
Clonality and Diversity in the Soft Rot Dickeya solani Phytopathogen


Abstract
:1. Introduction
2. Results
2.1. Collection of New D. solani Strains Isolated in Different Environments
2.2. D. solani Genomic Diversity
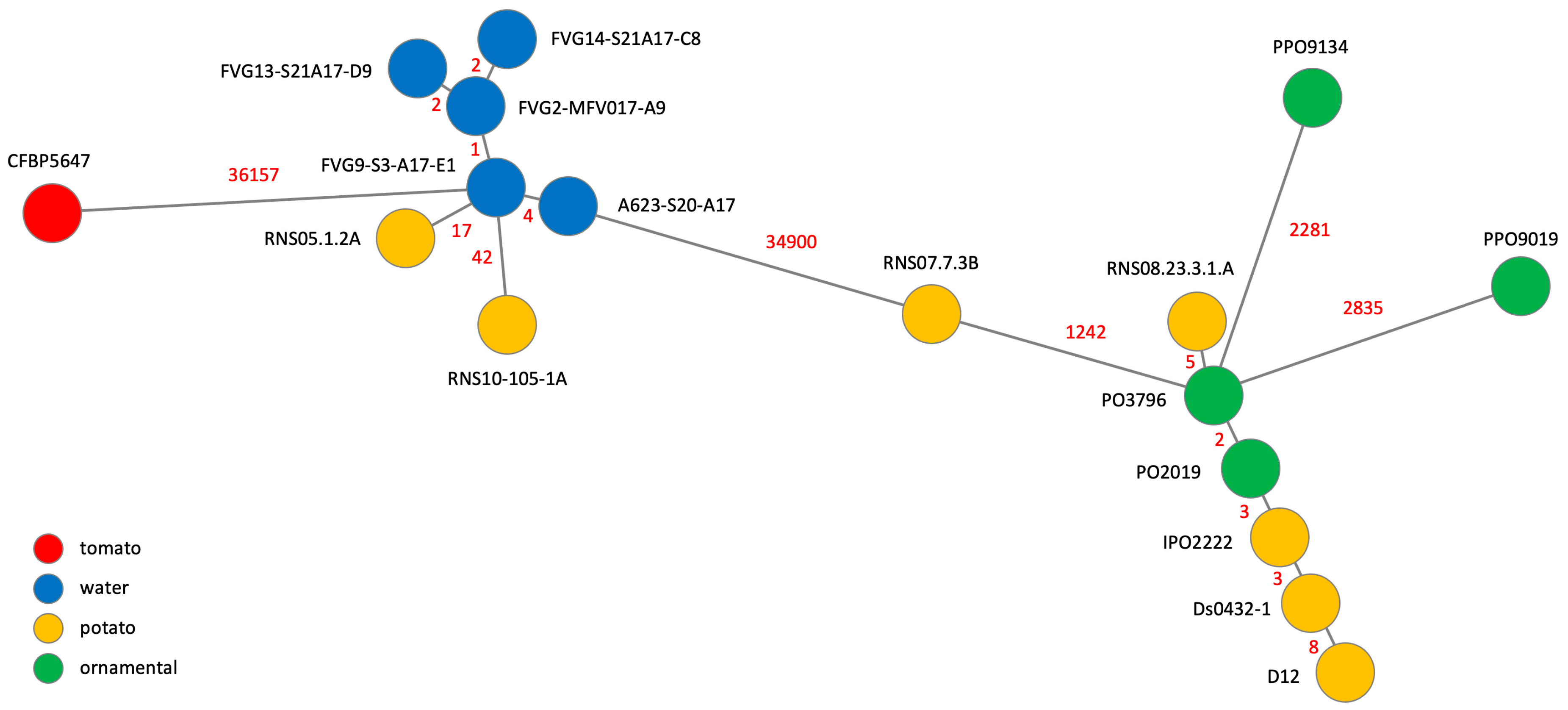
2.2.1. The Relatedness between D. solani Members
2.2.2. Relatedness of D. solani Strains by SNP Analysis
2.3. Genomic Comparison of the Three D. solani Clades
2.3.1. Genomic Synteny
2.3.2. Analysis of Clade-Specific Genes
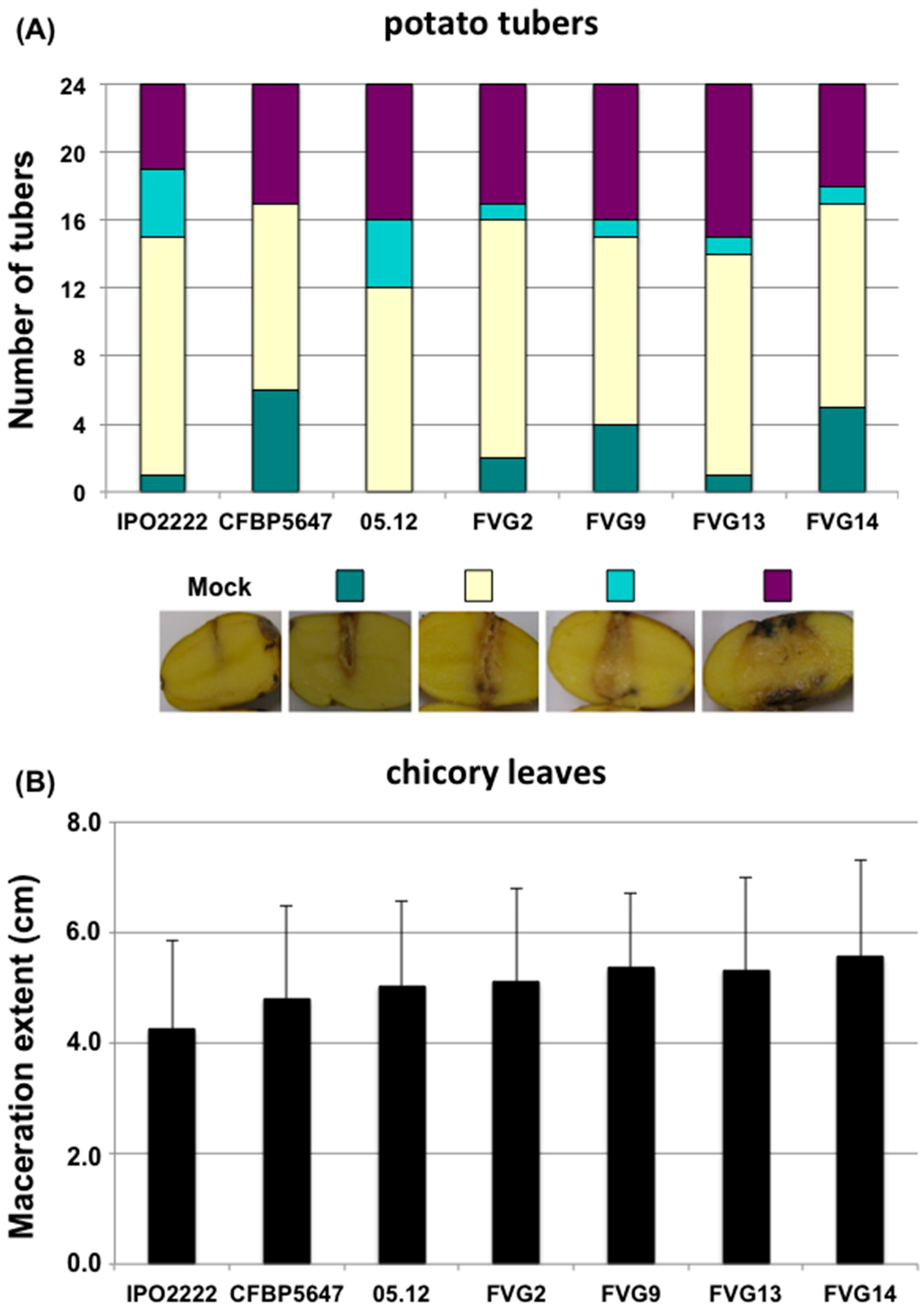
2.4. Virulence of New Isolates
3. Discussion
4. Materials and Methods
4.1. dnaX-leuS-recA Phylogeny of Dickeya Solani Strains from CIRM-CFBP
4.2. DNA Extraction, Genome Sequencing and Assembly
4.3. Genome Analysis
4.4. Minimum Spanning Tree Analysis
4.5. MAUVE
4.6. Virulence Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pédron, J.; Van Gijsegem, F. Diversity in the Bacterial Genus Dickeya Grouping Plant Pathogens and Waterways Isolates. OBM Genet. 2019, 3, 098. [Google Scholar] [CrossRef]
- Hugouvieux-Cotte-Pattat, N.; Pédron, J.; Van Gijsegem, F. Insight into biodiversity of the recently rearranged genus Dickeya. Front. Plant Sci. 2023, 14, 1168480. [Google Scholar] [CrossRef]
- Toth, I.K.; Barny, M.; Brurberg, M.B.; Condemine, G.; Czajkowski, R.; Elphinstone, J.G.; Helias, V.; Johnson, S.B.; Moleleki, L.N.; Pirhonen, M.; et al. Pectobacterium and Dickeya: Environment to Disease Development. In Plant Diseases Caused by Pectobacterium and Dickeya Species; Van Gijsegem, F., van der Wolf, J.M., Toth, I.K., Eds.; Springer Nature: Cham, Switzerland, 2021; pp. 39–84. [Google Scholar]
- Van Gijsegem, F.; Hugouvieux-Cotte-Pattat, N.; Kraepiel, Y.; Lojkowska, E.; Moleleki, L.; Gorshkov, V.; Yedidia, I. Molecular interactions of Pectobacterium and Dickeya with plants. In Plant Diseases Caused by Pectobacterium and Dickeya Species; Van Gijsegem, F., van der Wolf, J.M., Toth, I.K., Eds.; Springer Nature: Cham, Switzerland, 2021; pp. 85–148. [Google Scholar]
- Toth, I.K.; Barny, M.-A.; Czajkowski, R.; Elphinstone, J.G.; LI, X.S.; Pédron, J.; Pirhonen, M.; Van Gijsegem, F. Pectobacterium and Dickeya: Taxonomy and evolution. In Plant Diseases Caused by Pectobacterium and Dickeya Species; Van Gijsegem, F., van der Wolf, J.M., Toth, I.K., Eds.; Springer Nature: Cham, Switzerland, 2021; pp. 13–38. [Google Scholar]
- Wang, X.; He, S.W.; Guo, H.B.; Han, J.G.; Thin, K.K.; Gao, J.S.; Wang, Y.; Zhang, X.X. Dickeya oryzae sp. nov., isolated from the roots of rice. Int. J. Syst. Evol. Microbiol. 2020, 70, 4171–4178. [Google Scholar] [CrossRef]
- Hugouvieux-Cotte-Pattat, N.; Van Gijsegem, F. Diversity within the Dickeya zeae complex, identification of Dickeya zeae and Dickeya oryzae members, proposal of the novel species Dickeya parazeae sp. nov. Int. J. Syst. Evol. Microbiol. 2021, 71, 005059. [Google Scholar] [CrossRef]
- Alic, S.; Pédron, J.; Dreo, T.; Van Gijsegem, F. Genomic characterization of the new Dickeya fangzhongdai species regrouping plant pathogens and environmental isolates. BMC Genom. 2019, 20, 34. [Google Scholar] [CrossRef]
- Oulghazi, S.; Khayi, S.; Lafkih, N.; Massaoudi, Y.; El Karkouri, A.; El Hassouni, M.; Faure, D.; Moumni, M. First report of Dickeya dianthicola causing blackleg on potato in Morocco. Plant Dis. 2017, 101, 1671–1672. [Google Scholar] [CrossRef]
- Ge, T.; Jiang, H.; Tan, E.H.; Johnson, S.B.; Larkin, R.P.; Charkowski, A.O.; Secor, G.; Hao, J. Pangenomic analysis of Dickeya dianthicola strains related to the outbreak of blackleg and soft rot of potato in USA. Plant Dis. 2021, 105, 3946–3955. [Google Scholar] [CrossRef]
- Van der Wolf, J.M.; Nijhuis, E.H.; Kowalewska, M.J.; Saddler, G.S.; Parkinson, N.; Elphinstone, J.G.; Pritchard, L.; Toth, I.K.; Lojkowska, E.; Potrykus, M.; et al. Dickeya solani Sp. Nov., a Pectinolytic Plant-Pathogenic Bacterium Isolated from Potato (Solanum tuberosum). Int. J. Syst. Evol. Microbiol. 2014, 64, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Motyka-Pomagruk, A.; Zoledowska, S.; Misztak, A.E.; Sledz, W.; Mengoni, A.; Lojkowska, E. Comparative Genomics and Pangenome-Oriented Studies Reveal High Homogeneity of the Agronomically Relevant Enterobacterial Plant Pathogen Dickeya solani. BMC Genom. 2020, 21, 449. [Google Scholar] [CrossRef] [PubMed]
- Blin, P.; Robic, K.; Khayi, S.; Cigna, J.; Munier, E.; Dewaegeneire, P.; Laurent, A.; Jaszczyszyn, Y.; Hong, K.W.; Chan, K.G.; et al. Pattern and Causes of the Establishment of the Invasive Bacterial Potato Pathogen Dickeya solani and of the Maintenance of the Resident Pathogen D. dianthicola. Mol. Ecol. 2021, 30, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Khayi, S.; Chan, K.-G.; Faure, D. Patterns of Genomic Variations in the Plant Pathogen Dickeya solani. Microorganisms 2022, 10, 2254. [Google Scholar] [CrossRef]
- Pédron, J.; van der Wolf, J.M.; Portier, P.; Caullireau, E.; Van Gijsegem, F. The broad host range plant pathogen Dickeya dianthicola shows a high genetic diversity. Microorganisms 2022, 10, 1024. [Google Scholar] [CrossRef]
- Khayi, S.; Blin, P.; Pédron, J.; Chong, T.-M.; Chan, K.-G.; Moumni, M.; Hélias, V.; Van Gijsegem, F.; Faure, D. Population Genomics Reveals Additive and Replacing Horizontal Gene Transfers in the Emerging Pathogen Dickeya solani. BMC Genom. 2015, 16, 788. [Google Scholar] [CrossRef]
- Pédron, J.; Schaerer, S.; Kellenberger, I.; Van Gijsegem, F. Early Emergence of Dickeya solani Revealed by Analysis of Dickeya Diversity of Potato Blackleg and Soft Rot Causing Pathogens in Switzerland. Microorganisms 2021, 9, 1187. [Google Scholar] [CrossRef]
- Golanowska, M.; Potrykus, M.; Motyka-Pomagruk, A.; Kabza, M.; Bacci, G.; Galardini, M.; Bazzicalupo, M.; Makalowska, I.; Smalla, K.; Lojkowska, E.; et al. Comparison of highly and weakly virulent Dickeya solani strains, with a view on the pangenome and panregulon of this species. Front. Microbiol. 2018, 9, 1940. [Google Scholar] [CrossRef]
- Motyka-Pomagruk, A.; Babinska-Wensierska, W.; Sledz, W.; Kaczorowska, A.K.; Lojkowska, E. Phyloproteomic study by MALDI-TOF MS in view of intraspecies variation in a significant homogenous phytopathogen Dickeya solani. Sci. Rep. 2023, 13, 18863. [Google Scholar] [CrossRef]
- Morris, C.E.; Lacroix, C.; Chandeysson, C.; Guilbaud, C.; Monteil, C.; Piry, S.; Rochelle Newall, E.; Fiorini, S.; Van Gijsegem, F.; Barny, M.A.; et al. Comparative abundance and diversity of populations of the Pseudomonas syringae and Soft Rot Pectobacteriaceae species complexes throughout the Durance River catchment from its French Alps sources to its delta. Peer Community J. 2023, 3, e88. [Google Scholar] [CrossRef]
- Garlant, L.; Koskinen, P.; Rouhiainen, L.; Laine, P.; Paulin, L.; Auvinen, P.; Holm, L.; Pirhonen, M. Genome sequence of Dickeya solani, a new soft rot pathogen of potato, suggests its emergence may be related to a novel combination of non-ribosomal peptide/polyketide synthetase clusters. Diversity 2013, 5, 824–842. [Google Scholar] [CrossRef]
- Brual, T.; Effantin, G.; Baltenneck, J.; Attaiech, L.; Grosbois, C.; Royer, M.; Cigna, J.; Faure, D.; Hugouvieux-Cotte-Pattat, N.; Gueguen, E. A natural single nucleotide mutation in the small regulatory RNA ArcZ of Dickeya solani switches off the antimicrobial activities against yeast and bacteria. PLoS Genet. 2023, 19, e1010725. [Google Scholar] [CrossRef]
- Pédron, J.; Mondy, S.; Raoul des Essarts, Y.; Van Gijsegem, F.; Faure, D. Genomic and metabolic comparison with Dickeya dadantii 3937 reveals the emerging Dickeya solani potato pathogen to display distinctive metabolic activities and T5SS/T6SS-related toxin repertoire. BMC Genom. 2014, 15, 283. [Google Scholar] [CrossRef] [PubMed]
- Raoul des Essarts, Y.; Pédron, J.; Blin, P.; Van Dijk, E.; Faure, D.; Van Gijsegem, F. Common and distinctive adaptive traits expressed in Dickeya dianthicola and Dickeya solani pathogens when exploiting potato plant host. Environ. Microbiol. 2019, 21, 1004–1018. [Google Scholar] [CrossRef] [PubMed]
- Mavrodi, D.V.; Loper, J.E.; Paulsen, I.T.; Thomashow, L.S. Mobile genetic elements in the genome of the beneficial rhizobacterium Pseudomonas fluorescens Pf-5. BMC Microbiol. 2009, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, R.; Toussaint, A.; Ryan, M.P.; Pembroke, J.T.; Mergeay, M.; Adley, C.C. Tn4731 ICE family of bacterial mobile genetic elements. In Bacterial Integrative Mobile Genetic Elements; Roberts, A.P., Mullany., P., Eds.; Landes Biosciences: Austin, TX, USA, 2011. [Google Scholar]
- Costechareyre, D.; Balmand, S.; Condemine, G.; Rahbe, Y. Dickeya dadantii, a plant pathogenic bacterium producing Cyt-Like entomotoxins, causes cepticemia in the pea aphid Acyrthosiphon pisum. PLoS ONE 2012, 7, e30702. [Google Scholar] [CrossRef] [PubMed]
- Van der Wolf, J.M.; Acuña, I.; De Boer, S.H.; Brurberg, M.B.; Cahill, G.; Charkowski, A.O.; Coutinho, T.; Davey, T.; Dees, M.W.; Degefu, Y.; et al. Diseases Caused by Pectobacterium and Dickeya Species Around the World. In Plant Diseases Caused by Pectobacterium and Dickeya Species; Van Gijsegem, F., van der Wolf, J.M., Toth, I.K., Eds.; Springer Nature: Cham, Switzerland, 2021; pp. 215–261. ISBN 978-3-030-61458-4. [Google Scholar]
- Slawiak, M.; van Beckhoven, J.R.C.M.; Speksnijde, A.G.C.L.; Czajkowski, R.L.; Grabe, G.; van der Wolf, J.M. Biochemical and genetical analysis reveal a new clade of biovar 3 Dickeya spp. strains isolated from potato in Europe. Eur. J. Plant Pathol. 2009, 125, 245–261. [Google Scholar] [CrossRef]
- Toth, I.K.; van der Wolf, J.M.; Saddler, G.; Lojkowska, E.; Hélias, V.; Pirhonen, M.; Tsror (Lahkim), L.; Elphinstone, J.G. Dickeya species: An emerging problem for potato production in Europe. Plant Pathol. 2011, 60, 385–399. [Google Scholar] [CrossRef]
- Ben Moussa, H.; Bertrand, C.; Rochelle-Newall, E.; Fiorini, S.; Pedron, J.; Barny, M.A. The diversity of soft rot Pectobacteriaceae along the Durance river stream in the south-east of France revealed by multiple seasonal surveys. Phytopathology 2022, 112, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Jonkheer, E.M.; Brankovics, B.; Houwers, I.M.; van der Wolf, J.M.; Bonants, P.; Vreeburg, R.; Bollema, R.; de Haan, J.R.; Berke, L.; Smit, S.; et al. The Pectobacterium pangenome, with a focus on Pectobacterium brasiliense, shows a robust core and extensive exchange of genes from a shared gene pool. BMC Genom. 2021, 22, 265. [Google Scholar] [CrossRef] [PubMed]
- Lukjancenko, O.; Wassenaar, T.M.; Ussery, D.W. Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol. 2010, 60, 708–720. [Google Scholar] [CrossRef]
- Leal Sanabria, G.; Plasencia-Márquez, O.; Martínez Zubiaur, Y.; Silvestre-Vañó, M.; Pérez-López, E. First Report of Potato (Solanum tuberosum) Blackleg Disease Caused by Dickeya solani in Mayabeque, Cuba. Plant Dis. 2023, 34. [Google Scholar] [CrossRef]
- Rodríguez-Parra, J.A.; Moreno-López, J.A.; González-Almario, A. Dickeya solani, Pectobacterium atrosepticum and Pseudomonas asplenii: Causal agents of bacterial soft rot in cyclamen plants (Cyclamen persicum Mill.) in Colombia. Can. J. Plant Pathol. 2022, 44, 485–503. [Google Scholar] [CrossRef]
- Portier, P.; Pédron, J.; Taghouti, G.; Fischer-Le Saux, M.; Caullireau, E.; Bertrand, C.; Laurent, A.; Chawki, K.; Oulgazi, S.; Moumni, M.; et al. Elevation of Pectobacterium carotovorum subsp. odoriferum to species level as Pectobacterium odoriferum sp. nov., proposal of Pectobacterium brasiliense sp. nov. and Pectobacterium actinidiae sp. nov., emended description of Pectobacterium carotovorum and description of Pectobacterium versatile sp. nov., isolated from streams and symptoms on diverse plants. Int. J. Syst. Evol. Microbiol. 2019, 69, 3207–3216. [Google Scholar] [PubMed]
- Hélias, V.; Hamon, P.; Huchet, E.; Wolf, J.V.D.; Andrivon, D. Two new effective semiselective crystal violet pectate media for isolation of Pectobacterium and Dickeya. Plant Pathol. 2012, 61, 339–345. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Phillippy, A.M. Canu. Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Miele, V.; Penel, S.; Duret, L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinform. 2011, 12, 116. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. In Molecular Biology and Evolution; Oxford University Press: Oxford, UK, 2010; Volume 27, pp. 221–224. [Google Scholar]
- Ribeiro-Gonçalves, B.; Francisco, A.P.; Vaz, C.; Ramirez, M.; Carriço, J.A. PHYLOViZ Online: Web-based tool for visualization, phylogenetic inference, analysis and sharing of minimum spanning trees. Nucleic Acids Res. 2016, 44, W246–W251. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomes | Host/Habitat | Country of Isolation | Year of Isolation | # Contigs | # CDS * | Accession Number |
|---|---|---|---|---|---|---|
| CFBP 5647 | tomato | Guadeloupe, France | - | 1 | 4883 | GCA_032681045.1 |
| PO3796 | hyacinth | Netherlands | - | 131 | 4596 | GCA_033099915.1 |
| FVG2-MFV017-A9 | water | France | 2017 | 76 | 4815 | GCA_033100015.1 |
| FVG9-S3-A17-E1 | water | France | 2017 | 83 | 4803 | GCA_033099995.1 |
| FVG13-S21A17-D9 | water | France | 2017 | 73 | 4829 | GCA_033100035.1 |
| FVG14-S21A17-C8 | water | France | 2017 | 77 | 4811 | GCA_033099975.1 |
| IPO2222T | potato | Netherlands | 2007 | 1 | 4530 | GCA_001644705.1 |
| D12 | potato | Russia | 2010 | 30 | 4532 | GCA_014751545.1 |
| Ds0432-1 | potato | Finland | 2004 | 1 | 4498 | GCA_002846975.1 |
| RNS08.23.3.1.A | potato | France | 2008 | 1 | 4536 | GCA_000511285.2 |
| RNS07.7.3B | potato | France | 2007 | 29 | 4504 | GCA_001401695.1 |
| PPO9019 | muscari | Netherlands | 2006 | 2 | 4641 | GCA_002846995.1 |
| PPO9134 | hyacinth | Netherlands | - | 22 | 4546 | GCA_001417915.1 |
| RNS05.1.2A | potato | France | 2005 | 1 | 4868 | GCA_001401705.2 |
| RNS10-105-1A | potato | France | 2010 | 65 | 4712 | GCA_026891515.1 |
| A623-S20-A17 | water | France | 2017 | 3 | 4733 | GCA_020406975.1 |
| PO2019 | hyacinth | Netherlands | 2009 | 1 | 4600 | GCA_017161585.1 |
| Phage # | Length | # of Proteins | Coordinates | Closest Prophage |
|---|---|---|---|---|
| CFBP 5647 | ||||
| P1 | 42.9 Kb | 54 | 2038734-2081644 | Salmonella phage SEN5 |
| P2 | 62.8 Kb | 59 | 3531751-3594647 | Staphylococcus phage StauST398-4 |
| P3 | 36.4 Kb | 50 | 5003447-5038072 | Haemophilus phage SuMu |
| RNS05.1.2A | ||||
| P1 | 27.4 Kb | 35 | 42866-70288 | Peduovirus P24B2 |
| P2 | 45.9 Kb | 57 | 2129925-2175882 | Salmonella phage SEN5 |
| P3 | 43.9 Kb | 44 | 2520131-2564078 | Salmonella phage SEN5 |
| P4 | 38.1 Kb | 52 | 4215256-4253442 | Bacteriophage SfI |
| P5 | 47.6 Kb | 71 | 4736805-4784434 | Edwardsiella phage GF-2 |
| Genomic Regions | Genes ID | Coordinates | # Genes | Presence of HGT Features | Predicted Functions |
|---|---|---|---|---|---|
| CFBP 5647 (456 specific protein families) | |||||
| GR1 | 181-194 | 178626-188439 | 13 | yes | extrachromosomal |
| GR2 | 1260-1266 | 1382164-1386475 | 7 | yes | extrachromosomal |
| GR3 | 2855-2920 | 3090652-3158026 | 65 | yes | T4SS, T6SS, metabolism, PFG1-like cluster |
| GR4 | 3381-3386 | 3679697-3686573 | 6 | yes | Phage-related |
| GR5 | 4075-4153 | 4410800-4506413 | 78 | yes | Ice element |
| GR6 | 4313-4321 | 4669749-4682396 | 8 | yes | CRISPR |
| GR7 | 4396-4445 | 4760648-4805394 | 49 | yes | Efflux system, T4SS |
| GR8 | 4628-4678 | 5003447-5038082 | 50 | yes | Mu prophage |
| Core clade (IPO2222) (137 specific protein families) | |||||
| GR1 | 519-527 | 594202-604799 | 8 | no | Hypothetical |
| GR2 | 2362-2377 | 2609670-2622708 | 15 | yes | Hypothetical, Phage-related |
| GR3 | 2410-2414 | 2655792-2658817 | 5 | no | Cyt endotoxin |
| GR4 | 2765-2777 | 3011280-3023862 | 12 | yes | Extrachromosomal |
| GR5 | 3717-3722 | 4059817-4065224 | 6 | no | Metabolism, regulation |
| RNS05.1.2A clade (221 specific protein families) | |||||
| GR1 | 898-913 | 929496-951874 | 15 | no | Hypothetical |
| GR2 | 2051-2058 | 2171774-2176981 | 7 | yes | Phage-related |
| GR3 | 2237-2241 | 2348346-2352853 | 5 | no | Metabolism |
| GR4 | 2258-2264 | 2370494-2374210 | 6 | yes | Extrachromosomal |
| GR5 | 3791-3810 | 4049489-4069658 | 19 | yes | Plasmid-related |
| GR6 | 4187-4197 | 4443017-4458264 | 10 | no | Transport, hypothetical |
| GR7 | 4425-4486 | 4736909-4778212 | 61 | yes | Prophage P5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Gijsegem, F.; Portier, P.; Taghouti, G.; Pédron, J. Clonality and Diversity in the Soft Rot Dickeya solani Phytopathogen. Int. J. Mol. Sci. 2023, 24, 17553. https://doi.org/10.3390/ijms242417553
Van Gijsegem F, Portier P, Taghouti G, Pédron J. Clonality and Diversity in the Soft Rot Dickeya solani Phytopathogen. International Journal of Molecular Sciences. 2023; 24(24):17553. https://doi.org/10.3390/ijms242417553
Chicago/Turabian StyleVan Gijsegem, Frédérique, Perrine Portier, Géraldine Taghouti, and Jacques Pédron. 2023. "Clonality and Diversity in the Soft Rot Dickeya solani Phytopathogen" International Journal of Molecular Sciences 24, no. 24: 17553. https://doi.org/10.3390/ijms242417553
APA StyleVan Gijsegem, F., Portier, P., Taghouti, G., & Pédron, J. (2023). Clonality and Diversity in the Soft Rot Dickeya solani Phytopathogen. International Journal of Molecular Sciences, 24(24), 17553. https://doi.org/10.3390/ijms242417553