
Proteolytic Activation of the Epithelial Sodium Channel (ENaC): Its Mechanisms and Implications
 , , and
, , and
Abstract
:1. Introduction
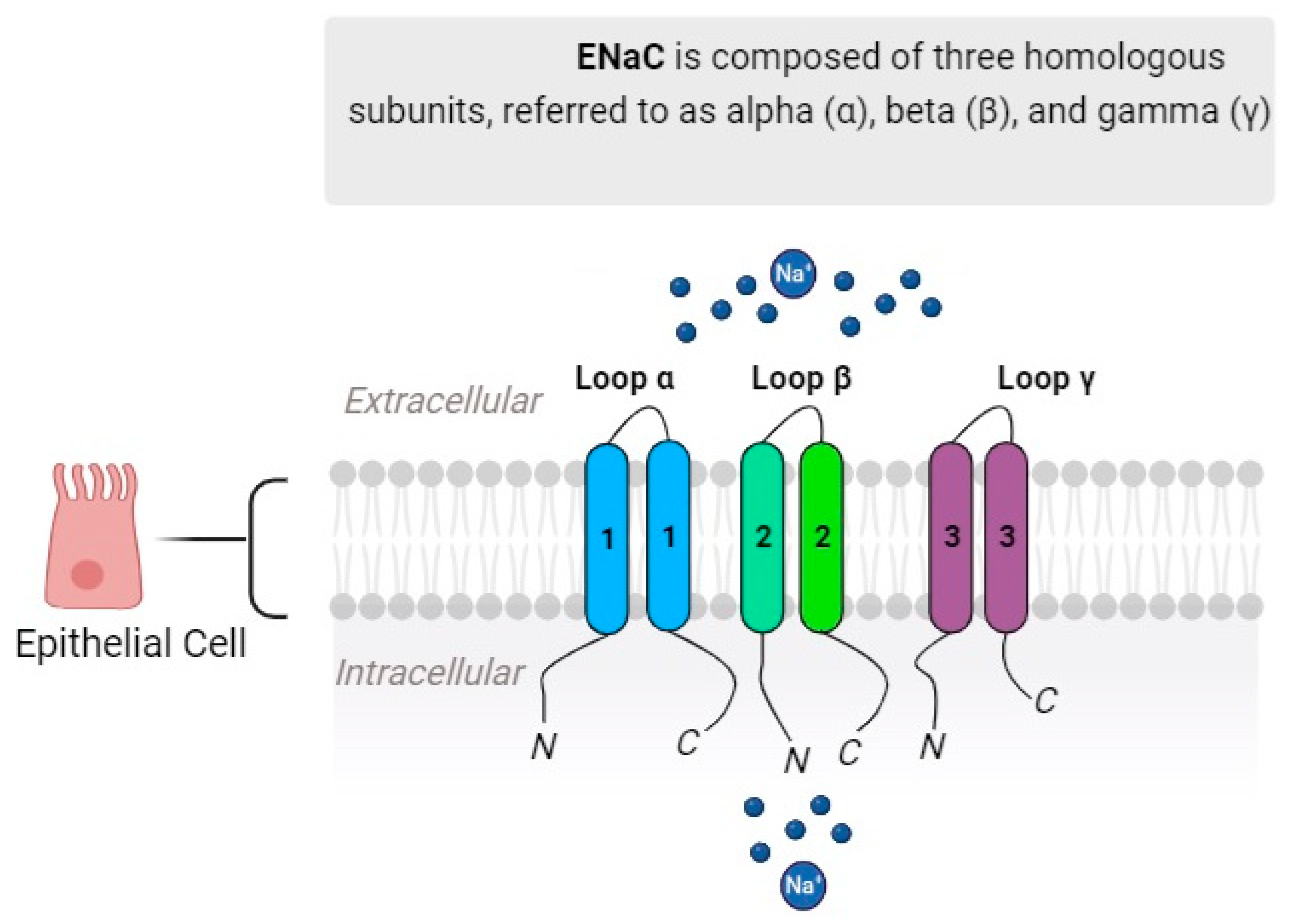
2. Proteolytic Activation of ENaC
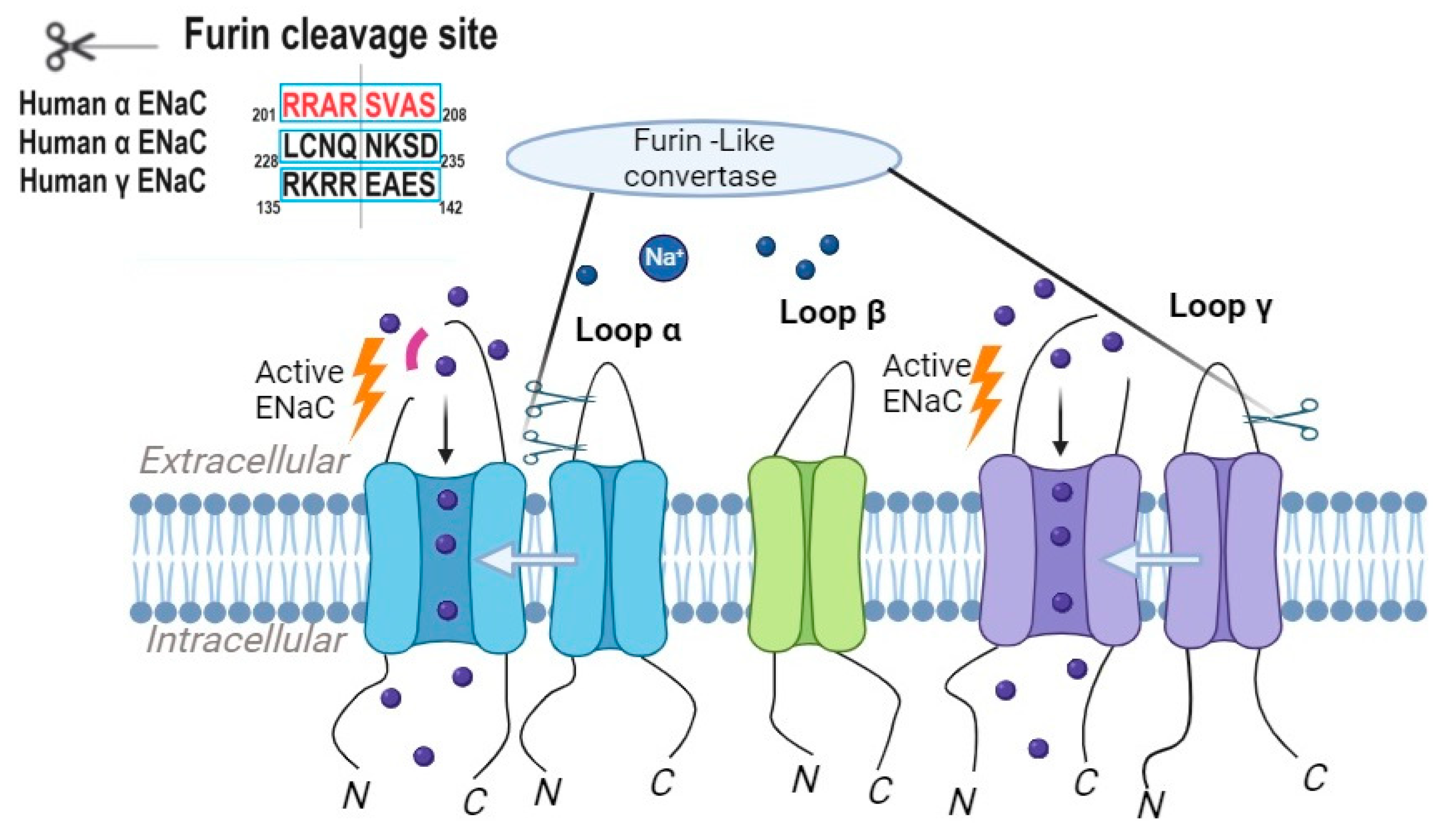
2.1. Furin-like Convertases
2.2. Tissue Kallikreins
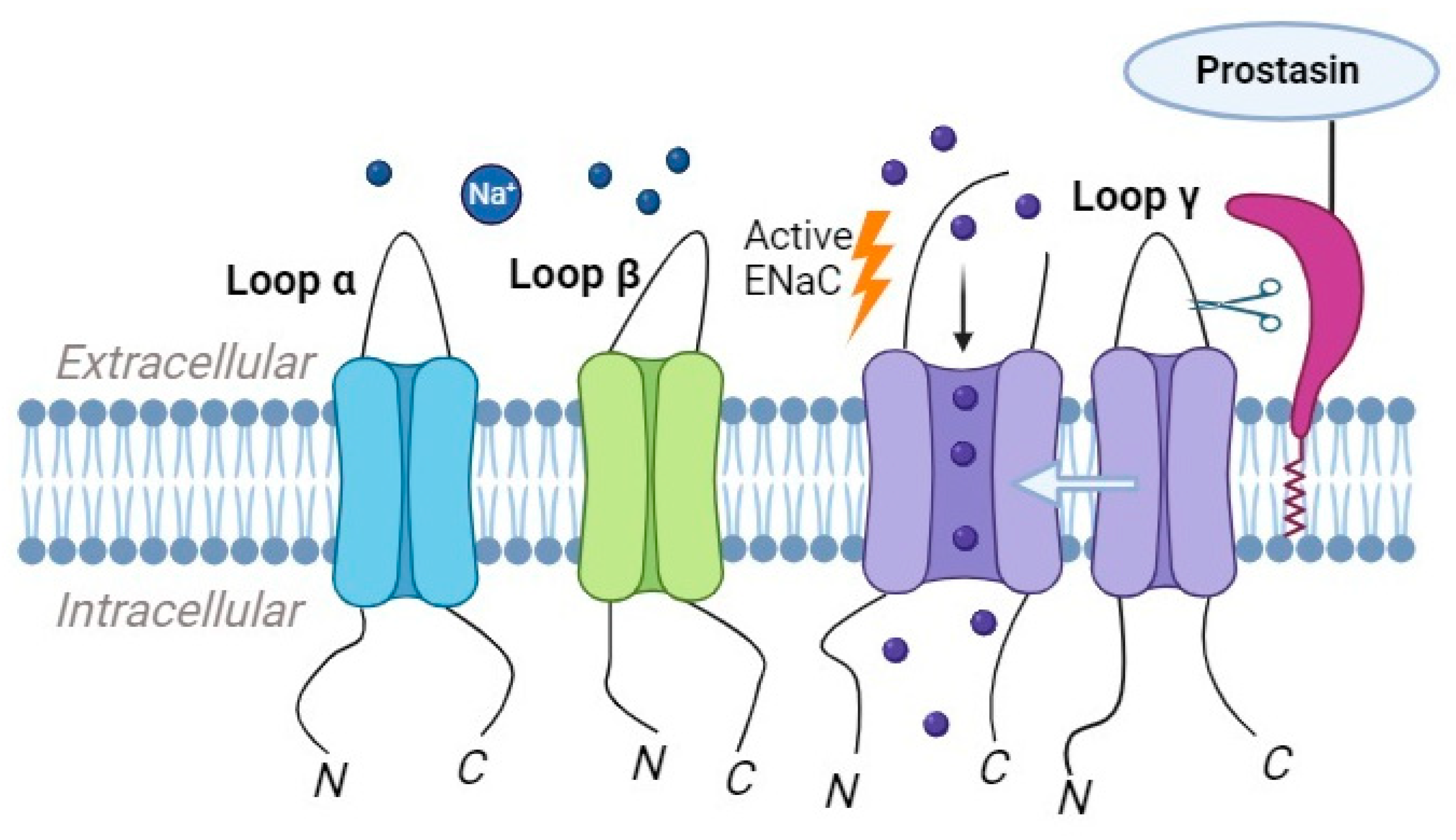
2.3. Prostasin
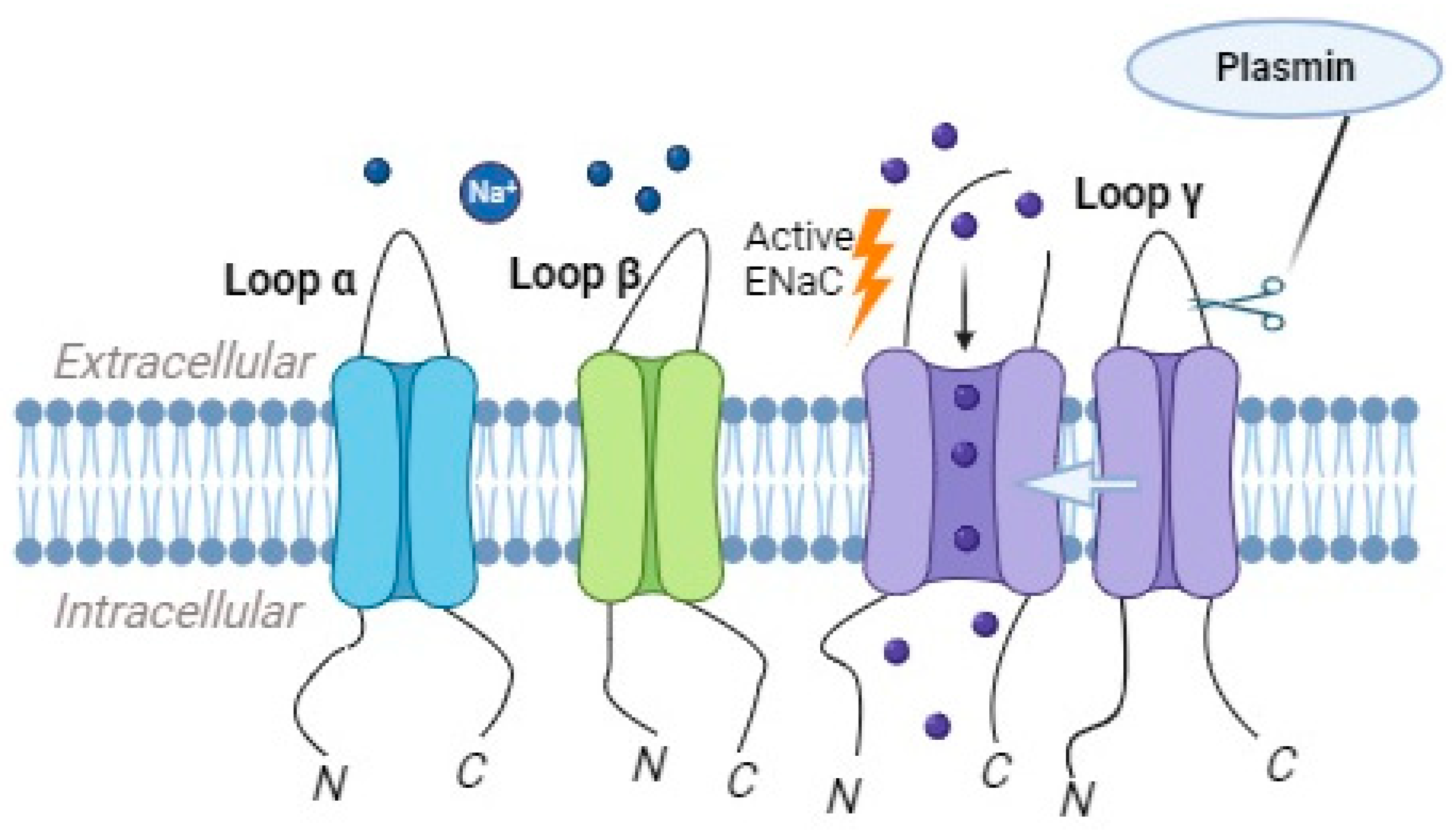
2.4. Plasmin as a Regulator of ENaC Activity
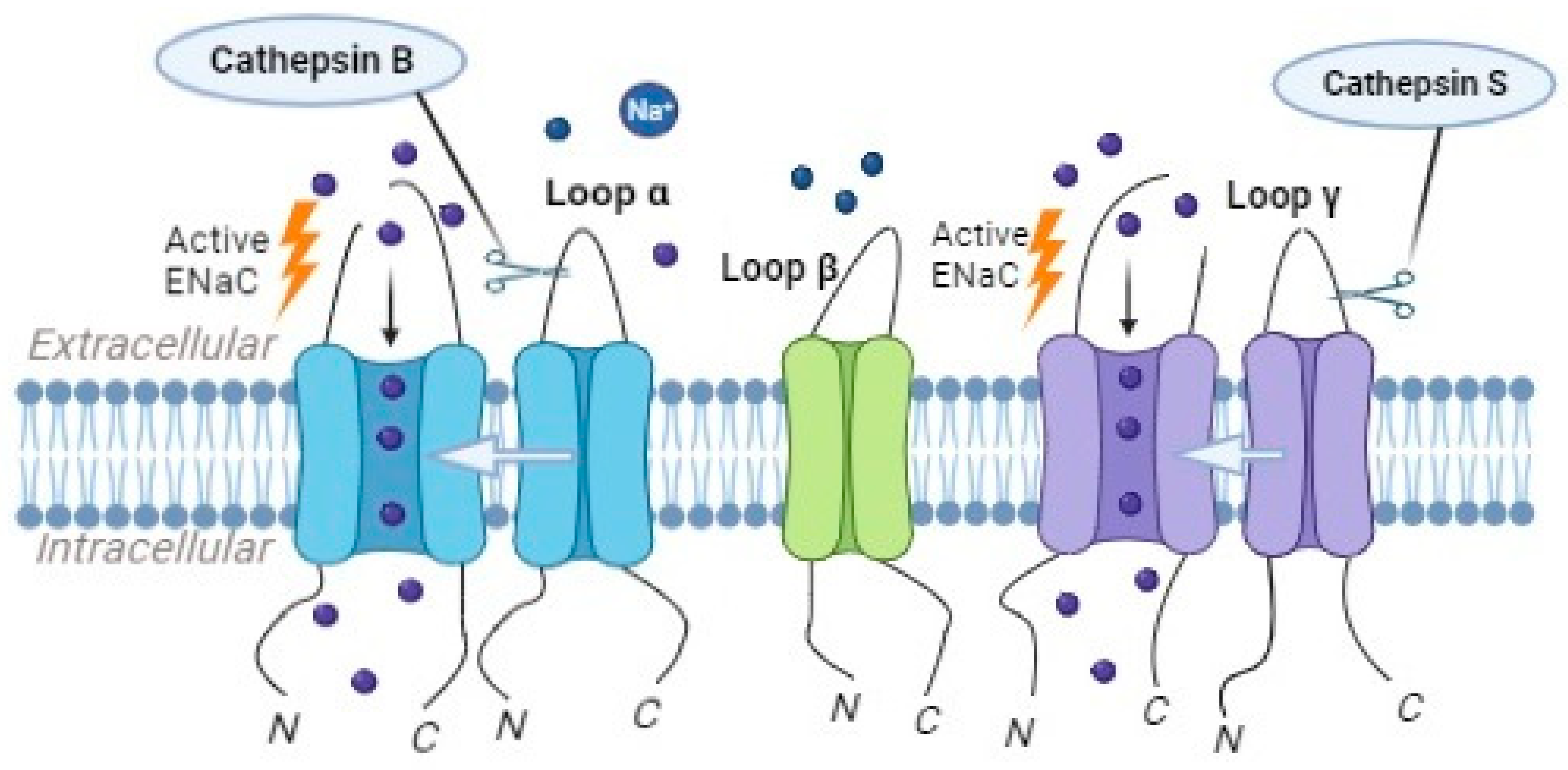
2.5. The Role of Different Cathepsins in ENaC Processing and Activation
3. Regulation of ENaC Activity by Endogenous Protease Inhibitors
3.1. Serine Protease Inhibitors
3.2. Kallikrein Inhibitors
3.3. Secretory Leukocyte Protease Inhibitor (SLPI)
3.4. Role of Cathepsins Endogenous Inhibitors on the Regulation of ENaC Activity
4. Physiological Implications of Proteolytic ENaC Activation
4.1. Blood Pressure Regulation
4.2. Pulmonary Function Regulation
4.3. Intestinal Sodium Absorption
5. Clinical Relevance and Pharmacological Prospects
6. Conclusions
Funding
Conflicts of Interest
References
- Blass, G.; Klemens, C.A.; Brands, M.W.; Palygin, O.; Staruschenko, A. Postprandial Effects on ENaC-Mediated Sodium Absorption. Sci. Rep. 2019, 9, 4296. [Google Scholar] [CrossRef]
- Shabbir, W.; Topcagic, N.; Aufy, M. Activation of autosomal recessive Pseudohypoaldosteronism1 ENaC with aldosterone. Eur. J. Pharmacol. 2021, 901, 174090. [Google Scholar] [CrossRef]
- Frindt, G.; Meyerson, J.R.; Satty, A.; Scandura, J.M.; Palmer, L.G. Expression of ENaC subunits in epithelia. J. Gen. Physiol. 2022, 154, e202213124. [Google Scholar] [CrossRef]
- Hanukoglu, I.; Hanukoglu, A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 2016, 579, 95–132. [Google Scholar] [CrossRef]
- Noreng, S.; Bharadwaj, A.; Posert, R.; Yoshioka, C.; Baconguis, I. Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife 2018, 7, e39340. [Google Scholar] [CrossRef]
- Ji, H.L.; Zhao, R.Z.; Chen, Z.X.; Shetty, S.; Idell, S.; Matalon, S. δ ENaC: A novel divergent amiloride-inhibitable sodium channel. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L1013–L1026. [Google Scholar] [CrossRef]
- Alvarez de la Rosa, D.; Canessa, C.M.; Fyfe, G.K.; Zhang, P. Structure and regulation of amiloride-sensitive sodium channels. Annu. Rev. Physiol. 2000, 62, 573–594. [Google Scholar] [CrossRef]
- Eaton, D.C.; Helms, M.N.; Koval, M.; Bao, H.F.; Jain, L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu. Rev. Physiol. 2009, 71, 403–423. [Google Scholar] [CrossRef]
- Garty, H.; Benos, D.J. Characteristics and regulatory mechanisms of the amiloride-blockable Na+ channel. Physiol. Rev. 1988, 68, 309–373. [Google Scholar] [CrossRef]
- Hummler, E.; Planes, C. Importance of ENaC-mediated sodium transport in alveolar fluid clearance using genetically-engineered mice. Cell Physiol. Biochem. 2010, 25, 63–70. [Google Scholar] [CrossRef]
- Kellenberger, S.; Schild, L. Epithelial sodium channel/degenerin family of ion channels: A variety of functions for a shared structure. Physiol. Rev. 2002, 82, 735–767. [Google Scholar] [CrossRef]
- Kleyman, T.R.; Sheng, S.; Kosari, F.; Kieber-Emmons, T. Mechanism of action of amiloride: A molecular prospective. Semin. Nephrol. 1999, 19, 524–532. [Google Scholar]
- Matalon, S.; Lazrak, A.; Jain, L.; Eaton, D.C. Invited review: Biophysical properties of sodium channels in lung alveolar epithelial cells. J. Appl. Physiol. 2002, 93, 1852–1859. [Google Scholar] [CrossRef]
- Matthay, M.A.; Clerici, C.; Saumon, G. Invited review: Active fluid clearance from the distal air spaces of the lung. J. Appl. Physiol. 2002, 93, 1533–1541. [Google Scholar] [CrossRef]
- Qadri, Y.J.; Rooj, A.K.; Fuller, C.M. ENaCs and ASICs as therapeutic targets. Am. J. Physiol. Cell Physiol. 2012, 302, C943–C965. [Google Scholar] [CrossRef]
- Rossier, B.C.; Pradervand, S.; Schild, L.; Hummler, E. Epithelial sodium channel and the control of sodium balance: Interaction between genetic and environmental factors. Annu. Rev. Physiol. 2002, 64, 877–897. [Google Scholar] [CrossRef]
- Soundararajan, R.; Pearce, D.; Hughey, R.P.; Kleyman, T.R. Role of epithelial sodium channels and their regulators in hypertension. J. Biol. Chem. 2010, 285, 30363–30369. [Google Scholar] [CrossRef]
- Stockand, J.D.; Staruschenko, A.; Pochynyuk, O.; Booth, R.E.; Silverthorn, D.U. Insight toward epithelial Na+ channel mechanism revealed by the acid-sensing ion channel 1 structure. IUBMB. Life 2008, 60, 620–628. [Google Scholar] [CrossRef]
- Teiwes, J.; Toto, R.D. Epithelial sodium channel inhibition in cardiovascular disease. A potential role for amiloride. Am. J. Hypertens. 2007, 20, 109–117. [Google Scholar] [CrossRef]
- Vuagniaux, G.; Vallet, V.; Jaeger, N.F.; Pfister, C.; Bens, M.; Farman, N.; Courtois-Coutry, N.; Vandewalle, A.; Rossier, B.C.; Hummler, E. Activation of the amiloride-sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J. Am. Soc. Nephrol. 2000, 11, 828–834. [Google Scholar] [CrossRef]
- Shabbir, W.; Topcagic, N.; Aufy, M.; Oz, M. CRISPR/Cas9 Mediated Knock Down of δ-ENaC Blunted the TNF-Induced Activation of ENaC in A549 Cells. Int. J. Mol. Sci. 2021, 22, 1858. [Google Scholar] [CrossRef]
- Lucas, R.; Yue, Q.; Alli, A.; Duke, B.J.; Al-Khalili, O.; Thai, T.L.; Hamacher, J.; Sridhar, S.; Lebedyeva, I.; Su, H.; et al. The Lectin-like Domain of TNF Increases ENaC Open Probability through a Novel Site at the Interface between the Second Transmembrane and C-terminal Domains of the α-Subunit. J. Biol. Chem. 2016, 291, 23440–23451. [Google Scholar] [CrossRef]
- Willam, A.; Aufy, M.; Tzotzos, S.; El-Malazi, D.; Poser, F.; Wagner, A.; Unterköfler, B.; Gurmani, D.; Martan, D.; Iqbal, S.M.; et al. TNF Lectin-Like Domain Restores Epithelial Sodium Channel Function in Frameshift Mutants Associated with Pseudohypoaldosteronism Type 1B. Front. Immunol. 2017, 8, 601. [Google Scholar] [CrossRef]
- Willam, A.; Aufy, M.; Tzotzos, S.; Evanzin, H.; Chytracek, S.; Geppert, S.; Fischer, B.; Fischer, H.; Pietschmann, H.; Czikora, I.; et al. Restoration of Epithelial Sodium Channel Function by Synthetic Peptides in Pseudohypoaldosteronism Type 1B Mutants. Front. Pharmacol. 2017, 8, 85. [Google Scholar] [CrossRef]
- Shabbir, W.; Tzotzos, S.; Bedak, M.; Aufy, M.; Willam, A.; Kraihammer, M.; Holzner, A.; Czikora, I.; Scherbaum-Hazemi, P.; Fischer, H.; et al. Glycosylation-dependent activation of epithelial sodium channel by solnatide. Biochem. Pharmacol. 2015, 98, 740–753. [Google Scholar] [CrossRef]
- Butterworth, M.B. Regulation of the epithelial sodium channel (ENaC) by membrane trafficking. Biochim. Biophys. Acta. 2010, 1802, 1166–1177. [Google Scholar] [CrossRef]
- Mutchler, S.M.; Kirabo, A.; Kleyman, T.R. Epithelial Sodium Channel and Salt-Sensitive Hypertension. Hypertension 2021, 77, 759–767. [Google Scholar] [CrossRef]
- Rotin, D.; Staub, O. Function and Regulation of the Epithelial Na+ Channel ENaC. Compr. Physiol. 2021, 11, 2017–2045. [Google Scholar]
- Madaio, M.P.; Czikora, I.; Kvirkvelia, N.; McMenamin, M.; Yue, Q.; Liu, T.; Toque, H.A.; Sridhar, S.; Covington, K.; Alaisami, R.; et al. The TNF-derived TIP peptide activates the epithelial sodium channel and ameliorates experimental nephrotoxic serum nephritis. Kidney Int. 2019, 95, 1359–1372. [Google Scholar] [CrossRef]
- Czikora, I.; Alli, A.A.; Sridhar, S.; Matthay, M.A.; Pillich, H.; Hudel, M.; Berisha, B.; Gorshkov, B.; Romero, M.J.; Gonzales, J.; et al. Epithelial Sodium Channel-α Mediates the Protective Effect of the TNF-Derived TIP Peptide in Pneumolysin-Induced Endothelial Barrier Dysfunction. Front. Immunol. 2017, 8, 842. [Google Scholar] [CrossRef]
- Martin-Malpartida, P.; Arrastia-Casado, S.; Farrera-Sinfreu, J.; Lucas, R.; Fischer, H.; Fischer, B.; Eaton, D.C.; Tzotzos, S.; Macias, M.J. Conformational ensemble of the TNF-derived peptide solnatide in solution. Comput. Struct. Biotechnol. J. 2022, 20, 2082–2090. [Google Scholar] [CrossRef]
- Yang, G.; Pillich, H.; White, R.; Czikora, I.; Pochic, I.; Yue, Q.; Hudel, M.; Gorshkov, B.; Verin, A.; Sridhar, S.; et al. Causes ENaC Dysfunction in Human Airway Epithelial Cells. Toxins 2018, 10, 79. [Google Scholar] [CrossRef]
- Lucas, R.; Hadizamani, Y.; Enkhbaatar, P.; Csanyi, G.; Caldwell, R.W.; Hundsberger, H.; Sridhar, S.; Lever, A.A.; Hudel, M.; Ash, D.; et al. Dichotomous Role of Tumor Necrosis Factor in Pulmonary Barrier Function and Alveolar Fluid Clearance. Front. Physiol. 2022, 12, 793251. [Google Scholar] [CrossRef]
- Czikora, I.; Alli, A.; Bao, H.F.; Kaftan, D.; Sridhar, S.; Apell, H.J.; Gorshkov, B.; White, R.; Zimmermann, A.; Wendel, A.; et al. A novel tumor necrosis factor-mediated mechanism of direct epithelial sodium channel activation. Am. J. Respir. Crit. Care Med. 2014, 190, 522–532. [Google Scholar] [CrossRef]
- Shabbir, W.; Scherbaum-Hazemi, P.; Tzotzos, S.; Fischer, B.; Fischer, H.; Pietschmann, H.; Lucas, R.; Lemmens-Gruber, R. Mechanism of action of novel lung edema therapeutic AP301 by activation of the epithelial sodium channel. Mol. Pharmacol. 2013, 84, 899–910. [Google Scholar] [CrossRef]
- Yang, G.; Hamacher, J.; Gorshkov, B.; White, R.; Sridhar, S.; Verin, A.; Chakraborty, T.; Lucas, R. The Dual Role of TNF in Pulmonary Edema. J. Cardiovasc. Dis. Res. 2010, 1, 29–36. [Google Scholar] [CrossRef]
- Hazemi, P.; Tzotzos, S.J.; Fischer, B.; Andavan, G.S.; Fischer, H.; Pietschmann, H.; Lucas, R.; Lemmens-Gruber, R. Essential structural features of TNF-α lectin-like domain derived peptides for activation of amiloride-sensitive sodium current in A549 cells. J. Med. Chem. 2010, 53, 8021–8029. [Google Scholar] [CrossRef]
- Mascher, D.; Tscherwenka, W.; Mascher, H.; Fischer, B. Sensitive determination of the peptide AP301--a motif of TNF-α--from human plasma using HPLC-MS/.MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 908, 18–22. [Google Scholar] [CrossRef]
- Schwameis, R.; Eder, S.; Pietschmann, H.; Fischer, B.; Mascher, H.; Tzotzos, S.; Fischer, H.; Lucas, R.; Zeitlinger, M.; Hermann, R. A FIM study to assess safety and exposure of inhaled single doses of AP301-A specific ENaC channel activator for the treatment of acute lung injury. J. Clin. Pharmacol. 2014, 54, 341–350. [Google Scholar] [CrossRef]
- Soundararajan, R.; Pearce, D.; Ziera, T. The role of the ENaC-regulatory complex in aldosterone-mediated sodium transport. Mol. Cell Endocrinol. 2012, 350, 242–247. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Callies, C.; Fels, J.; Oberleithner, H. The epithelial sodium channel (ENaC): Mediator of the aldosterone response in the vascular endothelium? Steroids 2010, 75, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, V.; Bertog, M.; Canonica, J.; Hummler, E.; Coleman, R.; Welling, P.A.; Korbmacher, C. Critical role of the mineralocorticoid receptor in aldosterone-dependent and aldosterone-independent regulation of ENaC in the distal nephron. Am. J. Physiol. Renal Physiol. 2021, 321, F257–F268. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Sun, W.; Gu, H.; Cheng, Y. Aldosterone-induced expression of ENaC-α is associated with activity of p65/p50 in renal epithelial cells. J. Nephrol. 2017, 30, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Tsilosani, A.; Gao, C.; Zhang, W. Aldosterone-Regulated Sodium Transport and Blood Pressure. Front. Physiol. 2022, 13, 770375. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Gao, Z.X.; Zhang, D.D.; Duan, X.P.; Terker, A.S.; Lin, D.H.; Ellison, D.H.; Wang, W.H. Effect of Angiotensin II on ENaC in the Distal Convoluted Tubule and in the Cortical Collecting Duct of Mineralocorticoid Receptor Deficient Mice. J. Am. Heart Assoc. 2020, 9, e014996. [Google Scholar] [CrossRef] [PubMed]
- Downs, C.A.; Johnson, N.M.; Coca, C.; Helms, M.N. Angiotensin II regulates δ-ENaC in human umbilical vein endothelial cells. Microvasc. Res. 2018, 116, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Zaika, O.; Mamenko, M.; Staruschenko, A.; Pochynyuk, O. Direct activation of ENaC by angiotensin II: Recent advances and new insights. Curr. Hypertens. Rep. 2013, 15, 17–24. [Google Scholar] [CrossRef]
- Jernigan, N.L.; Speed, J.; LaMarca, B.; Granger, J.P.; Drummond, H.A. Angiotensin II regulation of renal vascular ENaC proteins. Am. J. Hypertens. 2009, 22, 593–597. [Google Scholar] [CrossRef]
- Mamenko, M.; Zaika, O.; Ilatovskaya, D.V.; Staruschenko, A.; Pochynyuk, O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J. Biol. Chem. 2012, 287, 660–671. [Google Scholar] [CrossRef]
- Bankir, L.; Bichet, D.G.; Bouby, N. Vasopressin V2 receptors, ENaC, and sodium reabsorption: A risk factor for hypertension? Am. J. Physiol. Renal Physiol. 2010, 299, F917–F928. [Google Scholar] [CrossRef]
- Roos, K.P.; Bugaj, V.; Mironova, E.; Stockand, J.D.; Ramkumar, N.; Rees, S.; Kohan, D.E. Adenylyl cyclase VI mediates vasopressin-stimulated ENaC activity. J. Am. Soc. Nephrol. 2013, 24, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Vallet, V.; Chraibi, A.; Gaeggeler, H.P.; Horisberger, J.D.; Rossier, B.C. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 1997, 389, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Anand, D.; Hummler, E.; Rickman, O.J. ENaC activation by proteases. Acta Physiol. 2022, 235, e13811. [Google Scholar] [CrossRef] [PubMed]
- Kleyman, T.R.; Myerburg, M.M.; Hughey, R.P. Regulation of ENaCs by proteases: An increasingly complex story. Kidney Int. 2006, 70, 1391–1392. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, P.H.; Butterworth, M.B. Proteases, cystic fibrosis and the epithelial sodium channel (ENaC). Cell Tissue Res. 2013, 351, 309–323. [Google Scholar] [CrossRef]
- Gentzsch, M.; Dang, H.; Dang, Y.; Garcia-Caballero, A.; Suchindran, H.; Boucher, R.C.; Stutts, M.J. The cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelial Na+ channel. J. Biol. Chem. 2010, 285, 32227–32232. [Google Scholar] [CrossRef]
- Aufy, M.; Abdelaziz, R.F.; Hussein, A.M.; Topcagic, N.; Shamroukh, H.; Abdel-Maksoud, M.A.; Salem, T.Z.; Studenik, C.R. Impact of Enniatin B and Beauvericin on Lysosomal Cathepsin B Secretion and Apoptosis Induction. Int. J. Mol. Sci. 2023, 24, 2030. [Google Scholar] [CrossRef]
- Abdelaziz, R.F.; Hussein, A.M.; Kotob, M.H.; Weiss, C.; Chelminski, K.; Stojanovic, T.; Studenik, C.R.; Aufy, M. Enhancement of Radiation Sensitivity by Cathepsin L Suppression in Colon Carcinoma Cells. Int. J. Mol. Sci. 2023, 24, 17106. [Google Scholar] [CrossRef]
- Abdelaziz, R.F.; Hussein, A.M.; Kotob, M.H.; Weiss, C.; Chelminski, K.; Studenik, C.R.; Aufy, M. The Significance of Cathepsin B in Mediating Radiation Resistance in Colon Carcinoma Cell Line (Caco-2). Int. J. Mol. Sci. 2023, 24, 16146. [Google Scholar] [CrossRef]
- Sure, F.; Bertog, M.; Afonso, S.; Diakov, A.; Rinke, R.; Madej, M.G.; Wittmann, S.; Gramberg, T.; Korbmacher, C.; Ilyaskin, A.V. Transmembrane serine protease 2 (TMPRSS2) proteolytically activates the epithelial sodium channel (ENaC) by cleaving the channel’s γ-subunit. J. Biol. Chem. 2022, 298, 102004. [Google Scholar] [CrossRef]
- Haerteis, S.; Krappitz, M.; Bertog, M.; Krappitz, A.; Baraznenok, V.; Henderson, I.; Lindström, E.; Murphy, J.E.; Bunnett, N.W.; Korbmacher, C. Proteolytic activation of the epithelial sodium channel (ENaC) by the cysteine protease cathepsin-S. Pflugers Arch. 2012, 464, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, D.S.; Yu, L.; Ghazi, Z.M.; Thai, T.L.; Al-Khalili, O.; Ma, H.P.; Eaton, D.C.; Alli, A.A. ENaC activity is regulated by calpain-2 proteolysis of MARCKS proteins. Am. J. Physiol. Cell Physiol. 2017, 313, C42–C53. [Google Scholar] [CrossRef] [PubMed]
- Carattino, M.D.; Mueller, G.M.; Palmer, L.G.; Frindt, G.; Rued, A.C.; Hughey, R.P.; Kleyman, T.R. Prostasin interacts with the epithelial Na+ channel and facilitates cleavage of the γ-subunit by a second protease. Am. J. Physiol. Renal Physiol. 2014, 307, F1080–F1087. [Google Scholar] [CrossRef] [PubMed]
- Zachar, R.M.; Skjødt, K.; Marcussen, N.; Walter, S.; Toft, A.; Nielsen, M.R.; Jensen, B.L.; Svenningsen, P. The epithelial sodium channel γ-subunit is processed proteolytically in human kidney. J. Am. Soc. Nephrol. 2015, 26, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Tomita, K. Proteolytic activation of the epithelial sodium channel and therapeutic application of a serine protease inhibitor for the treatment of salt-sensitive hypertension. Clin. Exp. Nephrol. 2012, 16, 44–48. [Google Scholar] [CrossRef]
- Svenningsen, P.; Friis, U.G.; Bistrup, C.; Buhl, K.B.; Jensen, B.L.; Skøtt, O. Physiological regulation of epithelial sodium channel by proteolysis. Curr. Opin. Nephrol. Hypertens. 2011, 20, 529–533. [Google Scholar] [CrossRef]
- Larionov, A.; Dahlke, E.; Kunke, M.; Zanon Rodriguez, L.; Schiessl, I.M.; Magnin, J.L.; Kern, U.; Alli, A.A.; Mollet, G.; Schilling, O.; et al. Cathepsin B increases ENaC activity leading to hypertension early in nephrotic syndrome. J. Cell Mol. Med. 2019, 23, 6543–6553. [Google Scholar] [CrossRef]
- Alli, A.A.; Song, J.Z.; Al-Khalili, O.; Bao, H.F.; Ma, H.P.; Alli, A.A.; Eaton, D.C. Cathepsin B is secreted apically from Xenopus 2F3 cells and cleaves the epithelial sodium channel (ENaC) to increase its activity. J. Biol. Chem. 2012, 287, 30073–30083. [Google Scholar] [CrossRef]
- Kota, P.; Gentzsch, M.; Dang, Y.L.; Boucher, R.C.; Stutts, M.J. The N terminus of α-ENaC mediates ENaC cleavage and activation by furin. J. Gen. Physiol. 2018, 150, 1179–1187. [Google Scholar] [CrossRef]
- Sheng, S.; Carattino, M.D.; Bruns, J.B.; Hughey, R.P.; Kleyman, T.R. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am. J. Physiol. Renal Physiol. 2006, 290, F1488–F1496. [Google Scholar] [CrossRef]
- Harris, M.; Garcia-Caballero, A.; Stutts, M.J.; Firsov, D.; Rossier, B.C. Preferential assembly of epithelial sodium channel (ENaC) subunits in Xenopus oocytes: Role of furin-mediated endogenous proteolysis. J. Biol. Chem. 2008, 283, 7455–7463. [Google Scholar] [CrossRef] [PubMed]
- Douglas, L.E.J.; Reihill, J.A.; Ho, M.W.Y.; Axten, J.M.; Campobasso, N.; Schneck, J.L.; Rendina, A.R.; Wilcoxen, K.M.; Martin, S.L. A highly selective, cell-permeable furin inhibitor BOS-318 rescues key features of cystic fibrosis airway disease. Cell Chem. Biol. 2022, 29, 947–957.e8. [Google Scholar] [CrossRef] [PubMed]
- Douglas, L.E.J.; Reihill, J.A.; Montgomery, B.M.; Martin, S.L. Furin as a therapeutic target in cystic fibrosis airways disease. Eur. Respir. Rev. 2023, 32, 220256. [Google Scholar] [CrossRef] [PubMed]
- Reihill, J.A.; Walker, B.; Hamilton, R.A.; Ferguson, T.E.; Elborn, J.S.; Stutts, M.J.; Harvey, B.J.; Saint-Criq, V.; Hendrick, S.M.; Martin, S.L. Inhibition of protease-epithelial sodium channel signaling improves mucociliary function in cystic fibrosis airways. Am. J. Respir. Crit. Care Med. 2016, 194, 701710. [Google Scholar] [CrossRef] [PubMed]
- Picard, N.; Eladari, D.; El Moghrabi, S.; Planès, C.; Bourgeois, S.; Houillier, P.; Wang, Q.; Burnier, M.; Deschenes, G.; Knepper, M.A.; et al. Defective ENaC processing and function in tissue kallikrein-deficient mice. J. Biol. Chem. 2008, 283, 4602–4611. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Chao, J.; Palmer, L.G. Tissue kallikrein activation of the epithelial Na channel. Am. J. Physiol. Renal Physiol. 2012, 303, F540–F550. [Google Scholar] [CrossRef] [PubMed]
- Gondzik, V.; Weber, W.M.; Awayda, M.S. Coupling of epithelial Na+ and Cl- channels by direct and indirect activation by serine proteases. Am. J. Physiol. Cell Physiol. 2012, 303, C936–C946. [Google Scholar] [CrossRef]
- Antalis, T.M.; Buzza, M.S. Extracellular: Plasma Membrane Proteases—Serine Proteases. Encycl. Cell Biol. 2016, 650–660. [Google Scholar] [CrossRef]
- Miller, G.S.; List, K. The matriptase-prostasin proteolytic cascade in epithelial development and pathology. Cell Tissue Res. 2013, 351, 245–253. [Google Scholar] [CrossRef]
- Aggarwal, S.; Dabla, P.K.; Arora, S. Prostasin: An Epithelial Sodium Channel Regulator. J. Biomark. 2013, 2013, 179864. [Google Scholar] [CrossRef]
- Chen, L.M.; Skinner, M.L.; Kauffman, S.W.; Chao, J.; Chao, L.; Thaler, C.D.; Chai, K.X. Prostasin is a glycosylphosphatidylinositol-anchored active serine protease. J. Biol. Chem. 2001, 276, 21434–21442. [Google Scholar] [CrossRef] [PubMed]
- Keragala, C.B.; Medcalf, R.L. Plasminogen: An enigmatic zymogen. Blood 2021, 137, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Planès, C.; Randrianarison, N.H.; Charles, R.; Frateschi, S.; Cluzeaud, F.; Vuagniaux, G.; Soler, P.; Clerici, C.; Rossier, B.C.; Hummler, E. ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1. EMBO Mol. Med. 2010, 2, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Malsure, S.; Wang, Q.; Charles, R.P.; Sergi, C.; Perrier, R.; Christensen, B.M.; Maillard, M.; Rossier, B.C.; Hummler, E. Colon-specific deletion of epithelial sodium channel causes sodium loss and aldosterone resistance. J. Am. Soc. Nephrol. 2014, 25, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Frateschi, S.; Keppner, A.; Malsure, S.; Iwaszkiewicz, J.; Sergi, C.; Merillat, A.M.; Fowler-Jaeger, N.; Randrianarison, N.; Planès, C.; Hummler, E. Mutations of the serine protease CAP1/Prss8 lead to reduced embryonic viability, skin defects, and decreased ENaC activity. Am. J. Pathol. 2012, 181, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Leyvraz, C.; Charles, R.P.; Rubera, I.; Guitard, M.; Rotman, S.; Breiden, B.; Sandhoff, K.; Hummler, E. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J. Cell Biol. 2005, 170, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Crisante, G.; Battista, L.; Iwaszkiewicz, J.; Nesca, V.; Mérillat, A.M.; Sergi, C.; Zoete, V.; Frateschi, S.; Hummler, E. The CAP1/Prss8 catalytic triad is not involved in PAR2 activation and protease nexin-1 (PN-1) inhibition. FASEB J. 2014, 28, 4792–4805. [Google Scholar] [CrossRef]
- Frateschi, S.; Camerer, E.; Crisante, G.; Rieser, S.; Membrez, M.; Charles, R.P.; Beermann, F.; Stehle, J.C.; Breiden, B.; Sandhoff, K.; et al. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat. Commun. 2011, 2, 161. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Cell surface remodeling by plasmin: A new function for an old enzyme. J. Biomed. Biotechnol. 2012, 2012, 564259. [Google Scholar] [CrossRef]
- Deng, Q.; Kakizoe, Y.; Iwata, Y.; Nakagawa, T.; Miyasato, Y.; Nakagawa, M.; Nishiguchi, K.; Nagayoshi, Y.; Adachi, M.; Narita, Y.; et al. The serine protease plasmin plays detrimental roles in epithelial sodium channel activation and podocyte injury in Dahl salt-sensitive rats. Hypertens. Res. 2023, 46, 50–62. [Google Scholar] [CrossRef]
- Deschênes, G.; Wittner, M.; Stefano, A.; Jounier, S.; Doucet, A. Collecting duct is a site of sodium retention in PAN nephrosis: A rationale for amiloride therapy. J. Am. Soc. Nephrol. 2001, 12, 598–601. [Google Scholar] [PubMed]
- Deschênes, G.; Guigonis, V.; Doucet, A. Molecular mechanism of edema formation in nephrotic syndrome [in French]. Arch Pediatr. 2004, 11, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Svenningsen, P.; Bistrup, C.; Friis, U.G.; Bertog, M.; Haerteis, S.; Krueger, B.; Stubbe, J.; Jensen, O.N.; Thiesson, H.C.; Uhrenholt, T.R.; et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J. Am. Soc. Nephrol. 2009, 20, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Gonzales, E.C.; Shayestehfar, B.; Barton, C.H. Plasma levels and urinary excretion of fibrinolytic and protease inhibitory proteins in nephrotic syndrome. J. Lab. Clin. Med. 1994, 124, 118–124. [Google Scholar] [PubMed]
- Passero, C.J.; Mueller, G.M.; Rondon-Berrios, H.; Tofovic, S.P.; Hughey, R.P.; Kleyman, T.R. Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J. Biol. Chem. 2008, 283, 36586–36591. [Google Scholar] [CrossRef]
- Svenningsen, P.; Uhrenholt, T.R.; Palarasah, Y.; Skjødt, K.; Jensen, B.L.; Skøtt, O. Prostasin-dependent activation of epithelial Na+ channels by low plasmin concentrations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1733–R1741. [Google Scholar] [CrossRef]
- Gadau, J.; Peters, H.; Kastner, C.; Kühn, H.; Nieminen-Kelhä, M.; Khadzhynov, D.; Krämer, S.; Castrop, H.; Bachmann, S.; Theilig, F. Mechanisms of tubular volume retention in immune-mediated glomerulonephritis. Kidney Int. 2009, 75, 699–710. [Google Scholar] [CrossRef]
- Kim, S.W.; Wang, W.; Nielsen, J.; Praetorius, J.; Kwon, T.H.; Knepper, M.A.; Frøkiaer, J.; Nielsen, S. Increased expression and apical targeting of renal ENaC subunits in puromycin aminonucleoside-induced nephrotic syndrome in rats. Am. J. Physiol. Renal Physiol. 2004, 286, F922–F935. [Google Scholar] [CrossRef]
- Buhl, K.B.; Friis, U.G.; Svenningsen, P.; Gulaveerasingam, A.; Ovesen, P.; Frederiksen-Møller, B.; Jespersen, B.; Bistrup, C.; Jensen, B.L. Urinary plasmin activates collecting duct ENaC current in preeclampsia. Hypertension 2012, 60, 1346–1351. [Google Scholar] [CrossRef]
- Tan, C.D.; Hobbs, C.; Sameni, M.; Sloane, B.F.; Stutts, M.J.; Tarran, R. Cathepsin B contributes to Na+ hyperabsorption in cystic fibrosis airway epithelial cultures. J. Physiol. 2014, 592, 5251–5268. [Google Scholar] [CrossRef]
- Evans, T.I.; Joo, N.S.; Keiser, N.W.; Yan, Z.; Tyler, S.R.; Xie, W.; Zhang, Y.; Hsiao, J.J.; Cho, H.J.; Wright, M.E.; et al. Glandular Proteome Identifies Antiprotease Cystatin C as a Critical Modulator of Airway Hydration and Clearance. Am. J. Respir. Cell Mol. Biol. 2016, 54, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Coote, K.; Atherton-Watson, H.C.; Sugar, R.; Young, A.; MacKenzie-Beevor, A.; Gosling, M.; Bhalay, G.; Bloomfield, G.; Dunstan, A.; Bridges, R.J.; et al. Camostat attenuates airway epithelial sodium channel function in vivo through the inhibition of a channel-activating protease. J. Pharmacol. Exp. Ther. 2009, 329, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Essigke, D.; Ilyaskin, A.V.; Wörn, M.; Bohnert, B.N.; Xiao, M.; Daniel, C.; Amann, K.; Birkenfeld, A.L.; Szabo, R.; Bugge, T.H.; et al. Zymogen-locked mutant prostasin (Prss8) leads to incomplete proteolytic activation of the epithelial sodium channel (ENaC) and severely compromises triamterene tolerance in mice. Acta. Physiol. 2021, 232, e13640. [Google Scholar] [CrossRef] [PubMed]
- Koda, A.; Wakida, N.; Toriyama, K.; Yamamoto, K.; Iijima, H.; Tomita, K.; Kitamura, K. Urinary prostasin in humans: Relationships among prostasin, aldosterone and epithelial sodium channel activity. Hypertens. Res. 2009, 32, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Lugo, C.I.; Liu, L.P.; Bala, N.; Morales, A.G.; Gholam, M.F.; Abchee, J.C.; Elmoujahid, N.; Elshikha, A.S.; Avdiaj, R.; Searcy, L.A.; et al. Human Alpha-1 Antitrypsin Attenuates ENaC and MARCKS and Lowers Blood Pressure in Hypertensive Diabetic db/db Mice. Biomolecules 2022, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Rhaleb, N.E.; Yang, X.P.; Carretero, O.A. The kallikrein-kinin system as a regulator of cardiovascular and renal function. Compr. Physiol. 2011, 1, 971–993. [Google Scholar]
- Jovov, B.; Wills, N.K.; Donaldson, P.J.; Lewis, S.A. Vectorial secretion of a kallikrein-like enzyme by cultured renal cells. I. General properties. Am. J. Physiol. 1990, 259, C869–C882. [Google Scholar] [CrossRef]
- Margolius, H.S.; Chao, J. Amiloride inhibits mammalian renal kallikrein and a kallikrein-like enzyme from toad bladder and skin. J. Clin. Investig. 1980, 65, 1343–1350. [Google Scholar] [CrossRef]
- Bridges, R.J.; Newton, B.B.; Pilewski, J.M.; Devor, D.C.; Poll, C.T.; Hall, R.L. Na+ transport in normal and CF human bronchial epithelial cells is inhibited by BAY 39-9437. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L16–L23. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Hirsh, A.; Li, D.C.; Holloway, G.; Chao, J.; Boucher, R.C.; Gabriel, S.E. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 2002, 277, 8338–8345. [Google Scholar] [CrossRef]
- Liu, L.; Hering-Smith, K.S.; Schiro, F.R.; Hamm, L.L. Serine protease activity in m-1 cortical collecting duct cells. Hypertension 2002, 39, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Nugteren, S.; Samsom, J.N. Secretory Leukocyte Protease Inhibitor (SLPI) in mucosal tissues: Protects against inflammation, but promotes cancer. Cytokine Growth Factor Rev. 2021, 59, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Ferris, P.; Brown, R.; Delaney, R.; Dougan, C.; Doherty, D.; Small, D.; Mall, M.; Mcauley, D.; Connolly, B.; Weldon, S. Genetic deletion of SLPI results in increased protease activity and mucus clearance in a murine model of chronic lung disease. Eur. Respir. J. 2022, 60, 2558. [Google Scholar]
- Majchrzak-Gorecka, M.; Majewski, P.; Grygier, B.; Murzyn, K.; Cichy, J. Secretory leukocyte protease inhibitor (SLPI), a multifunctional protein in the host defense response. Cytokine Growth Factor Rev. 2016, 28, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.F.; Mitaera, T.; Fronius, M. COVID-19 and Liquid Homeostasis in the Lung-A Perspective through the Epithelial Sodium Channel (ENaC) Lens. Cells 2022, 11, 1801. [Google Scholar] [CrossRef] [PubMed]
- Negussie, A.B.; Dell, A.C.; Davis, B.A.; Geibel, J.P. Colonic Fluid and Electrolyte Transport 2022: An Update. Cells 2022, 11, 1712. [Google Scholar] [CrossRef] [PubMed]
- Adebamiro, A.; Cheng, Y.; Rao, U.S.; Danahay, H.; Bridges, R.J. A segment of gamma ENaC mediates elastase activation of Na+ transport. J. Gen. Physiol. 2007, 130, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Diakov, A.; Bera, K.; Mokrushina, M.; Krueger, B.; Korbmacher, C. Cleavage in the {gamma}-subunit of the epithelial sodium channel (ENaC) plays an important role in the proteolytic activation of near-silent channels. J. Physiol. 2008, 586, 4587–4608. [Google Scholar] [CrossRef]
- Haerteis, S.; Krueger, B.; Korbmacher, C.; Rauh, R. The delta-subunit of the epithelial sodium channel (ENaC) enhances channel activity and alters proteolytic ENaC activation. J. Biol. Chem. 2009, 284, 29024–29040. [Google Scholar] [CrossRef]
- Hughey, R.P.; Carattino, M.D.; Kleyman, T.R. Role of proteolysis in the activation of epithelial sodium channels. Curr. Opin. Nephrol. Hypertens. 2007, 16, 444–450. [Google Scholar] [CrossRef]
- Planès, C.; Caughey, G.H. Regulation of the epithelial Na+ channel by peptidases. Curr. Top. Dev. Biol. 2007, 78, 23–46. [Google Scholar] [PubMed]
- Artunc, F.; Bohnert, B.N.; Schneider, J.C.; Staudner, T.; Sure, F.; Ilyaskin, A.V.; Wörn, M.; Essigke, D.; Janessa, A.; Nielsen, N.V.; et al. Proteolytic activation of the epithelial sodium channel (ENaC) by factor VII activating protease (FSAP) and its relevance for sodium retention in nephrotic mice. Pflugers Arch. 2022, 474, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.J.; Reidel, B.; Tan, C.D.; Ghosh, A.; Alexis, N.E.; Donaldson, S.H.; Kesimer, M.; Ribeiro, C.M.P.; Tarran, R. SPLUNC1 degradation by the cystic fibrosis mucosal environment drives airway surface liquid dehydration. Eur. Respir. J. 2018, 52, 1800668. [Google Scholar] [CrossRef] [PubMed]
- Zachar, R.; Mikkelsen, M.K.; Skjødt, K.; Marcussen, N.; Zamani, R.; Jensen, B.L.; Svenningsen, P. The epithelial Na+ channel α- and γ-subunits are cleaved at predicted furin-cleavage sites, glycosylated and membrane associated in human kidney. Pflugers Arch. 2019, 471, 1383–1396. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protease | Function in ENaC Processing | Examples of Diseases/Conditions |
|---|---|---|
| Furin | Cleaves the pro-peptide of ENaC, allowing it to mature and be transported to the cell surface | Hypertension, Liddle syndrome |
| Cathepsin B | Enhances ENaC activity by cleaving the α-subunit | Asthma, Cystic fibrosis |
| Cathepsin S | Cleaves the γ-subunit, leading to increased ENaC activity | Cystic fibrosis, Lung diseases |
| Plasmin | Activates ENaC by cleaving the α-subunit | Hypertension, Edema |
| Kallikrein | Cleaves the γ-subunit, promoting ENaC activation | Hypertension, Edema |
| Trypsin | Activates ENaC by cleaving the α-subunit | Cystic fibrosis, Edema |
| Prostasin | Activates ENaC by cleaving the γ-subunit | Hypertension, Cystic fibrosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aufy, M.; Hussein, A.M.; Stojanovic, T.; Studenik, C.R.; Kotob, M.H. Proteolytic Activation of the Epithelial Sodium Channel (ENaC): Its Mechanisms and Implications. Int. J. Mol. Sci. 2023, 24, 17563. https://doi.org/10.3390/ijms242417563
Aufy M, Hussein AM, Stojanovic T, Studenik CR, Kotob MH. Proteolytic Activation of the Epithelial Sodium Channel (ENaC): Its Mechanisms and Implications. International Journal of Molecular Sciences. 2023; 24(24):17563. https://doi.org/10.3390/ijms242417563
Chicago/Turabian StyleAufy, Mohammed, Ahmed M. Hussein, Tamara Stojanovic, Christian R. Studenik, and Mohamed H. Kotob. 2023. "Proteolytic Activation of the Epithelial Sodium Channel (ENaC): Its Mechanisms and Implications" International Journal of Molecular Sciences 24, no. 24: 17563. https://doi.org/10.3390/ijms242417563
APA StyleAufy, M., Hussein, A. M., Stojanovic, T., Studenik, C. R., & Kotob, M. H. (2023). Proteolytic Activation of the Epithelial Sodium Channel (ENaC): Its Mechanisms and Implications. International Journal of Molecular Sciences, 24(24), 17563. https://doi.org/10.3390/ijms242417563