
Comparative Analysis of Transcriptomics and Metabolomics Reveals Defense Mechanisms in Melon Cultivars against Pseudoperonospora cubensis Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Response of Melon to P. cubensis Inoculation
2.2. Transcriptomic Variability in Melon Resistance and Susceptibility to P. cubensis
2.3. Basal Metabolite Variations between P. cubensis-Resistant and Susceptible Melon Cultivars
2.4. Differential Transcriptional Response in the Two Cultivars Following P. cubensis Inoculation
2.5. Differential Metabolic Response in the Two Cultivars after P. cubensis Inoculation
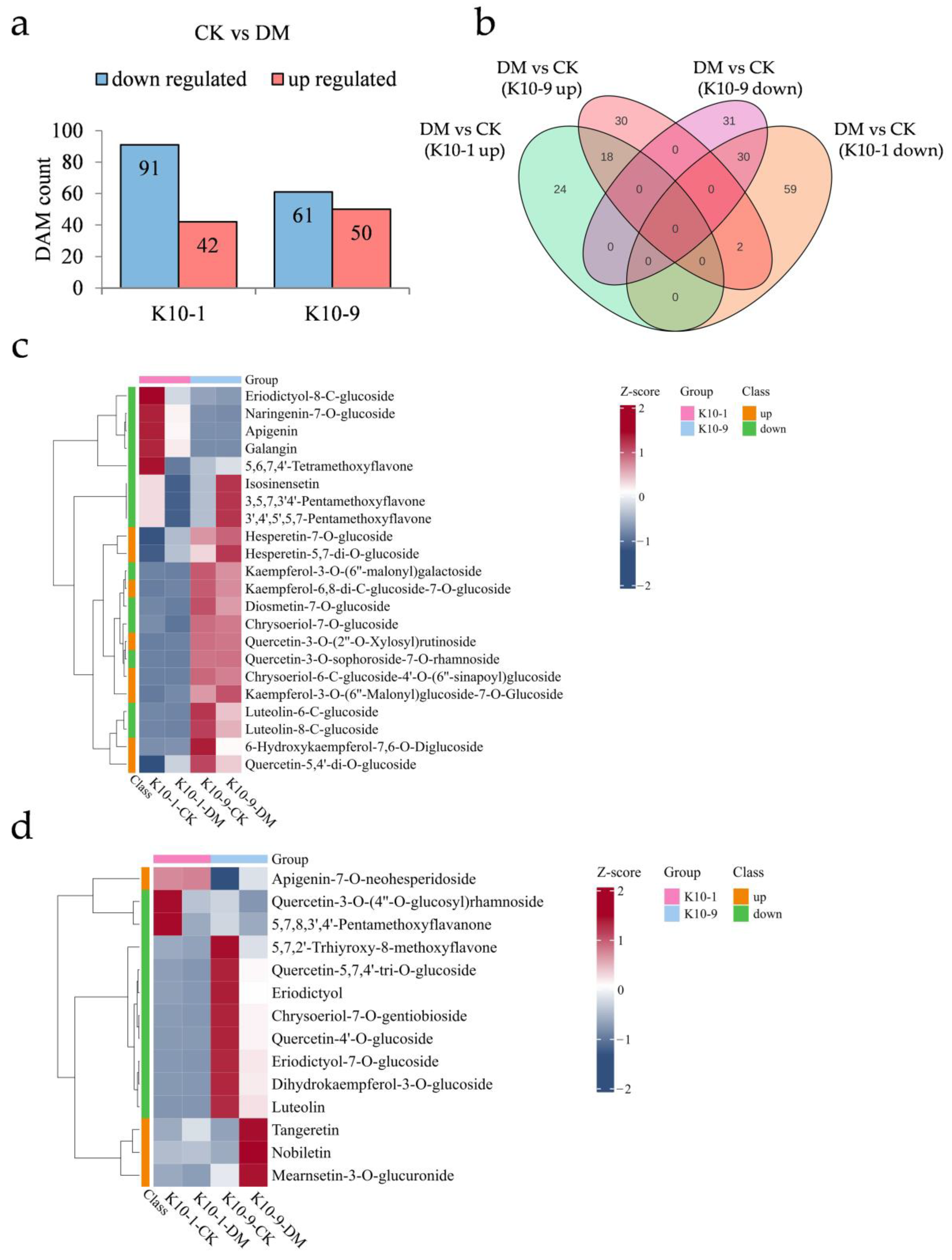
2.6. Flavonoids’ Contribution to Melon’s Resistance against DM
2.7. Activation of Lignin Biosynthesis Pathways in Both ‘K10-1′ and ‘K10-9′ Cultivars Post-P. cubensis Infection
3. Discussion
3.1. Different Basal Transcriptomes in Different Melon-Resistant Cultivars
3.2. Response of Melon to DM Infection: Inhibiting Flavonoid Biosynthesis and Augmentation of Lignin Synthesis
4. Materials and Methods
4.1. Plant Material and Treatment
4.2. RNA Sample Preparation and Transcriptome Sequencing
4.3. Gene Expression Quantification and Differential Analysis
4.4. Metabolite Extraction and UPLC-ESI-MS/MS Analysis
4.5. Data Processing and Metabolite Identification
4.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis
4.7. Determination of Total Flavonoid Content
4.8. Determination of Total Lignin Content and Lignin Composition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manchali, S.; Chidambara Murthy, K.N.; Vishnuvardana; Patil, B.S. Nutritional composition and health benefits of various botanical types of melon (Cucumis melo L.). Plants 2021, 10, 1755. [Google Scholar] [CrossRef]
- Zhang, X.; Ling, Y.; Yang, W.; Wei, M.; Wang, Z.; Li, M.; Yang, Y.; Liu, B.; Yi, H.; Guo, Y.-D.; et al. Fine mapping of a novel QTL DM9.1 conferring downy mildew resistance in melon. Front. Plant Sci. 2023, 14, 1202775. [Google Scholar] [CrossRef] [PubMed]
- Kitner, M.; Lebeda, A.; Sharma, R.; Runge, F.; Dvorak, P.; Tahir, A.; Choi, Y.J.; Sedlakova, B.; Thines, M. Coincidence of virulence shifts and population genetic changes of Pseudoperonospora cubensis in the Czech Republic. Plant Pathol. 2015, 64, 1467–1470. [Google Scholar] [CrossRef]
- Wallace, E.C.; D’Arcangelo, K.N.; Quesada-Ocampo, L.M. Population analyses reveal two host-adapted clades of Pseudoperonospora cubensis, the causal agent of cucurbit downy mildew, on commercial and wild cucurbits. Phytopathology 2020, 110, 1578–1587. [Google Scholar] [CrossRef] [PubMed]
- Keinath, A.P. Integrated management of downy mildew on slicing cucumber with fungicides and host resistance but not trellising. Plant Dis. 2019, 103, 2592–2598. [Google Scholar] [CrossRef] [PubMed]
- Keinath, A.P.; Miller, S.A.; Smart, C.D. Response of Pseudoperonospora cubensis to preventative fungicide applications varies by state and year. Plant Health Prog. 2019, 20, 142–146. [Google Scholar] [CrossRef]
- Thomas, A.; Neufeld, K.N.; Seebold, K.W.; Braun, C.A.; Schwarz, M.R.; Ojiambo, P.S. Resistance to fuopicolide and propamocarb and baseline sensitivity to ethaboxam among isolates of Pseudoperonospora cubensis from the eastern United States. Plant Dis. 2018, 102, 1619–1626. [Google Scholar] [CrossRef]
- Jeon, J.E.; Kim, J.G.; Fischer, C.R.; Mehta, N.; Dufour-Schroif, C.; Wemmer, K.; Mudgett, M.B.; Sattely, E. A pathogen-responsive gene cluster for highly modified fatty acids in tomato. Cell 2020, 180, 176–187. [Google Scholar] [CrossRef]
- Su, P.; Zhao, L.; Li, W.; Zhao, J.; Yan, J.; Ma, X.; Li, A.; Wang, H.; Kong, L. Integrated metabolo-transcriptomics and functional characterization reveals that the wheat auxin receptor TIR1 negatively regulates defense against Fusarium graminearumFA. J. Integr. Plant Biol. 2020, 63, 340–352. [Google Scholar] [CrossRef]
- Li, P.; Ruan, Z.; Fei, Z.; Yan, J.; Tang, G. Integrated transcriptome and metabolome analysis revealed that flavonoid biosynthesis may dominate the resistance of Zanthoxylum bungeanum against stem canker. J. Agric. Food Chem. 2021, 69, 6360–6378. [Google Scholar] [CrossRef]
- Xu, C.; Zhan, C.; Huang, S.; Xu, Q.; Tang, T.; Wang, Y.; Luo, J.; Zeng, X. Resistance to powdery mildew in qingke involves the accumulation of aromatic phenolamides through jasmonate-mediated activation of defense-related genes. Front. Plant Sci. 2022, 13, 900345. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Siskos, L.; Wang, C.; Schouten, H.J.; Visser, R.G.; Bai, Y. Breeding melon (Cucumis melo) with resistance to powdery mildew and downy mildew. Hortic. Plant J. 2022, 8, 545–561. [Google Scholar] [CrossRef]
- Kesh, H.; Kaushik, P. Advances in melon (Cucumis melo L.) breeding: An update. Sci. Hortic. 2021, 282, 110045. [Google Scholar] [CrossRef]
- Toporek, S.M.; Branham, S.E.; Keinath, A.P.; Wechter, W.P. QTL mapping of resistance to Pseudoperonospora cubensis clade 2, mating type A1, in Cucumis melo and dual-clade marker development. Theor. Appl. Genet. 2023, 136, 91. [Google Scholar] [CrossRef]
- Toporek, S.M.; Branham, S.E.; Katawczik, M.L.; Keinath, A.P.; Wechter, W.P. QTL mapping of resistance to Pseudoperonospora cubensis clade 1, mating type A2, in Cucumis melo. Theor. Appl. Genet. 2021, 134, 2577–2586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Zheng, J.; Chen, X.; Shi, X.; Wang, H.; Fu, Q. Comprehensive analysis of transcriptome and metabolome reveals the flavonoid metabolic pathway is associated with fruit peel coloration of melon. Molecules 2021, 26, 2830. [Google Scholar] [CrossRef]
- Nagashima, Y.; He, K.; Singh, J.; Metrani, R.; Crosby, K.M.; Jifon, J.; Jayaprakasha, G.K.; Patil, B.; Qian, X.; Koiwa, H. Transition of aromatic volatile and transcriptome profiles during melon fruit ripening. Plant Sci. 2021, 304, 110809. [Google Scholar] [CrossRef]
- Wang, X.; Song, S.; Wang, X.; Liu, J.; Dong, S. Transcriptomic and metabolomic analysis of seedling-stage soybean responses to PEG-simulated drought Stress. Int. J. Mol. Sci. 2022, 23, 6869. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, H.; You, J.; Yang, J.; Zhang, Q.; Zhao, J.; Aimaier, R.; Zhang, J.; Han, S.; Zhao, H.; et al. Transcriptome and metabolome analyses reveal that Bacillus subtilis BS-Z15 lipopeptides mycosubtilin homologue mediates plant defense responses. Front. Plant Sci. 2023, 13, 1088220. [Google Scholar] [CrossRef]
- Leyva-Pérez, M.O.; Jiménez-Ruiz, J.; Cabanás, G.C.; Valverde-Corredor, A.; Barroso, J.B.; Luque, F.; Mercado-Blanco, J. Tolerance of olive (Olea europaea) cv Frantoio to Verticillium dahliae relies on both basal and pathogen-induced differential transcriptomic responses. New Phytol. 2018, 217, 671–686. [Google Scholar] [CrossRef]
- Xiong, X.P.; Sun, S.C.; Zhu, Q.H.; Zhang, X.Y.; Li, Y.J.; Liu, F.; Xue, F.; Sun, J. The cotton lignin biosynthetic gene Gh4CL30 regulates lignification and phenolic content and contributes to Verticillium wilt resistance. Mol. Plant-Microbe Interact. 2021, 34, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Mekonnen, D.W.; Hartmann, M.; Yildiz, I.; Janowski, R.; Lange, B.; Geist, B.; Zeier, J.; Schäffner, A.R. UGT76B1, a promiscuous hub of small molecule-based immune signaling, glucosylates N-hydroxypipecolic acid, and balances plant immunity. Plant Cell 2021, 33, 714–734. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Nolan, T.M.; Song, G.; Liu, S.; Xie, Z.; Chen, J.; Schnable, P.S.; Walley, J.W.; Yin, Y. FERONIA receptor kinase contributes to plant immunity by suppressing jasmonic acid signaling in Arabidopsis thaliana. Curr. Biol. 2018, 28, 3316–3324. [Google Scholar] [CrossRef] [PubMed]
- González-Morales, S.; Solís-Gaona, S.; Valdés-Caballero, M.V.; Juárez-Maldonado, A.; Loredo-Treviño, A.; Benavides-Mendoza, A. Transcriptomics of biostimulation of plants under abiotic stress. Front. Genet. 2021, 12, 583888. [Google Scholar] [CrossRef] [PubMed]
- Kavi Kishor, P.B.; Tiozon, R.N.; Fernie, A.R.; Sreenivasulu, N. Abscisic acid and its role in the modulation of plant growth, development, and yield stability. Trends Plant Sci. 2022, 27, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.W.; Yang, S.H.; Shin, K.H.; Lee, S.C.; Kim, S.H. The AtLRK10L1.2, Arabidopsis ortholog of wheat LRK10, is involved in ABA-mediated signaling and drought resistance. Plant Cell Rep. 2015, 34, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Hu, Q.; Min, L.; Yang, X.; Jin, S.; Zhang, L.; Li, Y.; Ma, Y.; Qi, X.; Li, D.; Liu, H.; et al. Laccase GhLac1 modulates broad-spectrum biotic stress tolerance via manipulating phenylpropanoid pathway and jasmonic acid synthesis. Plant Physiol. 2018, 176, 1808–1823. [Google Scholar] [CrossRef]
- Gao, X.; Guo, P.; Wang, Z.; Chen, C.; Ren, Z. Transcriptome profiling reveals response genes for downy mildew resistance in cucumber. Planta 2021, 253, 112. [Google Scholar] [CrossRef]
- Postnikova, O.A.; Hult, M.; Shao, J.; Skantar, A.; Nemchinov, L.G. Transcriptome analysis of resistant and susceptible alfalfa cultivars infected with root-knot nematode Meloidogyne incognita. PLoS ONE 2015, 10, e0118269. [Google Scholar] [CrossRef]
- Jurgen, Z. Metabolic regulation of systemic acquired resistance. Curr. Opin. Plant Biol. 2021, 62, 102050. [Google Scholar]
- Holmes, E.C.; Chen, Y.C.; Mudgett, M.B.; Sattely, E.S. Arabidopsis UGT76B1 glycosylates N-hydroxy-pipecolic acid and inactivates systemic acquired resistance in tomato. Plant Cell 2021, 33, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Davoudi, M.; Song, M.; Zhang, M.; Chen, J.; Lou, Q. Long-distance control of pumpkin rootstock over cucumber scion under drought stress as revealed by transcriptome sequencing and mobile mRNAs identifications. Hortic. Res. 2022, 9, uhab033. [Google Scholar] [CrossRef] [PubMed]
- Ré, D.A.; Capella, M.; Bonaventure, G.; Chan, R.L. Arabidopsis AtHB7 and AtHB12 evolved divergently to fine tune processes associated with growth and responses to water stress. BMC Plant Biol. 2014, 14, 150. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.W.; Lee, D.K.; Jung, H.; Chung, P.J.; Kim, Y.S.; Choi, Y.D.; Suh, J.W.; Kim, J.K. Overexpression of OsTF1L, a rice HD-Zip transcription factor, promotes lignin biosynthesis and stomatal closure that improves drought tolerance. Plant Biotechnol. J. 2019, 17, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Carella, P.; Gogleva, A.; Hoey, D.J.; Bridgen, A.J.; Stolze, S.C.; Nakagami, H.; Schornack, S. Conserved biochemical defenses underpin host responses to oomycete infection in an early-divergent land plant lineage. Curr. Biol. 2019, 29, 2282–2294. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R. Evolution: An early role for flavonoids in defense against oomycete infection. Curr. Biol. 2019, 29, R688–R690. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Hu, L.; Li, Y.; Chen, X.; Zhang, Z.; Liu, B.; Li, P.; Gong, X.; Ma, F. MdUGT88F1-mediated phloridzin biosynthesis regulates apple development and Valsa canker resistance. Plant Physiol. 2019, 180, 2290–2305. [Google Scholar] [CrossRef]
- Sudheeran, P.K.; Ovadia, R.; Galsarker, O.; Maoz, I.; Sela, N.; Maurer, D.; Feygenberg, O.; Oren Shamir, M.; Alkan, N. Glycosylated flavonoids: Fruit’s concealed antifungal arsenal. New Phytol. 2020, 225, 1788–1798. [Google Scholar] [CrossRef]
- Bai, Q.; Duan, B.; Ma, J.; Fen, Y.; Sun, S.; Long, Q.; Lv, J.; Wan, D. Coexpression of PalbHLH1 and PalMYB90 genes from Populus alba enhances pathogen resistance in poplar by increasing the flavonoid content. Front. Plant Sci. 2020, 10, 1772. [Google Scholar] [CrossRef]
- Kursa, W.; Jamiołkowska, A.; Wyrostek, J.; Kowalski, R. Antifungal effect of plant extracts on the growth of the cereal pathogen Fusarium spp.—An In Vitro Study. Agronomy 2022, 12, 3204. [Google Scholar] [CrossRef]
- Shi, J.; Yan, X.; Sun, T.; Shen, Y.; Shi, Q.; Wang, W.; Bao, M.; Luo, H.; Nian, F. Homeostatic regulation of flavonoid and lignin biosynthesis in phenylpropanoid pathway of transgenic tobacco. Gene 2022, 809, 146017. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhang, J.; Tschaplinski, T.J.; Tuskan, G.A.; Chen, J.G.; Muchero, W. Regulation of lignin biosynthesis and its role in growth-defense tradeoffs. Front. Plant Sci. 2018, 9, 1427. [Google Scholar] [CrossRef]
- Ha, C.M.; Rao, X.; Saxena, G.; Dixon, R.A. Growth-defense trade-offs and yield loss in plants with engineered cell walls. New Phytol. 2021, 231, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; He, Y.; Kabahuma, M.; Chaya, T.; Kelly, A.; Borrego, E.; Bian, Y.; El Kasmi, F.; Yang, L.; Teixeira, P.; et al. A gene encoding maize caffeoyl-CoAO-methyltransferase confers quantitative resistance to multiple pathogens. Nat. Genet. 2017, 49, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Wang, Z.; Guo, Y.; Zhang, X. Comparative transcriptome profiling reveals the role of phytohormones and phenylpropanoid pathway in early-stage resistance against powdery mildew in watermelon (Citrullus lanatus L.). Front. Plant Sci. 2022, 13, 1016822. [Google Scholar] [CrossRef]
- Garcia-Mas, J.; Benjak, A.; Sanseverino, W.; Bourgeois, M.; Mir, G.; González, V.M.; Hénaff, E.; Câmara, F.; Cozzuto, L.; Lowy, E.; et al. The genome of melon (Cucumis melo L.). Proc. Natl. Acad. Sci. USA 2012, 109, 11872–11877. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Shraim, A.; Ahmed, T.; Rahman, M.; Hijji, Y.M. Determination of total flavonoid content by aluminum chloride assay: A critical evaluation. LWT 2021, 150, 111932. [Google Scholar] [CrossRef]
- Barnes, W.J.; Anderson, C.T. Acetyl bromide soluble lignin (ABSL) assay for total lignin quantification from plant biomass. Bio-protocol 2017, 7, e2149. [Google Scholar] [CrossRef]
- Chen, F.; Zhuo, C.; Xiao, X.; Pendergast, T.H.; Devos, K.M. A rapid thioacidolysis method for biomass lignin composition and tricin analysis. Biotechnol. Biofuels 2021, 14, 18. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, Y.; Xiong, X.; Yang, W.; Liu, B.; Shen, Y.; Xu, L.; Lu, F.; Li, M.; Guo, Y.; Zhang, X. Comparative Analysis of Transcriptomics and Metabolomics Reveals Defense Mechanisms in Melon Cultivars against Pseudoperonospora cubensis Infection. Int. J. Mol. Sci. 2023, 24, 17552. https://doi.org/10.3390/ijms242417552
Ling Y, Xiong X, Yang W, Liu B, Shen Y, Xu L, Lu F, Li M, Guo Y, Zhang X. Comparative Analysis of Transcriptomics and Metabolomics Reveals Defense Mechanisms in Melon Cultivars against Pseudoperonospora cubensis Infection. International Journal of Molecular Sciences. 2023; 24(24):17552. https://doi.org/10.3390/ijms242417552
Chicago/Turabian StyleLing, Yueming, Xianpeng Xiong, Wenli Yang, Bin Liu, Yue Shen, Lirong Xu, Fuyuan Lu, Meihua Li, Yangdong Guo, and Xuejun Zhang. 2023. "Comparative Analysis of Transcriptomics and Metabolomics Reveals Defense Mechanisms in Melon Cultivars against Pseudoperonospora cubensis Infection" International Journal of Molecular Sciences 24, no. 24: 17552. https://doi.org/10.3390/ijms242417552
APA StyleLing, Y., Xiong, X., Yang, W., Liu, B., Shen, Y., Xu, L., Lu, F., Li, M., Guo, Y., & Zhang, X. (2023). Comparative Analysis of Transcriptomics and Metabolomics Reveals Defense Mechanisms in Melon Cultivars against Pseudoperonospora cubensis Infection. International Journal of Molecular Sciences, 24(24), 17552. https://doi.org/10.3390/ijms242417552