J. Nutr. 1934, 8, 597–608. [Google Scholar] [CrossRef]
- Schwarz, K.; Foltz, C.M. Selenium as an integral part of factor 3 against dietary necrotic liver degeneration. J. Am. Chem. Soc. 1957, 79, 3292–3293. [Google Scholar] [CrossRef]
- McCoy, K.E.; Weswig, P.H. Some selenium responses in the rat not related to vitamin E. J. Nutr. 1969, 98, 383–389. [Google Scholar] [CrossRef]
- Thompson, J.N.; Scott, M.L. Role of selenium in the nutrition of the chick. J. Nutr. 1969, 97, 335–342. [Google Scholar] [CrossRef]
- Hatfield, D.L.; Tsuji, P.A.; Carlson, B.A.; Gladyshev, V.N. Selenium and selenocysteine: Roles in cancer, health, and development. Trends Biochem. Sci. 2014, 39, 112–120. [Google Scholar] [CrossRef]
- Shini, S.; Sultan, A.; Bryden, W.L. Selenium biochemistry and bioavailability: Implications for animal agriculture. Agriculture 2015, 54, 1277–1288. [Google Scholar] [CrossRef]
- Flohe, L.; Gunzler, W.A.; Schock, H.H. Glutathione peroxidase: A selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef]
- Chen, X.D.; Zhao, Z.P.; Zhao, J.C.; Lei, X.G. Evolution, regulation, and function of porcine selenogenome. Free Radic. Biol. Med. 2018, 127, 116–123. [Google Scholar] [CrossRef]
- Carr, S.N.; Crites, B.R.; Pate, J.L.; Hughes, C.H.K.; Matthews, J.C.; Bridges, P.J. Form of supplemental selenium affects the expression of mRNA transcripts encoding selenoproteins, and proteins regulating cholesterol uptake, in the corpus luteum of grazing beef cows. Animals 2022, 12, 313. [Google Scholar] [CrossRef]
- Li, Q.; Chen, K.C.; Bridges, P.J.; Matthews, J.C. Pituitary and liver selenoprotein transcriptome profiles of grazing steers and their sensitivity to the form of selenium in vitamin-mineral mixes. Front. Anim. Sci. 2022, 3, 911094. [Google Scholar] [CrossRef]
- Novoselov, S.V.; Kryukov, G.V.; Xu, X.M.; Carlson, B.A.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein H is a nucleolar thioredoxin-like protein with a unique expression pattern. J. Biol. Chem. 2007, 282, 11960–11968. [Google Scholar] [CrossRef]
- Lu, C.; Qiu, F.; Zhou, H.; Peng, Y.; Hao, W.; Xu, J.; Yuan, J.; Wang, S.; Qiang, B.; Xu, C.; et al. Identification and characterization of selenoprotein K: An antioxidant in cardiomyocytes. FEBS Lett. 2006, 580, 5189–5197. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.A.; Bellinger, F.P.; Berry, M.J. The neuroprotective functions of selenoprotein M and its role in cytosolic calcium regulation. Antioxid. Redox Signal. 2010, 12, 809–818. [Google Scholar] [CrossRef]
- Takebe, G.; Yarimizu, J.; Saito, Y.; Hayashi, T.; Nakamura, H.; Yodoi, J.; Nagasawa, S.; Takahashi, K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J. Biol. Chem. 2002, 277, 41254–41258. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Kumar, R.A.; Koc, A.; Sun, Z.; Gladyshev, V.N. Selenoprotein R is a zinc-containing stereo-specific methionine sulfoxide reductase. Proc. Natl. Acad. Sci. USA 2002, 99, 4245–4250. [Google Scholar] [CrossRef] [PubMed]
- Beilstein, M.A.; Vendeland, S.C.; Barofsky, E.; Jensen, O.N.; Whanger, P.D. Selenoprotein W of rat muscle binds glutathione and an unknown small molecular weight moiety. J. Inorg. Biochem. 1996, 61, 117–124. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Ammerman, C.B.; Miller, S.M. Selenium in ruminant nutrition: A review. J. Dairy. Sci. 1975, 58, 1561–1577. [Google Scholar] [CrossRef]
- NASEM. Nutrient Requirements of Beef Cattle: Eighth Revised Edition; The National Academies Press: Washington, DC, USA, 2016. [Google Scholar]
- Boyne, R.; Arthur, J.R. Alterations of neutrophil function in selenium-deficient cattle. J. Comp. Pathol. 1979, 89, 151–158. [Google Scholar] [CrossRef]
- Gleed, P.T.; Allen, W.M.; Mallinson, C.B.; Rowlands, G.J.; Sansom, B.F.; Vagg, M.J.; Caswell, R.D. Effects of selenium and copper supplementation on the growth of beef steers. Vet. Rec. 1983, 113, 388–392. [Google Scholar] [CrossRef] [PubMed]
- McClure, T.J.; Eamens, G.J.; Healy, P.J. Improved fertility in dairy cows after treatment with selenium pellets. Aust. Vet. J. 1986, 63, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Erskine, R.J.; Eberhart, R.J.; Grasso, P.J.; Scholz, R.W. Induction of Escherichia coli mastitis in cows fed selenium-deficient or selenium-supplemented diets. Am. J. Vet. Res. 1989, 50, 2093–2100. [Google Scholar]
- Enjalbert, F.; Lebreton, P.; Salat, O. Effects of copper, zinc and selenium status on performance and health in commercial dairy and beef herds: Retrospective study. J. Anim. Physiol. Anim. Nutr. 2006, 90, 459–466. [Google Scholar] [CrossRef]
- Arthur, J.R.; McKenzie, R.C.; Beckett, G.J. Selenium in the immune system. J. Nutr. 2003, 133, 1457S–1459S. [Google Scholar] [CrossRef] [PubMed]
- Sgoifo Rossi, C.A.; Compiani, R.; Baldi, G.; Muraro, M.; Marden, J.P.; Rossi, R.; Pastorelli, G.; Corino, C.; Dell’Orto, V. Organic selenium supplementation improves growth parameters, immune and antioxidant status of newly received beef cattle. J. Anim. Feed Sci. 2017, 26, 100–108. [Google Scholar] [CrossRef]
- Carr, S.N.; Jia, Y.; Crites, B.R.; Hamilton, C.H.; Burris, W.R.; Edwards, J.L.; Matthews, J.C.; Bridges, P.J. Form of supplemental selenium in vitamin-mineral premixes differentially affects early luteal and gestational concentrations of progesterone, and postpartum concentrations of prolactin in beef cows. Animals 2020, 10, 967. [Google Scholar] [CrossRef]
- Cerny, K.L.; Anderson, L.; Burris, W.R.; Rhoads, M.; Matthews, J.C.; Bridges, P.J. Form of supplemental selenium fed to cycling cows affects systemic concentrations of progesterone but not those of estradiol. Theriogenology 2016, 85, 800–806. [Google Scholar] [CrossRef]
- Spencer, T.E.; Bazer, F.W. Biology of progesterone action during pregnancy recognition and maintenance of pregnancy. Front. Biosci. 2002, 7, d1879–d1898. [Google Scholar] [CrossRef]
- Carter, F.; Forde, N.; Duffy, P.; Wade, M.; Fair, T.; Crowe, M.A.; Evans, A.C.O.; Kenny, D.A.; Roche, J.F.; Lonergan, P. Effect of increasing progesterone concentration from Day 3 of pregnancy on subsequent embryo survival and development in beef heifers. Reprod. Fertil. Dev. 2008, 20, 368–375. [Google Scholar] [CrossRef]
- Garrett, J.E.; Geisert, R.D.; Zavy, M.T.; Morgan, G.L. Evidence for maternal regulation of early conceptus growth and development in beef cattle. J. Reprod. Fertil. 1988, 84, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.E.; Lamming, G.E. Relationship between maternal endocrine environment, early embryo development and inhibition of the luteolytic mechanism in cows. Reprod 2001, 121, 175–180. [Google Scholar] [CrossRef]
- Crites, B.R.; Carr, S.N.; Anderson, L.H.; Matthews, J.C.; Bridges, P.J. Form of dietary selenium affects mRNA encoding interferon-stimulated and progesterone-induced genes in the bovine endometrium and conceptus length at maternal recognition of pregnancy. J. Anim. Sci. 2022, 100, skac137. [Google Scholar] [CrossRef] [PubMed]
- Mansouri-Attia, N.; Aubert, J.; Reinaud, P.; Giraud-Delville, C.; Taghouti, G.; Galio, L.; Everts, R.E.; Degrelle, S.; Richard, C.; Hue, I.; et al. Gene expression profiles of bovine caruncular and intercaruncular endometrium at implantation. Physiol. Genom. 2009, 39, 14–27. [Google Scholar] [CrossRef]
- Haeger, J.D.; Hambruch, N.; Pfarrer, C. The bovine placenta in vivo and in vitro. Theriogenology 2016, 86, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Ott, T.L. Symposium review: Immunological detection of the bovine conceptus during early pregnancy. J. Dairy. Sci. 2019, 102, 3766–3777. [Google Scholar] [CrossRef]
- Walker, C.G.; Meier, S.; Littlejohn, M.D.; Lehnert, K.; Roche, J.R.; Mitchell, M.D. Modulation of the maternal immune system by the pre-implantation embryo. BMC Genom. 2010, 11, 474. [Google Scholar] [CrossRef]
- Khanam, A.; Platel, K. Bioaccessibility of selenium, selenomethionine and selenocysteine from foods and influence of heat processing on the same. Food Chem. 2016, 194, 1293–1299. [Google Scholar] [CrossRef]
- Daniels, L.A. Selenium metabolism and bioavailability. Biol. Trace Elem. Res. 1996, 54, 185–199. [Google Scholar] [CrossRef]
- Matthews, J.C.; Zhang, Z.; Patterson, J.D.; Bridges, P.J.; Stromberg, A.J.; Boling, J.A. Hepatic transcriptome profiles differ among maturing beef heifers supplemented with inorganic, organic, or mixed (50% inorganic:50% organic) forms of dietary selenium. Biol. Trace Elem. Res. 2014, 160, 321–339. [Google Scholar] [CrossRef]
- Cerny, K.; Garbacik, S.; Skees, C.; Burris, W.; Matthews, J.; Bridges, P. Gestational form of selenium in free-choice mineral mixes affects transcriptome profiles of the neonatal calf testis, including those of steroidogenic and spermatogenic pathways. Biol. Trace Elem. Res. 2016, 169, 56–68. [Google Scholar] [CrossRef]
- Crites, B.R.; Carr, S.N.; Matthews, J.C.; Bridges, P.J. Form of dietary selenium affects mRNA encoding cholesterol biosynthesis and immune response elements in the early luteal phase bovine corpus luteum. J. Anim. Sci. 2022, 100, skac135. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef]
- Silva, J.F.; Ocarino, N.M.; Serakides, R. Thyroid hormones and female reproduction. Biol. Reprod. 2018, 99, 907–921. [Google Scholar] [CrossRef]
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [CrossRef]
- Kirovski, D.; Dodovski, P.; Savić, Đ.; Vujanac, I.; Prodanović, R.; Mirilović, M.; Sladojević, Ž.; Đorđević, A. Placental iodothyronine deiodinases expression in pregnant cows exposed to propylthiouracil (PTU) and thyroid axis activity of their calves. Act. Vet. 2016, 66, 61–75. [Google Scholar] [CrossRef]
- Awadeh, F.T.; Kincaid, R.L.; Johnson, K.A. Effect of level and source of dietary selenium on concentrations of thyroid hormones and immunoglobulins in beef cows and calves. J. Anim. Sci. 1998, 76, 1204–1215. [Google Scholar] [CrossRef]
- Gereben, B.; Zeöld, A.; Dentice, M.; Salvatore, D.; Bianco, A.C. Activation and inactivation of thyroid hormone by deiodinases: Local action with general consequences. Cell Mol. Life Sci. 2008, 65, 570–590. [Google Scholar] [CrossRef]
- Forhead, A.J.; Fowden, A.L. Thyroid hormones in fetal growth and prepartum maturation. J. Endocrinol. 2014, 221, R87–R103. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, J.S.; Murray, R.W.; Foley, R.C. Observations on the morphogenesis and histochemistry of the bovine preattachment placenta between 16 and 33 days of gestation. Anat. Rec. 1958, 132, 321–341. [Google Scholar] [CrossRef]
- Assis Neto, A.C.; Pereira, F.T.; Santos, T.C.; Ambrosio, C.E.; Leiser, R.; Miglino, M.A. Morpho-physical recording of bovine conceptus (Bos indicus) and placenta from days 20 to 70 of pregnancy. Reprod. Domest. Anim. 2010, 45, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.E.; Sandra, O.; Wolf, E. Genes involved in conceptus-endometrial interactions in ruminants: Insights from reductionism and thoughts on holistic approaches. Reproduction 2008, 135, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.E.; Johnson, G.A.; Burghardt, R.C.; Bazer, F.W. Progesterone and placental hormone actions on the uterus: Insights from domestic animals. Biol. Reprod. 2004, 71, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Bazer, F.W.; Spencer, T.E.; Johnson, G.A. Interferons and uterine receptivity. Semin. Reprod. Med. 2009, 27, 90–102. [Google Scholar] [CrossRef]
- Bazer, F.W.; Burghardt, R.C.; Johnson, G.A.; Spencer, T.E.; Wu, G. Interferons and progesterone for establishment and maintenance of pregnancy: Interactions among novel cell signaling pathways. Reprod. Biol. 2008, 8, 179–211. [Google Scholar] [CrossRef] [PubMed]
- Forde, N.; Carter, F.; Spencer, T.; Bazer, F.; Sandra, O.; Mansouri-Attia, N.; Okumu, L.; McGettigan, P.; Mehta, J.; McBride, R. Conceptus-induced changes in the endometrial transcriptome: How soon does the cow know she is pregnant? Biol. Reprod. 2011, 85, 144–156. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Shirozu, T.; Sasaki, K.; Kawahara, M.; Yanagawa, Y.; Nagano, M.; Yamauchi, N.; Takahashi, M. Expression dynamics of bovine MX genes in the endometrium and placenta during early to mid pregnancy. J. Reprod. Dev. 2016, 62, 29–35. [Google Scholar] [CrossRef]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef]
- Lerner, M.R.; Boyle, J.A.; Mount, S.M.; Wolin, S.L.; Steitz, J.A. Are snRNPs involved in splicing? Nature 1980, 283, 220–224. [Google Scholar] [CrossRef]
- von Rango, U. Fetal tolerance in human pregnancy--a crucial balance between acceptance and limitation of trophoblast invasion. Immunol. Lett. 2008, 115, 21–32. [Google Scholar] [CrossRef]
- Groebner, A.E.; Schulke, K.; Schefold, J.C.; Fusch, G.; Sinowatz, F.; Reichenbach, H.D.; Wolf, E.; Meyer, H.H.; Ulbrich, S.E. Immunological mechanisms to establish embryo tolerance in early bovine pregnancy. Reprod. Fertil. Dev. 2011, 23, 619–632. [Google Scholar] [CrossRef]
- Kamat, M.M.; Vasudevan, S.; Maalouf, S.A.; Townson, D.H.; Pate, J.L.; Ott, T.L. Changes in myeloid lineage cells in the uterus and peripheral blood of dairy heifers during early pregnancy. Biol. Reprod. 2016, 95, 68. [Google Scholar] [CrossRef] [PubMed]
- Vacca, P.; Cantoni, C.; Vitale, M.; Prato, C.; Canegallo, F.; Fenoglio, D.; Ragni, N.; Moretta, L.; Mingari, M.C. Crosstalk between decidual NK and CD14+ myelomonocytic cells results in induction of Tregs and immunosuppression. Proc. Natl. Acad. Sci. USA 2010, 107, 11918–11923. [Google Scholar] [CrossRef]
- Gerloff, B.J. Effect of selenium supplementation on dairy cattle. J. Anim. Sci. 1992, 70, 3934–3940. [Google Scholar] [CrossRef] [PubMed]
- Dargatz, D.A.; Ross, P.F. Blood selenium concentrations in cows and heifers on 253 cow-calf operations in 18 states. J. Anim. Sci. 1996, 74, 2891–2895. [Google Scholar] [CrossRef]
- Wahlen, R.; Evans, L.; Turner, J.; Hearn, R. The use of collision/reaction cell ICP-MS for the determination of elements in blood and serum samples. Spectroscopy 2005, 20, 84–89. [Google Scholar]
- Bogdanova, E.A.; Barsova, E.V.; Shagina, I.A.; Scheglov, A.; Anisimova, V.; Vagner, L.L.; Lukyanov, S.A.; Shagin, D.A. Normalization of full-length-enriched cDNA. Methods Mol. Biol. 2011, 729, 85–98. [Google Scholar] [CrossRef]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alnebert, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed]
- Sayols, S.; Scherzinger, D.; Klein, H. dupRadar: A Bioconductor package for the assessment of PCR artifacts in RNA-Seq data. BMC Bioinform. 2016, 17, 428. [Google Scholar] [CrossRef] [PubMed]
- Daley, T.; Smith, A.D. Predicting the molecular complexity of sequencing libraries. Nat. Methods 2013, 10, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Rockett, J.C.; Hellmann, G.M. Confirming microarray data—Is it really necessary? Genomics 2004, 83, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ulbrich, S.E.; Meyer, S.U.; Zitta, K.; Hiendleder, S.; Sinowatz, F.; Bauersachs, S.; Büttner, M.; Fröhlich, T.; Arnold, G.J.; Reichenbach, H.D.; et al. Bovine endometrial metallopeptidases MMP14 and MMP2 and the metallopeptidase inhibitor TIMP2 participate in maternal preparation of pregnancy. Mol. Cell. Endocrinol. 2011, 332, 48–57. [Google Scholar] [CrossRef] [PubMed]

| Gene 2 | Gene Name | qPCR 3 | |||
|---|---|---|---|---|---|
| ISe | MIX | SEM | p-Value 4 | ||
| Iodothyronine deiodinases | |||||
| Dio1 | Iodothyronine deiodinase 1 | Unable to be detected | |||
| Dio2 | Iodothyronine deiodinase 2 | 1.15 | 0.53 | 0.25 | <0.05 |
| Dio3 | Iodothyronine deiodinase 3 | 1.03 | 0.73 | 0.10 | <0.05 |
| Glutathione peroxidases | |||||
| Gpx1 | Glutathione peroxidase 1 | 1.01 | 0.90 | 0.09 | 0.40 |
| Gpx2 | Glutathione peroxidase 2 | 1.41 | 0.91 | 0.36 | 0.68 |
| Gpx3 | Glutathione peroxidase 3 | 1.14 | 1.26 | 0.16 | 0.66 |
| Gpx4 | Glutathione peroxidase 4 | 1.05 | 1.15 | 0.14 | 0.66 |
| Gpx6 | Glutathione peroxidase 6 | 1.11 | 0.96 | 0.24 | 0.67 |
| Thioredoxin reductases | |||||
| Txnrd1 | Thioredoxin reductase 1 | 1.08 | 1.00 | 0.15 | 0.69 |
| Txnrd2 | Thioredoxin reductase 2 | 1.04 | 0.99 | 0.11 | 0.75 |
| Txnrd3 | Thioredoxin reductase 3 | 1.02 | 0.95 | 0.09 | 0.58 |
| Other selenoproteins | |||||
| Selenof | Selenoprotein F | 1.05 | 1.15 | 0.15 | 0.66 |
| Selenoh | Selenoprotein H | 1.06 | 0.79 | 0.12 | 0.18 |
| Selenoi | Selenoprotein I | 1.01 | 1.02 | 0.07 | 0.97 |
| Selenok | Selenoprotein K | 1.03 | 1.12 | 0.12 | 0.61 |
| Selenom | Selenoprotein M | 6.59 | 5.36 | 2.90 | 0.77 |
| Selenon | Selenoprotein N | 1.01 | 0.96 | 0.07 | 0.56 |
| Selenoo | Selenoprotein O | 1.05 | 1.12 | 1.10 | 0.63 |
| Selenop | Selenoprotein P | 1.04 | 1.09 | 1.10 | 0.71 |
| Selenor | Selenoprotein R | 1.11 | 1.11 | 0.18 | 0.68 |
| Selenos | Selenoprotein S | 1.07 | 1.11 | 0.14 | 0.85 |
| Selenot | Selenoprotein T | 1.01 | 1.03 | 0.06 | 0.84 |
| Selenov | Selenoprotein V | 1.10 | 1.14 | 0.21 | 0.90 |
| Selenow | Selenoprotein W | 1.02 | 0.93 | 0.09 | 0.47 |
| Selenophosphate synthetase | |||||
| Sephs2 | Selenophosphate synthetase 2 | 1.02 | 1.15 | 0.08 | 0.25 |
| Selenoprotein P receptors | |||||
| Lrp2 | LDL receptor related protein 2 | 1.02 | 0.78 | 0.11 | 0.17 |
| Lrp8 | LDL receptor related protein 8 | 1.05 | 0.85 | 0.11 | 0.26 |
| Tfrc | Transferrin receptor | 1.10 | 0.93 | 0.17 | 0.62 |
| Gene ID | Gene Description | Fold Change | p-Value 2 |
|---|---|---|---|
| Upregulated in MIX | |||
| CCDC152 | Coiled-coil domain containing 152 | 4.03 | 0.0111 |
| CD79B | CD79b molecule | 3.75 | 0.0329 |
| TM4SF5 | Transmembrane 4 L six family member 5 | 3.39 | 0.0366 |
| C22orf31 | Chromosome 22 open reading frame 31 | 3.38 | 0.0152 |
| SLC17A7 | Solute carrier family 17 member 7 | 3.34 | 0.0338 |
| CCL21 | C-C motif chemokine ligand 21 | 2.93 | 0.0359 |
| FAM180B | Family with sequence similarity 180 member B | 2.88 | 0.0103 |
| COL17A1 | Collagen type XVII alpha 1 chain | 2.35 | 0.0019 |
| CCR9 | C–C motif chemokine receptor 9 | 2.34 | 0.0184 |
| OMG | Oligodendrocyte myelin glycoprotein | 2.33 | 0.0429 |
| Downregulated in MIX | |||
| DPYSL4 | Dihydropyrimidinase-like 4 | −4.07 | 0.0108 |
| KLK5 | Kallikrein-related peptidase 5 | −4.03 | 0.0004 |
| GRM3 | Glutamate metabotropic receptor 3 | −3.70 | 0.0004 |
| NIPAL4 | NIPA-like domain containing 4 | −3.59 | 0.0020 |
| NALCN | Sodium leak channel, nonselective | −3.26 | 0.0134 |
| PTGER1 | Prostaglandin E receptor 1 | −3.16 | 0.0178 |
| COL28A1 | Collagen type XXVIII alpha 1 chain | −3.12 | 0.0105 |
| SLC26A8 | Solute carrier family 26 member 8 | −3.12 | 0.0026 |
| SLC34A3 | Solute carrier family 34 member 3 | −3.11 | 0.0237 |
| DMKN | Dermokine | −2.93 | <0.0001 |
| Canonical Pathway 2 | Gene Symbols | Ratio 3 | Z-Score | p-Value 4 |
|---|---|---|---|---|
| ATM Signaling | Up: ATR, CBX1, CBX3, CBX5, CCNB2, CDK2, HP1BP3, MDC1, PPP2R1B, RAD50, RAD51, RBBP8, RNF8, SMC2, SMC3, SMC1A, ZNF420 Down: BID, BRAT1, MAPK12, MAPK13, PPM1L, PPP2R1A, TP73, TRRAP | 0.25 (25/100) | 0.471 | <0.0001 |
| Spliceosomal Cycle | UP: BCAS2, CDC5L, CTNNBL1, CWC15, DDX23, DHX38, ISY1-RAB43, MAGOHB, RBM8A, SLU7, SNRNP200, ZNF830 Down: PRPF19, SF3B3 | 0.29 (14/49) | 2.673 | <0.0001 |
| Role of PKR in Interferon Induction and Antiviral Response | Up: APAF1, DNAJC3, HMGB1, HSP90AA1, HSP90AB1, HSP90B1, HSPA4, METAP2, MSR1, REL Down: ATF3, BID, FADD, FOS, HSPA6, IRF1, IRF9, MAPK12, MAPK13, PDGFRB, PYCARD, RELA, STAT2, STAT3, TARBP2, TNFRSF1A | 0.19 (26/136) | −3.128 | <0.0001 |
| CLEAR Signaling Pathway | Up: ATP6V1C1, ATP6V1E1, BECN1, BMPR1B, HPS5, ITPR2, MAP4K3, NRBF2, PPP2R1B, RRAGB, VPS26A, YWHAE Down: ATP6V0A1, ATP6V0D1, BLOC1S3, BMP6, CRTC2, CTNS, CTSA, FLT1, GBA1, GLB1, KCNIP3, MAPK7, MAPK12, MAPK13, MLST8, PDGFRB, PML, PPM1L, PPP2R1A, PRKAG1, PRKCB, RAB7A, RPTOR, SEC13, SESN2, TCIRG1, TGFA, TGFBR3, TNFRSF1A, TRPM1, TSC2 | 0.15 (43/285) | −0.762 | <0.0001 |
| Autophagy | Up: ATG3, ATG10, ATR, BECN1, CALM1, CDKN1B, GNAI3, NRBF2, PIK3C2A, PPP2R1B, RB1CC1, SESN1, STX17, VPS41 Down: ATG2A, BMP6, FOS, IRS1, IRS2, MAPK12, MLST8, PI4K2A, PPM1L, PPP2R1A, PRKAG1, RAB7A, RAB7B, RIPK1, RPTOR, SLC7A5, TGFA, TNFRSF1A, TSC2, ULK1, WIPI2 | 0.16 35/216 | 0.845 | <0.0001 |
| Molecular and Cellular Functions 2 | Z-Score 3 | p-Value 4 |
|---|---|---|
| Cell death and survival (732 molecules) | ||
| Necrosis | −1.392 | <0.0001 |
| Apoptosis | −2.252 | <0.0001 |
| Cell death of tumor cell lines | −1.757 | <0.0001 |
| Cell survival | 1.029 | <0.0001 |
| Apoptosis of tumor cell lines | −1.425 | <0.0001 |
| Cell viability | 1.112 | <0.0001 |
| Cell viability of tumor cell lines | 1.278 | <0.0001 |
| Necrosis of epithelial tissue | −1.682 | <0.0001 |
| Cell death of osteosarcoma cells | −1.671 | <0.0001 |
| Cell death of bone cancer cell lines | −0.434 | <0.0001 |
| Cell death of blood cells | −1.469 | <0.0001 |
| Cell death of sarcoma cell lines | −0.943 | <0.0001 |
| Colony survival of tumor cell lines | 1.183 | <0.0001 |
| Necrosis of tumor | −1.881 | <0.0001 |
| Cell death of breast cancer cell lines | −2.267 | <0.0001 |
| Apoptosis of peritoneal macrophages | −0.574 | <0.0001 |
| Cellular response to therapeutics (149 molecules) | ||
| Radiosensitivity of cells | −1.38 | <0.0001 |
| Gene expression (462 molecules) | ||
| Expression of RNA | −3.621 | <0.0001 |
| Transcription of RNA | −2.971 | <0.0001 |
| Transcription | −2.877 | <0.0001 |
| Transcription of DNA | −2.485 | <0.0001 |
| Activation of DNA endogenous promoter | −2.373 | <0.0001 |
| Transactivation | 1.115 | <0.0001 |
| Transactivation of RNA | 0.804 | <0.0001 |
| Repression of RNA | 0.646 | <0.0001 |
| RNA post-translational modification (92 molecules) | ||
| Processing of RNA | −1.066 | <0.0001 |
| Processing of mRNA | −1.635 | <0.0001 |
| Splicing of RNA | −1.339 | <0.0001 |
| Splicing of mRNA | −1.682 | <0.0001 |
| Cellular assembly and organization (505 molecules) | ||
| Development of cytoplasm | −1.738 | <0.0001 |
| Cohesion of sister chromatids | 0.854 | <0.0001 |
| Formation of centriole | 0.921 | <0.0001 |
| Remodeling of chromatin | 0.900 | <0.0001 |
| Formation of cellular protrusions | −0.647 | <0.0001 |
| Replication of centriole | 0.921 | <0.0001 |
| RNA-Seq 3 | qPCR 3 | |||||||
|---|---|---|---|---|---|---|---|---|
| Gene 2 | Gene Name | ISe | MIX | p-Value | ISe | MIX | SEM | p-Value 4 |
| Ido1 | Indoleamine 2,3-dioxygenase 1 | 1.00 | 0.68 | 0.02 | Not corroborated | |||
| Ifit1 | Interferon-induced protein with tetratricopeptide repeats 1 | 1.00 | 0.83 | 0.04 | Not corroborated | |||
| Ifit2 | Interferon induced protein with tetratricopeptide repeats 2 | 1.00 | 0.71 | 0.01 | 1.11 | 0.54 | 0.17 | 0.04 |
| Ifit3 * | Interferon induced protein with tetratricopeptide repeats 3 | 1.00 | 0.78 | 0.02 | 1.08 | 0.65 | 0.13 | 0.04 |
| Irf1 * | Interferon regulatory factor 1 | 1.00 | 0.79 | 0.02 | 1.05 | 0.75 | 0.11 | 0.08 |
| Irf9 | Interferon regulatory factor 9 | 1.00 | 0.83 | <0.01 | 1.01 | 0.95 | 0.06 | 0.47 |
| Isg15 * | ISG15 ubiquitin like modifier | 1.00 | 0.78 | <0.01 | 1.04 | 0.76 | 0.09 | 0.05 |
| Isg20 | Interferon stimulated exonuclease, transcript variant 1 | 1.00 | 0.74 | <0.01 | 1.06 | 0.76 | 0.12 | 0.11 |
| Oas2 * | 2′-5′-oligoadenylate synthetase 2 | 1.00 | 0.81 | 0.01 | 1.03 | 0.67 | 0.10 | 0.01 |
| Rsad1 | Radical S-adenosyl methionine domain containing 1 | 1.00 | 0.83 | <0.01 | Not corroborated | |||
| Rsad2 * | Radical S-adenosyl methionine domain containing 2 | 1.00 | 0.75 | <0.01 | 1.05 | 0.57 | 0.14 | 0.01 |
| Scara5 | Scavenger receptor class A member 5 | 1.00 | 0.87 | 0.09 | 1.04 | 0.71 | 0.09 | 0.02 |
| Scarb1 | Scavenger receptor class B member 1 | 1.00 | 0.83 | 0.01 | 1.04 | 0.81 | 0.09 | 0.12 |
| Stat1 | Signal transducer and activator of transcription 1 | 1.00 | 0.92 | 0.16 | Not corroborated | |||
| Stat2 | Signal transducer and activator of transcription 2 | 1.00 | 0.84 | 0.02 | Not corroborated | |||
| Tgfb1 | Transforming growth factor beta 1 | 1.00 | 0.87 | 0.06 | Not corroborated | |||
| Timp2 | TIMP metallopeptidase inhibitor 2 | 1.00 | 0.86 | 0.26 | 1.02 | 0.93 | 0.08 | 0.48 |
| Timp3 | TIMP metallopeptidase inhibitor 3 | 1.00 | 0.83 | <0.01 | 1.02 | 0.79 | 0.08 | 0.06 |
| Trim56 | Tripartite motif containing 56 | 1.00 | 0.83 | 0.02 | 1.04 | 0.55 | 0.09 | <0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carr, S.N.; Crites, B.R.; Shinde, H.; Bridges, P.J. Transcriptomic Changes in Response to Form of Selenium on the Interferon-Tau Signaling Mechanism in the Caruncular Tissue of Beef Heifers at Maternal Recognition of Pregnancy. Int. J. Mol. Sci. 2023, 24, 17327. https://doi.org/10.3390/ijms242417327
Carr SN, Crites BR, Shinde H, Bridges PJ. Transcriptomic Changes in Response to Form of Selenium on the Interferon-Tau Signaling Mechanism in the Caruncular Tissue of Beef Heifers at Maternal Recognition of Pregnancy. International Journal of Molecular Sciences. 2023; 24(24):17327. https://doi.org/10.3390/ijms242417327
Chicago/Turabian StyleCarr, Sarah N., Benjamin R. Crites, Harshraj Shinde, and Phillip J. Bridges. 2023. "Transcriptomic Changes in Response to Form of Selenium on the Interferon-Tau Signaling Mechanism in the Caruncular Tissue of Beef Heifers at Maternal Recognition of Pregnancy" International Journal of Molecular Sciences 24, no. 24: 17327. https://doi.org/10.3390/ijms242417327
APA StyleCarr, S. N., Crites, B. R., Shinde, H., & Bridges, P. J. (2023). Transcriptomic Changes in Response to Form of Selenium on the Interferon-Tau Signaling Mechanism in the Caruncular Tissue of Beef Heifers at Maternal Recognition of Pregnancy. International Journal of Molecular Sciences, 24(24), 17327. https://doi.org/10.3390/ijms242417327