
Autism Spectrum Disorder: Neurodevelopmental Risk Factors, Biological Mechanism, and Precision Therapy

Abstract
:1. Introduction
2. Technological Advances in Animal Models of Autism
2.1. Genetic Animal Models
2.2. Syndromic ASD Animal Models Caused by CNVs
2.3. Idiopathic Animal Model
2.4. Environmental Models
3. Importance of Multidisciplinary Assessment of Animal Models of Autism
3.1. ASD Core Behavioral Testing
3.2. Neuropathology
3.3. Neuroimaging
3.4. Neurochemistry
4. Biological Mechanisms and Neural Circuitry of ASD
4.1. Activity-Dependent Gene Transcription
4.2. mRNA Translation and Non-Coding RNA
4.3. Synaptic Signaling Pathway
4.4. Epigenetic Post-Translational Modifications
4.5. Immunology and Neuroinflammation
4.6. Internal Neural Loops in the Brain
5. Targeting Molecules in Neural Circuitry May Be the Prospect of Autism Spectrum Disorder Treatment
5.1. Nonpharmacological Therapies
5.2. Pharmacological Therapies
5.3. Cell Therapies
5.4. Neurotransmitter Manipulations
5.5. Targeted Translation and Epigenetic Regulation
5.6. Other Biological Targets
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Malwane, M.I.; Nguyen, E.B.; Trejo, S., Jr.; Kim, E.Y.; Cucalon-Calderon, J.R. A Delayed Diagnosis of Autism Spectrum Disorder in the Setting of Complex Attention Deficit Hyperactivity Disorder. Cureus 2022, 14, e258252022. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chen, L.; Dai, Y.; Jia, F.; Hao, Y.; Li, L.; Zhang, J.; Wu, L.; Ke, X.; Yi, M.; et al. Vitamin A Status Is More Commonly Associated with Symptoms and Neurodevelopment in Boys with Autism Spectrum Disorders-A Multicenter Study in China. Front. Nutr. 2022, 9, 851980. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.; Allison, C.; Baron-Cohen, S. Autistic adults have poorer quality healthcare and worse health based on self-report data. Mol. Autism 2022, 13, 23. [Google Scholar] [CrossRef]
- Hofer, J.; Hoffmann, F.; Kamp-Becker, I.; Poustka, L.; Roessner, V.; Stroth, S.; Wolff, N.; Bachmann, C.J. Pathways to a diagnosis of autism spectrum disorder in Germany: A survey of parents. Child Adolesc. Psychiatry Ment. Health 2019, 13, 16. [Google Scholar] [CrossRef]
- Mintal, K.; Toth, A.; Hormay, E.; Kovacs, A.; Laszlo, K.; Bufa, A.; Marosvolgyi, T.; Kocsis, B.; Varga, A.; Vizvari, Z.; et al. Novel probiotic treatment of autism spectrum disorder associated social behavioral symptoms in two rodent models. Sci. Rep. 2022, 12, 5399. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, Y.X.; Gu, L.J.; Cheng, Y. Understanding autism spectrum disorders with animal models: Applications, insights, and perspectives. Zool. Res. 2021, 42, 800–824. [Google Scholar] [CrossRef]
- Born, G.; Grayton, H.M.; Langhorst, H.; Dudanova, I.; Rohlmann, A.; Woodward, B.W.; Collier, D.A.; Fernandes, C.; Missler, M. Genetic targeting of NRXN2 in mice unveils role in excitatory cortical synapse function and social behaviors. Front. Synaptic Neurosci. 2015, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Grayton, H.M.; Missler, M.; Collier, D.A.; Fernandes, C. Altered social behaviours in neurexin 1alpha knockout mice resemble core symptoms in neurodevelopmental disorders. PLoS ONE 2013, 8, e67114. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Sudhof, T.C. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Radyushkin, K.; Hammerschmidt, K.; Boretius, S.; Varoqueaux, F.; El-Kordi, A.; Ronnenberg, A.; Winter, D.; Frahm, J.; Fischer, J.; Brose, N.; et al. Neuroligin-3-deficient mice: Model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 2009, 8, 416–425. [Google Scholar] [CrossRef]
- Isshiki, M.; Tanaka, S.; Kuriu, T.; Tabuchi, K.; Takumi, T.; Okabe, S. Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat. Commun. 2014, 5, 4742. [Google Scholar] [CrossRef] [Green Version]
- Ellegood, J.; Lerch, J.P.; Henkelman, R.M. Brain abnormalities in a Neuroligin3 R451C knockin mouse model associated with autism. Autism Res. 2011, 4, 368–376. [Google Scholar] [CrossRef]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef] [Green Version]
- Schmeisser, M.J.; Ey, E.; Wegener, S.; Bockmann, J.; Stempel, A.V.; Kuebler, A.; Janssen, A.L.; Udvardi, P.T.; Shiban, E.; Spilker, C.; et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 2012, 486, 256–260. [Google Scholar] [CrossRef]
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.M.; Kajiwara, Y.; Buxbaum, J.D.; et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 2015, 11, 1400–1413. [Google Scholar] [CrossRef] [Green Version]
- Jacot-Descombes, S.; Keshav, N.U.; Dickstein, D.L.; Wicinski, B.; Janssen, W.G.M.; Hiester, L.L.; Sarfo, E.K.; Warda, T.; Fam, M.M.; Harony-Nicolas, H.; et al. Altered synaptic ultrastructure in the prefrontal cortex of Shank3-deficient rats. Mol. Autism 2020, 11, 89. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef]
- Hampson, D.R.; Blatt, G.J. Autism spectrum disorders and neuropathology of the cerebellum. Front. Neurosci. 2015, 9, 420. [Google Scholar] [CrossRef]
- Hevner, R.F. Brain overgrowth in disorders of RTK-PI3K-AKT signaling: A mosaic of malformations. Semin. Perinatol. 2015, 39, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, R.M.; Kordich, J.J.; Chan, C.Y.; Sasi, N.K.; Celano, S.L.; Sisson, K.A.; Van Baren, M.; Kortus, M.G.; Aguiar, D.J.; Martin, K.R.; et al. Chemical Biology Screening Identifies a Vulnerability to Checkpoint Kinase Inhibitors in TSC2-Deficient Renal Angiomyolipomas. Front. Oncol. 2022, 12, 852859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Marro, S.G.; Zhang, Y.; Arendt, K.L.; Patzke, C.; Zhou, B.; Fair, T.; Yang, N.; Sudhof, T.C.; Wernig, M.; et al. The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci. Transl. Med. 2018, 10, eaar4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, D.; Usdin, K. Molecular analysis of FMR1 alleles for fragile X syndrome diagnosis and patient stratification. Expert Rev. Mol. Diagn. 2020, 20, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Lannom, M.C.; Nielsen, J.; Nawaz, A.; Shilikbay, T.; Ceman, S. FMRP and MOV10 regulate Dicer1 expression and dendrite development. PLoS ONE 2021, 16, e0260005. [Google Scholar] [CrossRef]
- Nageshappa, S.; Carromeu, C.; Trujillo, C.A.; Mesci, P.; Espuny-Camacho, I.; Pasciuto, E.; Vanderhaeghen, P.; Verfaillie, C.M.; Raitano, S.; Kumar, A.; et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol. Psychiatry 2016, 21, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Marchetto, M.C.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Rietveld, L.; Stuss, D.P.; McPhee, D.; Delaney, K.R. Genotype-specific effects of Mecp2 loss-of-function on morphology of Layer V pyramidal neurons in heterozygous female Rett syndrome model mice. Front. Cell. Neurosci. 2015, 9, 145. [Google Scholar] [CrossRef] [Green Version]
- Bach, S.; Shovlin, S.; Moriarty, M.; Bardoni, B.; Tropea, D. Rett Syndrome and Fragile X Syndrome: Different Etiology with Common Molecular Dysfunctions. Front. Cell. Neurosci. 2021, 15, 764761. [Google Scholar] [CrossRef]
- Wilkinson, B.; Grepo, N.; Thompson, B.L.; Kim, J.; Wang, K.; Evgrafov, O.V.; Lu, W.; Knowles, J.A.; Campbell, D.B. The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl. Psychiatry 2015, 5, e568. [Google Scholar] [CrossRef]
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 2017, 19, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Gompers, A.L.; Su-Feher, L.; Ellegood, J.; Copping, N.A.; Riyadh, M.A.; Stradleigh, T.W.; Pride, M.C.; Schaffler, M.D.; Wade, A.A.; Catta-Preta, R.; et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci. 2017, 20, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; de la Iglesia, H.O.; Catterall, W.A. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Llamosas, N.; Arora, V.; Vij, R.; Kilinc, M.; Bijoch, L.; Rojas, C.; Reich, A.; Sridharan, B.; Willems, E.; Piper, D.R.; et al. SYNGAP1 Controls the Maturation of Dendrites, Synaptic Function, and Network Activity in Developing Human Neurons. J. Neurosci. 2020, 40, 7980–7994. [Google Scholar] [CrossRef]
- Berryer, M.H.; Chattopadhyaya, B.; Xing, P.; Riebe, I.; Bosoi, C.; Sanon, N.; Antoine-Bertrand, J.; Levesque, M.; Avoli, M.; Hamdan, F.F.; et al. Decrease of SYNGAP1 in GABAergic cells impairs inhibitory synapse connectivity, synaptic inhibition and cognitive function. Nat. Commun. 2016, 7, 13340. [Google Scholar] [CrossRef] [Green Version]
- Oz, S.; Ivashko-Pachima, Y.; Gozes, I. The ADNP derived peptide, NAP modulates the tubulin pool: Implication for neurotrophic and neuroprotective activities. PLoS ONE 2012, 7, e51458. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, D.; Liao, H.; Zhang, S.; Guo, W.; Chen, L.; Lu, L.; Huang, T.; Cai, Y.D. Exploring the Genomic Patterns in Human and Mouse Cerebellums Via Single-Cell Sequencing and Machine Learning Method. Front. Genet. 2022, 13, 857851. [Google Scholar] [CrossRef]
- Manjarrez, J.R.; Sun, L.; Prince, T.; Matts, R.L. Hsp90-dependent assembly of the DBC2/RhoBTB2-Cullin3 E3-ligase complex. PLoS ONE 2014, 9, e90054. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.K.; Yoon, J.H.; Song, M. Autism Spectrum Disorder Genes: Disease-Related Networks and Compensatory Strategies. Front. Mol. Neurosci. 2022, 15, 922840. [Google Scholar] [CrossRef]
- Bao, Z.; Fang, K.; Miao, Z.; Li, C.; Yang, C.; Yu, Q.; Zhang, C.; Miao, Z.; Liu, Y.; Ji, J. Human Cerebral Organoid Implantation Alleviated the Neurological Deficits of Traumatic Brain Injury in Mice. Oxidative Med. Cell. Longev. 2021, 2021, 6338722. [Google Scholar] [CrossRef]
- Echevarria-Cooper, D.M.; Hawkins, N.A.; Misra, S.N.; Huffman, A.M.; Thaxton, T.; Thompson, C.H.; Ben-Shalom, R.; Nelson, A.D.; Lipkin, A.M.; George, A.L., Jr.; et al. Cellular and behavioral effects of altered NaV1.2 sodium channel ion permeability in Scn2aK1422E mice. Hum. Mol. Genet. 2022, 31, 2964–2988. [Google Scholar] [CrossRef] [PubMed]
- Crook, O.M.; Mulvey, C.M.; Kirk, P.D.W.; Lilley, K.S.; Gatto, L. A Bayesian mixture modelling approach for spatial proteomics. PLoS Comput. Biol. 2018, 14, e1006516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaswani, A.R.; Weykopf, B.; Hagemann, C.; Fried, H.U.; Brustle, O.; Blaess, S. Correct setup of the substantia nigra requires Reelin-mediated fast, laterally-directed migration of dopaminergic neurons. Elife 2019, 8, e41623. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Frank, J.; Gilles, M.; Lang, M.; Treutlein, J.; Streit, F.; Wolf, I.A.C.; Peus, V.; Scharnholz, B.; Send, T.S.; et al. Impact on birth weight of maternal smoking throughout pregnancy mediated by DNA methylation. BMC Genom. 2018, 19, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.; Kim, K.; Serraz, B.; Cho, Y.S.; Kim, D.; Kang, M.; Lee, E.J.; Lee, H.; Bae, Y.C.; Paoletti, P.; et al. Early correction of synaptic long-term depression improves abnormal anxiety-like behavior in adult GluN2B-C456Y-mutant mice. PLoS Biol. 2020, 18, e3000717. [Google Scholar] [CrossRef]
- Qiu, C.; Albayram, O.; Kondo, A.; Wang, B.; Kim, N.; Arai, K.; Tsai, C.Y.; Bassal, M.A.; Herbert, M.K.; Washida, K.; et al. Cis P-tau underlies vascular contribution to cognitive impairment and dementia and can be effectively targeted by immunotherapy in mice. Sci. Transl. Med. 2021, 13, eaaz7615. [Google Scholar] [CrossRef]
- Villa, C.; Combi, R.; Conconi, D.; Lavitrano, M. Patient-Derived Induced Pluripotent Stem Cells (iPSCs) and Cerebral Organoids for Drug Screening and Development in Autism Spectrum Disorder: Opportunities and Challenges. Pharmaceutics 2021, 13, 280. [Google Scholar] [CrossRef]
- Drakulic, D.; Djurovic, S.; Syed, Y.A.; Trattaro, S.; Caporale, N.; Falk, A.; Ofir, R.; Heine, V.M.; Chawner, S.; Rodriguez-Moreno, A.; et al. Copy number variants (CNVs): A powerful tool for iPSC-based modelling of ASD. Mol. Autism 2020, 11, 42. [Google Scholar] [CrossRef]
- Gass, N.; Weber-Fahr, W.; Sartorius, A.; Becker, R.; Didriksen, M.; Stensbol, T.B.; Bastlund, J.F.; Meyer-Lindenberg, A.; Schwarz, A.J. An acetylcholine alpha7 positive allosteric modulator rescues a schizophrenia-associated brain endophenotype in the 15q13.3 microdeletion, encompassing CHRNA7. Eur. Neuropsychopharmacol. 2016, 26, 1150–1160. [Google Scholar] [CrossRef] [Green Version]
- Gupta, C.; Chandrashekar, P.; Jin, T.; He, C.; Khullar, S.; Chang, Q.; Wang, D. Bringing machine learning to research on intellectual and developmental disabilities: Taking inspiration from neurological diseases. J. Neurodev. Disord. 2022, 14, 28. [Google Scholar] [CrossRef]
- Rein, B.; Yan, Z. 16p11.2 Copy Number Variations and Neurodevelopmental Disorders. Trends Neurosci. 2020, 43, 886–901. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the Cores of Autism. Cell 2019, 178, 1287–1298. [Google Scholar] [CrossRef]
- Nicholson, M.W.; Ting, C.Y.; Chan, D.Z.H.; Cheng, Y.C.; Lee, Y.C.; Hsu, C.C.; Huang, C.Y.; Hsieh, P.C.H. Utility of iPSC-Derived Cells for Disease Modeling, Drug Development, and Cell Therapy. Cells 2022, 11, 1853. [Google Scholar] [CrossRef]
- Wang, Y.; Billon, C.; Walker, J.K.; Burris, T.P. Therapeutic Effect of a Synthetic RORalpha/gamma Agonist in an Animal Model of Autism. ACS Chem. Neurosci. 2016, 7, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Meyza, K.Z.; Blanchard, D.C. The BTBR mouse model of idiopathic autism—Current view on mechanisms. Neurosci. Biobehav. Rev. 2017, 76, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Karvat, G.; Kimchi, T. Acetylcholine elevation relieves cognitive rigidity and social deficiency in a mouse model of autism. Neuropsychopharmacology 2014, 39, 831–840. [Google Scholar] [CrossRef] [Green Version]
- Wohr, M. Effect of social odor context on the emission of isolation-induced ultrasonic vocalizations in the BTBR T+tf/J mouse model for autism. Front. Neurosci. 2015, 9, 73. [Google Scholar]
- Guo, Y.P.; Commons, K.G. Serotonin neuron abnormalities in the BTBR mouse model of autism. Autism Res. 2017, 10, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Reshetnikov, V.V.; Ayriyants, K.A.; Ryabushkina, Y.A.; Sozonov, N.G.; Bondar, N.P. Sex-specific behavioral and structural alterations caused by early-life stress in C57BL/6 and BTBR mice. Behav. Brain Res. 2021, 414, 113489. [Google Scholar] [CrossRef]
- McQuaid, R.J.; Audet, M.C.; Jacobson-Pick, S.; Anisman, H. Environmental enrichment influences brain cytokine variations elicited by social defeat in mice. Psychoneuroendocrinology 2013, 38, 987–996. [Google Scholar] [CrossRef]
- Mahmood, U.; Ahn, S.; Yang, E.J.; Choi, M.; Kim, H.; Regan, P.; Cho, K.; Kim, H.S. Dendritic spine anomalies and PTEN alterations in a mouse model of VPA-induced autism spectrum disorder. Pharm. Res 2018, 128, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Seung, H.; Kim, K.C.; Gonzales, E.L.T.; Oh, H.A.; Yang, S.M.; Ko, M.J.; Han, S.H.; Banerjee, S.; Shin, C.Y. Agmatine rescues autistic behaviors in the valproic acid-induced animal model of autism. Neuropharmacology 2017, 113, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Chen, X.; Jiang, X.; Niu, J.; Cui, C.; Chen, Z.; Sun, J. Betaine ameliorates prenatal valproic-acid-induced autism-like behavioral abnormalities in mice by promoting homocysteine metabolism. Psychiatry Clin. Neurosci. 2019, 73, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Murakami, T.; Kawashima, M.; Hasegawa-Baba, Y.; Mizukami, S.; Imatanaka, N.; Akahori, Y.; Yoshida, T.; Shibutani, M. Maternal Exposure to Valproic Acid Primarily Targets Interneurons Followed by Late Effects on Neurogenesis in the Hippocampal Dentate Gyrus in Rat Offspring. Neurotox. Res. 2017, 31, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prevention or Amelioration of Autism-Like Symptoms in Animal Models: Will it Bring Us Closer to Treating Human ASD? Int. J. Mol. Sci. 2019, 20, 1074. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Pozzo-Miller, L. Dysfunction of the corticostriatal pathway in autism spectrum disorders. J. Neurosci. Res. 2020, 98, 2130–2147. [Google Scholar] [CrossRef]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [Green Version]
- Chanda, S.; Aoto, J.; Lee, S.J.; Wernig, M.; Sudhof, T.C. Pathogenic mechanism of an autism-associated neuroligin mutation involves altered AMPA-receptor trafficking. Mol. Psychiatry 2016, 21, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Lyons-Warren, A.M.; McCormack, M.C.; Holder, J.L., Jr. Sensory Processing Phenotypes in Phelan-McDermid Syndrome and SYNGAP1-Related Intellectual Disability. Brain Sci. 2022, 12, 137. [Google Scholar] [CrossRef]
- AlOlaby, R.R.; Zafarullah, M.; Barboza, M.; Peng, G.; Varian, B.J.; Erdman, S.E.; Lebrilla, C.; Tassone, F. Differential Methylation Profile in Fragile X Syndrome-Prone Offspring Mice after in Utero Exposure to Lactobacillus Reuteri. Genes 2022, 13, 1300. [Google Scholar] [CrossRef]
- Sgritta, M.; Dooling, S.W.; Buffington, S.A.; Momin, E.N.; Francis, M.B.; Britton, R.A.; Costa-Mattioli, M. Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 2019, 101, 246–259.e6. [Google Scholar] [CrossRef]
- Ruiz-Falco Rojas, M.L.; Feucht, M.; Macaya, A.; Wilken, B.; Hahn, A.; Maamari, R.; Hirschberg, Y.; Ridolfi, A.; Kingswood, J.C. Real-World Evidence Study on the Long-Term Safety of Everolimus in Patients with Tuberous Sclerosis Complex: Final Analysis Results. Front. Pharm. 2022, 13, 802334. [Google Scholar] [CrossRef]
- Cohen, A.L.; Mulder, B.P.F.; Prohl, A.K.; Soussand, L.; Davis, P.; Kroeck, M.R.; McManus, P.; Gholipour, A.; Scherrer, B.; Bebin, E.M.; et al. Tuberous Sclerosis Complex Autism Center of Excellence Network Study, G. Tuber Locations Associated with Infantile Spasms Map to a Common Brain Network. Ann. Neurol. 2021, 89, 726–739. [Google Scholar] [CrossRef]
- Ligsay, A.; El-Deeb, M.; Salcedo-Arellano, M.J.; Schloemerkemper, N.; Grayson, J.S.; Hagerman, R. General Anesthetic Use in Fragile X Spectrum Disorders. J. Neurosurg. Anesth. 2019, 31, 285–290. [Google Scholar] [CrossRef]
- Ayaz, G.; Turan, G.; Olgun, C.E.; Kars, G.; Karakaya, B.; Yavuz, K.; Demiralay, O.D.; Can, T.; Muyan, M.; Yasar, P. A prelude to the proximity interaction mapping of CXXC5. Sci. Rep. 2021, 11, 17587. [Google Scholar] [CrossRef]
- Zhang, W.J.; Shi, L.L.; Zhang, L. Dysregulated cortical synaptic plasticity under methyl-CpG binding protein 2 deficiency and its implication in motor impairments. World J. Psychiatry 2022, 12, 673–682. [Google Scholar] [CrossRef]
- Arciniegas Ruiz, S.M.; Eldar-Finkelman, H. Glycogen Synthase Kinase-3 Inhibitors: Preclinical and Clinical Focus on CNS-A Decade Onward. Front. Mol. Neurosci. 2021, 14, 792364. [Google Scholar] [CrossRef]
- Leyh, J.; Paeschke, S.; Mages, B.; Michalski, D.; Nowicki, M.; Bechmann, I.; Winter, K. Classification of Microglial Morphological Phenotypes Using Machine Learning. Front. Cell. Neurosci. 2021, 15, 701673. [Google Scholar] [CrossRef]
- Yasin, H.; Gibson, W.T.; Langlois, S.; Stowe, R.M.; Tsang, E.S.; Lee, L.; Poon, J.; Tran, G.; Tyson, C.; Wong, C.K.; et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J. Hum. Genet. 2019, 64, 271–280. [Google Scholar] [CrossRef]
- Ostrowski, P.J.; Zachariou, A.; Loveday, C.; Beleza-Meireles, A.; Bertoli, M.; Dean, J.; Douglas, A.G.L.; Ellis, I.; Foster, A.; Graham, J.M.; et al. The CHD8 overgrowth syndrome: A detailed evaluation of an emerging overgrowth phenotype in 27 patients. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 557–564. [Google Scholar] [CrossRef]
- Ogiwara, I.; Miyamoto, H.; Tatsukawa, T.; Yamagata, T.; Nakayama, T.; Atapour, N.; Miura, E.; Mazaki, E.; Ernst, S.J.; Cao, D.; et al. Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun. Biol. 2018, 1, 96. [Google Scholar] [CrossRef] [PubMed]
- Kruth, K.A.; Grisolano, T.M.; Ahern, C.A.; Williams, A.J. SCN2A channelopathies in the autism spectrum of neuropsychiatric disorders: A role for pluripotent stem cells? Mol. Autism 2020, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.J.; Campbell, A.J.; Cottrell, J.R.; Moller, R.S.; Wagner, F.F.; Auldridge, A.L.; Bernier, R.A.; Catterall, W.A.; Chung, W.K.; Empfield, J.R.; et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018, 41, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Gazina, E.V.; Leaw, B.T.; Richards, K.L.; Wimmer, V.C.; Kim, T.H.; Aumann, T.D.; Featherby, T.J.; Churilov, L.; Hammond, V.E.; Reid, C.A.; et al. ’Neonatal’ Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Hum. Mol. Genet. 2015, 24, 1457–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ouyang, P.; Zheng, Y.; Mi, L.; Zhao, J.; Ning, Y.; Guo, W. A Selective Review of the Excitatory-Inhibitory Imbalance in Schizophrenia: Underlying Biology, Genetics, Microcircuits, and Symptoms. Front. Cell Dev. Biol. 2021, 9, 664535. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, L.; Duan, J.; Zeng, Q.; Chen, L.; Fang, Y.; Hu, J.; Cao, D.; Liao, J. Phenotypes in Children with SYNGAP1 Encephalopathy in China. Front. Neurosci. 2021, 15, 761473. [Google Scholar] [CrossRef]
- Bahry, J.A.; Fedder-Semmes, K.N.; Sceniak, M.P.; Sabo, S.L. An Autism-Associated de novo Mutation in GluN2B Destabilizes Growing Dendrites by Promoting Retraction and Pruning. Front. Cell. Neurosci. 2021, 15, 692232. [Google Scholar] [CrossRef]
- Marballi, K.K.; Gallitano, A.L. Immediate Early Genes Anchor a Biological Pathway of Proteins Required for Memory Formation, Long-Term Depression and Risk for Schizophrenia. Front. Behav. Neurosci. 2018, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Micheletti, S.; Vivanti, G.; Renzetti, S.; Martelli, P.; Calza, S.; Fazzi, E. Imitation in Angelman syndrome: The role of social engagement. Sci. Rep. 2020, 10, 16398. [Google Scholar] [CrossRef]
- Philippe, A. Alternatives to Gold Standard Diagnostic Tools for Distinguishing “Natural Kinds” on the Autism Spectrum. Front. Psychiatry 2022, 13, 862410. [Google Scholar] [CrossRef]
- Loureiro, L.O.; Howe, J.L.; Reuter, M.S.; Iaboni, A.; Calli, K.; Roshandel, D.; Pritisanac, I.; Moses, A.; Forman-Kay, J.D.; Trost, B.; et al. A recurrent SHANK3 frameshift variant in Autism Spectrum Disorder. NPJ Genom. Med. 2021, 6, 91. [Google Scholar] [CrossRef]
- Steinman, K.J.; Spence, S.J.; Ramocki, M.B.; Proud, M.B.; Kessler, S.K.; Marco, E.J.; Green Snyder, L.; D’Angelo, D.; Chen, Q.; Chung, W.K.; et al. 16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. A 2016, 170, 2943–2955. [Google Scholar] [CrossRef]
- Pucilowska, J.; Vithayathil, J.; Tavares, E.J.; Kelly, C.; Karlo, J.C.; Landreth, G.E. The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J. Neurosci. 2015, 35, 3190–3200. [Google Scholar] [CrossRef] [Green Version]
- Francisco, A.A.; Horsthuis, D.J.; Popiel, M.; Foxe, J.J.; Molholm, S. Atypical response inhibition and error processing in 22q11.2 Deletion Syndrome and schizophrenia: Towards neuromarkers of disease progression and risk. Neuroimage Clin. 2020, 27, 102351. [Google Scholar] [CrossRef]
- Cruz, E.; Descalzi, G.; Steinmetz, A.; Scharfman, H.E.; Katzman, A.; Alberini, C.M. CIM6P/IGF-2 Receptor Ligands Reverse Deficits in Angelman Syndrome Model Mice. Autism Res. 2021, 14, 29–45. [Google Scholar] [CrossRef]
- Steinmetz, A.B.; Stern, S.A.; Kohtz, A.S.; Descalzi, G.; Alberini, C.M. Insulin-Like Growth Factor II Targets the mTOR Pathway to Reverse Autism-Like Phenotypes in Mice. J. Neurosci. 2018, 38, 1015–1029. [Google Scholar] [CrossRef] [Green Version]
- Avraham, Y.; Berry, E.M.; Donskoy, M.; Ahmad, W.A.; Vorobiev, L.; Albeck, A.; Mankuta, D. Beta-carotene as a novel therapy for the treatment of “Autistic like behavior” in animal models of Autism. Behav. Brain Res. 2019, 364, 469–479. [Google Scholar] [CrossRef]
- Schwartzer, J.J.; Onore, C.E.; Rose, D.; Ashwood, P. C57BL/6J bone marrow transplant increases sociability in BTBR T(+) Itpr3(tf)/J mice. Brain Behav. Immun. 2017, 59, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Charlier, B.; Coglianese, A.; De Rosa, F.; de Grazia, U.; Operto, F.F.; Coppola, G.; Filippelli, A.; Dal Piaz, F.; Izzo, V. The Effect of Plasma Protein Binding on the Therapeutic Monitoring of Antiseizure Medications. Pharmaceutics 2021, 13, 1208. [Google Scholar] [CrossRef]
- Danzer, S.C. Valproic Acid Leads New Neurons Down the Wrong Path. Epilepsy Curr. 2019, 19, 132–133. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; He, M.; Xiao, L.; Sun, Y.; Ding, J.; Li, W.; Guo, B.; Wang, L.; Wang, Y.; Gao, C.; et al. Prenatal GABAB Receptor Agonist Administration Corrects the Inheritance of Autism-Like Core Behaviors in Offspring of Mice Prenatally Exposed to Valproic Acid. Front. Psychiatry 2022, 13, 835993. [Google Scholar] [CrossRef] [PubMed]
- Mowery, T.M.; Wilson, S.M.; Kostylev, P.V.; Dina, B.; Buchholz, J.B.; Prieto, A.L.; Garraghty, P.E. Embryological exposure to valproic acid disrupts morphology of the deep cerebellar nuclei in a sexually dimorphic way. Int. J. Dev. Neurosci. 2015, 40, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Thabault, M.; Turpin, V.; Maisterrena, A.; Jaber, M.; Egloff, M.; Galvan, L. Cerebellar and Striatal Implications in Autism Spectrum Disorders: From Clinical Observations to Animal Models. Int. J. Mol. Sci. 2022, 23, 2294. [Google Scholar] [CrossRef] [PubMed]
- Codagnone, M.G.; Podesta, M.F.; Uccelli, N.A.; Reines, A. Differential Local Connectivity and Neuroinflammation Profiles in the Medial Prefrontal Cortex and Hippocampus in the Valproic Acid Rat Model of Autism. Dev. Neurosci. 2015, 37, 215–231. [Google Scholar] [CrossRef]
- Al-Amin, M.M.; Rahman, M.M.; Khan, F.R.; Zaman, F.; Mahmud Reza, H. Astaxanthin improves behavioral disorder and oxidative stress in prenatal valproic acid-induced mice model of autism. Behav. Brain Res. 2015, 286, 112–121. [Google Scholar] [CrossRef]
- Yadav, S.; Tiwari, V.; Singh, M.; Yadav, R.K.; Roy, S.; Devi, U.; Gautam, S.; Rawat, J.K.; Ansari, M.N.; Saeedan, A.S.; et al. Comparative efficacy of alpha-linolenic acid and gamma-linolenic acid to attenuate valproic acid-induced autism-like features. J. Physiol. Biochem. 2017, 73, 187–198. [Google Scholar] [CrossRef]
- Ha, S.; Park, H.; Mahmood, U.; Ra, J.C.; Suh, Y.H.; Chang, K.A. Human adipose-derived stem cells ameliorate repetitive behavior, social deficit and anxiety in a VPA-induced autism mouse model. Behav. Brain Res. 2017, 317, 479–484. [Google Scholar] [CrossRef]
- Laue, H.E.; Coker, M.O.; Madan, J.C. The Developing Microbiome From Birth to 3 Years: The Gut-Brain Axis and Neurodevelopmental Outcomes. Front. Pediatr. 2022, 10, 815885. [Google Scholar] [CrossRef]
- Kirsten, T.B.; Casarin, R.C.; Bernardi, M.M.; Felicio, L.F. Pioglitazone abolishes cognition impairments as well as BDNF and neurotensin disturbances in a rat model of autism. Biol. Open 2019, 8, bio041327. [Google Scholar] [CrossRef] [Green Version]
- Patterson, P.H. Modeling autistic features in animals. Pediatr. Res. 2011, 69, 34R–40R. [Google Scholar] [CrossRef]
- Qian, K.; Koike, T.; Tamada, K.; Takumi, T.; Schuller, B.W.; Yamamoto, Y. Sensing the Sounds of Silence: A Pilot Study on the Detection of Model Mice of Autism Spectrum Disorder from Ultrasonic Vocalisations. In Proceedings of the Annual International Conference of the IEEE Engineering in Medicine & Biology Society, Guadalajara, Mexico, 26–30 July 2021; pp. 68–71. [Google Scholar]
- Zieminska, E.; Ruszczynska, A.; Augustyniak, J.; Toczylowska, B.; Lazarewicz, J.W. Zinc and Copper Brain Levels and Expression of Neurotransmitter Receptors in Two Rat ASD Models. Front. Mol. Neurosci. 2021, 14, 656740. [Google Scholar] [CrossRef]
- Kuo, H.Y.; Liu, F.C. Valproic acid induces aberrant development of striatal compartments and corticostriatal pathways in a mouse model of autism spectrum disorder. FASEB J. 2017, 31, 4458–4471. [Google Scholar] [CrossRef] [Green Version]
- Cheaha, D.; Bumrungsri, S.; Chatpun, S.; Kumarnsit, E. Characterization of in utero valproic acid mouse model of autism by local field potential in the hippocampus and the olfactory bulb. Neurosci. Res. 2015, 98, 28–34. [Google Scholar] [CrossRef]
- Barrett, C.E.; Hennessey, T.M.; Gordon, K.M.; Ryan, S.J.; McNair, M.L.; Ressler, K.J.; Rainnie, D.G. Developmental disruption of amygdala transcriptome and socioemotional behavior in rats exposed to valproic acid prenatally. Mol. Autism 2017, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Sunnetci, E.; Durankus, F.; Albayrak, Y.; Erdogan, M.A.; Atasoy, O.; Erbas, O. Effects of the Prenatal Administration of Tetanus Toxoid on the Sociability and Explorative Behaviors of Rat Offspring: A Preliminary Study. Clin. Psychopharmacol. Neurosci. 2021, 19, 84–92. [Google Scholar] [CrossRef]
- Hughes, E.M.; Calcagno, P.; Clarke, M.; Sanchez, C.; Smith, K.; Kelly, J.P.; Finn, D.P.; Roche, M. Prenatal exposure to valproic acid reduces social responses and alters mRNA levels of opioid receptor and pre-pro-peptide in discrete brain regions of adolescent and adult male rats. Brain Res. 2020, 1732, 146675. [Google Scholar] [CrossRef]
- Pang, Q.Q.; Kim, J.H.; Choi, J.M.; Song, J.L.; Lee, S.; Cho, E.J. Cirsium japonicum var. Maackii Improves Cognitive Impairment under Amyloid Beta25-35-Induced Alzheimer’s Disease Model. BioMed Res. Int. 2022, 2022, 4513998. [Google Scholar] [CrossRef]
- Yang, X.; Yin, H.; Wang, X.; Sun, Y.; Bian, X.; Zhang, G.; Li, A.; Cao, A.; Li, B.; Ebrahimi-Fakhari, D.; et al. Social Deficits and Cerebellar Degeneration in Purkinje Cell Scn8a Knockout Mice. Front. Mol. Neurosci. 2022, 15, 822129. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Cao, X.C.; Peng, J.Y.; Huang, S.L.; Chen, C.; Jia, S.Z.; Ni, J.Z.; Song, G.L. Reversal of Lipid Metabolism Dysregulation by Selenium and Folic Acid Co-Supplementation to Mitigate Pathology in Alzheimer’s Disease. Antioxidants 2022, 11, 829. [Google Scholar] [CrossRef]
- Coleman, P.D.; Romano, J.; Lapham, L.; Simon, W. Cell counts in cerebral cortex of an autistic patient. J. Autism Dev. Disord. 1985, 15, 245–255. [Google Scholar] [CrossRef]
- Gupta, A.; Bansal, R.; Alashwal, H.; Kacar, A.S.; Balci, F.; Moustafa, A.A. Neural Substrates of the Drift-Diffusion Model in Brain Disorders. Front. Comput. Neurosci. 2021, 15, 678232. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Flory, M.; Kuchna, I.; Nowicki, K.; Ma, S.Y.; Imaki, H.; Wegiel, J.; Frackowiak, J.; Kolecka, B.M.; Wierzba-Bobrowicz, T.; et al. Neuronal nucleus and cytoplasm volume deficit in children with autism and volume increase in adolescents and adults. Acta Neuropathol. Commun. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova, M.F.; Sokhadze, E.M.; Casanova, E.L.; Li, X. Transcranial Magnetic Stimulation in Autism Spectrum Disorders: Neuropathological Underpinnings and Clinical Correlations. Semin. Pediatr. Neurol. 2020, 35, 100832. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Mirnics, K. Immune system disturbances in schizophrenia. Biol. Psychiatry 2014, 75, 316–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fetit, R.; Hillary, R.F.; Price, D.J.; Lawrie, S.M. The neuropathology of autism: A systematic review of post-mortem studies of autism and related disorders. Neurosci. Biobehav. Rev. 2021, 129, 35–62. [Google Scholar] [CrossRef]
- Van Kooten, I.A.; Palmen, S.J.; von Cappeln, P.; Steinbusch, H.W.; Korr, H.; Heinsen, H.; Hof, P.R.; van Engeland, H.; Schmitz, C. Neurons in the fusiform gyrus are fewer and smaller in autism. Brain 2008, 131, 987–999. [Google Scholar] [CrossRef] [Green Version]
- Lew, C.H.; Groeniger, K.M.; Hanson, K.L.; Cuevas, D.; Greiner, D.M.Z.; Hrvoj-Mihic, B.; Bellugi, U.; Schumann, C.M.; Semendeferi, K. Serotonergic innervation of the amygdala is increased in autism spectrum disorder and decreased in Williams syndrome. Mol. Autism 2020, 11, 12. [Google Scholar] [CrossRef]
- Molnar-Szakacs, I.; Kupis, L.; Uddin, L.Q. Neuroimaging Markers of Risk and Pathways to Resilience in Autism Spectrum Disorder. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 200–210. [Google Scholar] [CrossRef]
- Wolff, J.J.; Jacob, S.; Elison, J.T. The journey to autism: Insights from neuroimaging studies of infants and toddlers. Dev. Psychopathol. 2018, 30, 479–495. [Google Scholar] [CrossRef]
- Lewis, J.D.; Evans, A.C.; Pruett, J.R., Jr.; Botteron, K.N.; McKinstry, R.C.; Zwaigenbaum, L.; Estes, A.M.; Collins, D.L.; Kostopoulos, P.; Gerig, G.; et al. Infant Brain Imaging Study, N. The Emergence of Network Inefficiencies in Infants with Autism Spectrum Disorder. Biol. Psychiatry 2017, 82, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Hazlett, H.C.; Gu, H.; Munsell, B.C.; Kim, S.H.; Styner, M.; Wolff, J.J.; Elison, J.T.; Swanson, M.R.; Zhu, H.; Botteron, K.N.; et al. Data Coordinating, C.; Image Processing, C.; Statistical, A. Early brain development in infants at high risk for autism spectrum disorder. Nature 2017, 542, 348–351. [Google Scholar] [CrossRef]
- Di Martino, A.; Yan, C.G.; Li, Q.; Denio, E.; Castellanos, F.X.; Alaerts, K.; Anderson, J.S.; Assaf, M.; Bookheimer, S.Y.; Dapretto, M.; et al. The autism brain imaging data exchange: Towards a large-scale evaluation of the intrinsic brain architecture in autism. Mol. Psychiatry 2014, 19, 659–667. [Google Scholar] [CrossRef]
- Fernandez, M.; Mollinedo-Gajate, I.; Penagarikano, O. Neural Circuits for Social Cognition: Implications for Autism. Neuroscience 2018, 370, 148–162. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Favia, M.; Spera, I.; Campanella, A.; Lanza, M.; Castegna, A. Metabolic Features of Brain Function with Relevance to Clinical Features of Alzheimer and Parkinson Diseases. Molecules 2022, 27, 951. [Google Scholar] [CrossRef]
- Fontaine, C.J.; Pinar, C.; Yang, W.; Pang, A.F.; Suesser, K.E.; Choi, J.S.J.; Christie, B.R. Impaired Bidirectional Synaptic Plasticity in Juvenile Offspring Following Prenatal Ethanol Exposure. Alcohol Clin. Exp. Res. 2019, 43, 2153–2166. [Google Scholar] [CrossRef]
- Stojanovic, T.; Capo, I.; Aronica, E.; Adle-Biassette, H.; Hoger, H.; Sieghart, W.; Kovacs, G.G.; Milenkovic, I. The alpha1, alpha2, alpha3, and gamma2 subunits of GABAA receptors show characteristic spatial and temporal expression patterns in rhombencephalic structures during normal human brain development. J. Comp. Neurol. 2016, 524, 1805–1824. [Google Scholar] [CrossRef]
- Horder, J.; Petrinovic, M.M.; Mendez, M.A.; Bruns, A.; Takumi, T.; Spooren, W.; Barker, G.J.; Kunnecke, B.; Murphy, D.G. Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl. Psychiatry 2018, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Puts, N.A.J.; Wodka, E.L.; Harris, A.D.; Crocetti, D.; Tommerdahl, M.; Mostofsky, S.H.; Edden, R.A.E. Reduced GABA and altered somatosensory function in children with autism spectrum disorder. Autism Res. 2017, 10, 608–619. [Google Scholar] [CrossRef] [Green Version]
- Al-Otaish, H.; Al-Ayadhi, L.; Bjorklund, G.; Chirumbolo, S.; Urbina, M.A.; El-Ansary, A. Relationship between absolute and relative ratios of glutamate, glutamine and GABA and severity of autism spectrum disorder. Metab. Brain Dis. 2018, 33, 843–854. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Shuang, M.; Lu, T.; Wang, Z.; Zhang, T.; Yue, W.; Jia, M.; Ruan, Y.; Liu, J.; et al. Association study and mutation sequencing of genes on chromosome 15q11-q13 identified GABRG3 as a susceptibility gene for autism in Chinese Han population. Transl. Psychiatry 2018, 8, 152. [Google Scholar] [CrossRef] [Green Version]
- Mahdavi, M.; Kheirollahi, M.; Riahi, R.; Khorvash, F.; Khorrami, M.; Mirsafaie, M. Meta-Analysis of the Association between GABA Receptor Polymorphisms and Autism Spectrum Disorder (ASD). J. Mol. Neurosci. 2018, 65, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Reis de Assis, D.; Szabo, A.; Requena Osete, J.; Puppo, F.; O’Connell, K.S.; Akkouh, I.A.; Hughes, T.; Frei, E.; Andreassen, O.A.; Djurovic, S. Using iPSC Models to Understand the Role of Estrogen in Neuron-Glia Interactions in Schizophrenia and Bipolar Disorder. Cells 2021, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Eltokhi, A.; Santuy, A.; Merchan-Perez, A.; Sprengel, R. Glutamatergic Dysfunction and Synaptic Ultrastructural Alterations in Schizophrenia and Autism Spectrum Disorder: Evidence from Human and Rodent Studies. Int. J. Mol. Sci. 2020, 22, 59. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Kulangara, K.; Antoniello, K.; Markram, H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. USA 2007, 104, 13501–13506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yennawar, M.; White, R.S.; Jensen, F.E. AMPA Receptor Dysregulation and Therapeutic Interventions in a Mouse Model of CDKL5 Deficiency Disorder. J. Neurosci. 2019, 39, 4814–4828. [Google Scholar] [CrossRef] [Green Version]
- Gibson, J.M.; Howland, C.P.; Ren, C.; Howland, C.; Vernino, A.; Tsai, P.T. A Critical Period for Development of Cerebellar-Mediated Autism-Relevant Social Behavior. J. Neurosci. 2022, 42, 2804–2823. [Google Scholar] [CrossRef]
- Soda, T.; Mapelli, L.; Locatelli, F.; Botta, L.; Goldfarb, M.; Prestori, F.; D’Angelo, E. Hyperexcitability and Hyperplasticity Disrupt Cerebellar Signal Transfer in the IB2 KO Mouse Model of Autism. J. Neurosci. 2019, 39, 2383–2397. [Google Scholar]
- Chung, C.; Ha, S.; Kang, H.; Lee, J.; Um, S.M.; Yan, H.; Yoo, Y.E.; Yoo, T.; Jung, H.; Lee, D.; et al. Early Correction of N-Methyl-D-Aspartate Receptor Function Improves Autistic-like Social Behaviors in Adult Shank2(-/-) Mice. Biol. Psychiatry 2019, 85, 534–543. [Google Scholar] [CrossRef]
- Marro, S.G.; Chanda, S.; Yang, N.; Janas, J.A.; Valperga, G.; Trotter, J.; Zhou, B.; Merrill, S.; Yousif, I.; Shelby, H.; et al. Neuroligin-4 Regulates Excitatory Synaptic Transmission in Human Neurons. Neuron 2019, 103, 617–626.e6. [Google Scholar] [CrossRef]
- Jennings, L.; Basiri, R. Amino Acids, B Vitamins, and Choline May Independently and Collaboratively Influence the Incidence and Core Symptoms of Autism Spectrum Disorder. Nutrients 2022, 14, 2896. [Google Scholar] [CrossRef]
- Goncalves, S.; Nunes-Costa, D.; Cardoso, S.M.; Empadinhas, N.; Marugg, J.D. Enzyme Promiscuity in Serotonin Biosynthesis, From Bacteria to Plants and Humans. Front. Microbiol. 2022, 13, 873555. [Google Scholar] [CrossRef]
- Tan, Z.; Wei, H.; Song, X.; Mai, W.; Yan, J.; Ye, W.; Ling, X.; Hou, L.; Zhang, S.; Yan, S.; et al. Positron Emission Tomography in the Neuroimaging of Autism Spectrum Disorder: A Review. Front. Neurosci. 2022, 16, 806876. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, J.; Xu, Q.; Hong, Q.; Zhu, J.; Chi, X. Research Progress in Vitamin A and Autism Spectrum Disorder. Behav. Neurol. 2021, 2021, 5417497. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, H.; Wang, P.; Cui, W.; Xu, K.; Chen, D.; Hu, M.; Li, Z.; Geng, X.; Wei, S. Oxytocin and serotonin in the modulation of neural function: Neurobiological underpinnings of autism-related behavior. Front. Neurosci. 2022, 16, 919890. [Google Scholar] [CrossRef]
- Sjaarda, C.P.; Hecht, P.; McNaughton, A.J.M.; Zhou, A.; Hudson, M.L.; Will, M.J.; Smith, G.; Ayub, M.; Liang, P.; Chen, N.; et al. Interplay between maternal Slc6a4 mutation and prenatal stress: A possible mechanism for autistic behavior development. Sci. Rep. 2017, 7, 8735. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Feng, D.; Chu, M.L.; Fish, I.; Lovera, S.; Sands, Z.A.; Kelm, S.; Valade, A.; Wood, M.; Ceska, T.; et al. Crystal structure of dopamine D1 receptor in complex with G protein and a non-catechol agonist. Nat. Commun. 2021, 12, 3305. [Google Scholar] [CrossRef]
- Suárez-Pereira, I.; García-Domínguez, I.; Bravo, L.; Santiago, M.; García-Revilla, J.; Espinosa-Oliva, A.M.; Alonso-Bellido, I.M.; López-Martín, C.; Pérez-Villegas, E.M.; Armengol, J.A.; et al. The Absence of Caspase-8 in the Dopaminergic System Leads to Mild Autism-like Behavior. Front. Cell Dev. Biol. 2022, 10, 839715. [Google Scholar] [CrossRef]
- Su, T.; Pei, L. Acupuncture and oxytocinergic system: The promising treatment for autism. Transl. Neurosci. 2021, 12, 96–102. [Google Scholar] [CrossRef]
- Vinithakumari, A.A.; Padhi, P.; Hernandez, B.; Lin, S.J.; Dunkerson-Kurzhumov, A.; Showman, L.; Breitzman, M.; Stokes, C.; Sulaiman, Y.; Tangudu, C.; et al. Clostridioides difficile Infection Dysregulates Brain Dopamine Metabolism. Microbiol. Spectr. 2022, 10, e0007322. [Google Scholar] [CrossRef]
- Mandic-Maravic, V.; Grujicic, R.; Milutinovic, L.; Munjiza-Jovanovic, A.; Pejovic-Milovancevic, M. Dopamine in Autism Spectrum Disorders-Focus on D2/D3 Partial Agonists and Their Possible Use in Treatment. Front. Psychiatry 2021, 12, 787097. [Google Scholar] [CrossRef]
- Gunaydin, L.A.; Grosenick, L.; Finkelstein, J.C.; Kauvar, I.V.; Fenno, L.E.; Adhikari, A.; Lammel, S.; Mirzabekov, J.J.; Airan, R.D.; Zalocusky, K.A.; et al. Natural neural projection dynamics underlying social behavior. Cell 2014, 157, 1535–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, K.U.S.; Meli, N.; Blaess, S. The Development of the Mesoprefrontal Dopaminergic System in Health and Disease. Front. Neural Circuits 2021, 15, 746582. [Google Scholar] [CrossRef] [PubMed]
- DiCarlo, G.E.; Aguilar, J.I.; Matthies, H.J.; Harrison, F.E.; Bundschuh, K.E.; West, A.; Hashemi, P.; Herborg, F.; Rickhag, M.; Chen, H.; et al. Autism-linked dopamine transporter mutation alters striatal dopamine neurotransmission and dopamine-dependent behaviors. J. Clin. Investig. 2019, 129, 3407–3419. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Zviagin, E.; Skouta, R. FDA-Approved Oximes and Their Significance in Medicinal Chemistry. Pharmaceuticals 2022, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Dhulkifle, H.; Agouni, A.; Zeidan, A.; Al-Kuwari, M.S.; Parray, A.; Tolefat, M.; Korashy, H.M. Influence of the Aryl Hydrocarbon Receptor Activating Environmental Pollutants on Autism Spectrum Disorder. Int. J. Mol. Sci. 2021, 22, 9258. [Google Scholar] [CrossRef]
- Li, H.; Gao, J.; Chang, Y.; Li, K.; Wang, L.; Ju, C.; Zhang, F. JWX-A0108, a positive allosteric modulator of alpha7 nAChR, attenuates cognitive deficits in APP/PS1 mice by suppressing NF-kappaB-mediated inflammation. Int. Immunopharmacol. 2021, 96, 107726. [Google Scholar] [CrossRef]
- Bleuze, L.; Triaca, V.; Borreca, A. FMRP-Driven Neuropathology in Autistic Spectrum Disorder and Alzheimer’s disease: A Losing Game. Front. Mol. Biosci. 2021, 8, 699613. [Google Scholar] [CrossRef]
- Yang, T.; Xiao, T.; Sun, Q.; Wang, K. The current agonists and positive allosteric modulators of alpha7 nAChR for CNS indications in clinical trials. Acta Pharm. Sin. B 2017, 7, 611–622. [Google Scholar] [CrossRef]
- Pejhan, S.; Rastegar, M. Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules 2021, 11, 75. [Google Scholar] [CrossRef]
- Kim, W.; Zhao, F.; Wu, R.; Qin, S.; Nowsheen, S.; Huang, J.; Zhou, Q.; Chen, Y.; Deng, M.; Guo, G.; et al. ZFP161 regulates replication fork stability and maintenance of genomic stability by recruiting the ATR/ATRIP complex. Nat. Commun. 2019, 10, 5304. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Han, T.; Guo, R.; Chen, P.; Peng, C.; Prag, G.; Hu, R. An Integrative Synthetic Biology Approach to Interrogating Cellular Ubiquitin and Ufm Signaling. Int. J. Mol. Sci. 2020, 21, 4231. [Google Scholar] [CrossRef]
- Deutsch, S.I.; Luyo, Z.N.M.; Burket, J.A. Targeted NMDA Receptor Interventions for Autism: Developmentally Determined Expression of GluN2B and GluN2A-Containing Receptors and Balanced Allosteric Modulatory Approaches. Biomolecules 2022, 12, 181. [Google Scholar] [CrossRef]
- Zahra, A.; Wang, Y.; Wang, Q.; Wu, J. Shared Etiology in Autism Spectrum Disorder and Epilepsy with Functional Disability. Behav. Neurol. 2022, 2022, 5893519. [Google Scholar] [CrossRef]
- Kalinowska, M.; van der Lei, M.B.; Kitiashvili, M.; Mamcarz, M.; Oliveira, M.M.; Longo, F.; Klann, E. Deletion of Fmr1 in parvalbumin-expressing neurons results in dysregulated translation and selective behavioral deficits associated with fragile X syndrome. Mol. Autism 2022, 13, 29. [Google Scholar] [CrossRef]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef]
- Fang, Y.; Zhong, Q.; Wang, Y.; Gu, C.; Liu, S.; Li, A.; Yan, Q. CPEB3 functions as a tumor suppressor in colorectal cancer via JAK/STAT signaling. Aging 2020, 12, 21404–21422. [Google Scholar] [CrossRef]
- Parras, A.; Anta, H.; Santos-Galindo, M.; Swarup, V.; Elorza, A.; Nieto-Gonzalez, J.L.; Pico, S.; Hernandez, I.H.; Diaz-Hernandez, J.I.; Belloc, E.; et al. Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 2018, 560, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Fioriti, L.; Myers, C.; Huang, Y.Y.; Li, X.; Stephan, J.S.; Trifilieff, P.; Colnaghi, L.; Kosmidis, S.; Drisaldi, B.; Pavlopoulos, E.; et al. The Persistence of Hippocampal-Based Memory Requires Protein Synthesis Mediated by the Prion-like Protein CPEB3. Neuron 2015, 86, 1433–1448. [Google Scholar] [CrossRef] [Green Version]
- Bludau, A.; Royer, M.; Meister, G.; Neumann, I.D.; Menon, R. Epigenetic Regulation of the Social Brain. Trends Neurosci. 2019, 42, 471–484. [Google Scholar] [CrossRef]
- Yoon, S.H.; Choi, J.; Lee, W.J.; Do, J.T. Genetic and Epigenetic Etiology Underlying Autism Spectrum Disorder. J. Clin. Med. 2020, 9, 966. [Google Scholar] [CrossRef] [Green Version]
- Thomas, K.T.; Zakharenko, S.S. MicroRNAs in the Onset of Schizophrenia. Cells 2021, 10, 2679. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, Z.M.; Tan, W.; Wang, X.; Li, Y.; Bai, B.; Li, Y.; Zhang, S.F.; Yan, H.L.; Chen, Z.L.; et al. Partial loss of psychiatric risk gene Mir137 in mice causes repetitive behavior and impairs sociability and learning via increased Pde10a. Nat. Neurosci. 2018, 21, 1689–1703. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.C.; Lin, L.S.; Long, S.; Ke, X.Y.; Fukunaga, K.; Lu, Y.M.; Han, F. Signalling pathways in autism spectrum disorder: Mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2022, 7, 229. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, Y.; Zhu, J.; Li, Y.; Chen, J.; Lin, Y. Integrative Analysis of lncRNA-miRNA-mRNA Regulatory Network Reveals the Key lncRNAs Implicated Potentially in the Differentiation of Adipocyte in Goats. Front. Physiol. 2022, 13, 900179. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Feng, Z.; Du, L.; Huang, Y.; Ge, J.; Deng, Y.; Mei, Z. The Potential Role of MicroRNA-124 in Cerebral Ischemia Injury. Int. J. Mol. Sci. 2019, 21, 120. [Google Scholar] [CrossRef] [Green Version]
- Falese, J.P.; Donlic, A.; Hargrove, A.E. Targeting RNA with small molecules: From fundamental principles towards the clinic. Chem. Soc. Rev. 2021, 50, 2224–2243. [Google Scholar] [CrossRef]
- Suchocki, T.; Czech, B.; Dunislawska, A.; Slawinska, A.; Derebecka, N.; Wesoly, J.; Siwek, M.; Szyda, J. SNP prioritization in targeted sequencing data associated with humoral immune responses in chicken. Poult. Sci. 2021, 100, 101433. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; Ubieta, L.T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Kuehner, J.N.; Bruggeman, E.C.; Wen, Z.; Yao, B. Epigenetic Regulations in Neuropsychiatric Disorders. Front. Genet. 2019, 10, 268. [Google Scholar] [CrossRef] [Green Version]
- Ziats, M.N.; Rennert, O.M. Aberrant expression of long noncoding RNAs in autistic brain. J. Mol. Neurosci. 2013, 49, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Kwan, V.; Unda, B.K.; Singh, K.K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 2016, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Durak, O.; Gao, F.; Kaeser-Woo, Y.J.; Rueda, R.; Martorell, A.J.; Nott, A.; Liu, C.Y.; Watson, L.A.; Tsai, L.H. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 2016, 19, 1477–1488. [Google Scholar] [CrossRef]
- Caracci, M.O.; Avila, M.E.; De Ferrari, G.V. Synaptic Wnt/GSK3beta Signaling Hub in Autism. Neural Plast. 2016, 2016, 9603751. [Google Scholar] [CrossRef] [Green Version]
- Remnestal, J.; Bergstrom, S.; Olofsson, J.; Sjostedt, E.; Uhlen, M.; Blennow, K.; Zetterberg, H.; Zettergren, A.; Kern, S.; Skoog, I.; et al. Association of CSF proteins with tau and amyloid beta levels in asymptomatic 70-year-olds. Alzheimer’s Res. Ther. 2021, 13, 54. [Google Scholar] [CrossRef]
- Modi, M.E.; Sahin, M. Tau: A Novel Entry Point for mTOR-Based Treatments in Autism Spectrum Disorder? Neuron 2020, 106, 359–361. [Google Scholar] [CrossRef]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell Physiol. 2019, 234, 2373–2385. [Google Scholar] [CrossRef]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef]
- Zang, G.; Fang, L.; Chen, L.; Wang, C. Ameliorative effect of nicergoline on cognitive function through the PI3K/AKT signaling pathway in mouse models of Alzheimer’s disease. Mol. Med. Rep. 2018, 17, 7293–7300. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.; Man, H.Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.S.; Tso, W.W.Y.; Ip, J.J.K.; Mak, C.C.Y.; Leung, G.K.C.; Tsang, M.H.Y.; Ying, D.; Pei, S.L.C.; Lee, S.L.; Yang, W.; et al. Identification of mutations in the PI3K-AKT-mTOR signalling pathway in patients with macrocephaly and developmental delay and/or autism. Mol. Autism 2017, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, C.; Ahn, Y.; Michalski, B.; Rho, J.M.; Fahnestock, M. Decreased mTOR signaling pathway in human idiopathic autism and in rats exposed to valproic acid. Acta Neuropathol. Commun. 2015, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Homma, T.; Osaki, T. Superoxide Radicals in the Execution of Cell Death. Antioxidants 2022, 11, 501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Miao, Y.; Liu, Q.; Li, S.; He, J. Changes of pro-inflammatory and anti-inflammatory macrophages after peripheral nerve injury. RSC Adv. 2020, 10, 38767–38773. [Google Scholar] [CrossRef] [PubMed]
- Trovato, F.; Parra, R.; Pracucci, E.; Landi, S.; Cozzolino, O.; Nardi, G.; Cruciani, F.; Pillai, V.; Mosti, L.; Cwetsch, A.W.; et al. Modelling genetic mosaicism of neurodevelopmental disorders in vivo by a Cre-amplifying fluorescent reporter. Nat. Commun. 2020, 11, 6194. [Google Scholar] [CrossRef]
- Lieberman, O.J.; McGuirt, A.F.; Tang, G.; Sulzer, D. Roles for neuronal and glial autophagy in synaptic pruning during development. Neurobiol. Dis. 2019, 122, 49–63. [Google Scholar] [CrossRef]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834. [Google Scholar] [CrossRef]
- Sharma, A.; Mehan, S. Targeting PI3K-AKT/mTOR signaling in the prevention of autism. Neurochem. Int. 2021, 147, 105067. [Google Scholar] [CrossRef]
- Mazzaro, N.; Barini, E.; Spillantini, M.G.; Goedert, M.; Medini, P.; Gasparini, L. Tau-Driven Neuronal and Neurotrophic Dysfunction in a Mouse Model of Early Tauopathy. J. Neurosci. 2016, 36, 2086–2100. [Google Scholar] [CrossRef] [Green Version]
- Vithayathil, J.; Pucilowska, J.; Friel, D.; Landreth, G.E. Chronic impairment of ERK signaling in glutamatergic neurons of the forebrain does not affect spatial memory retention and LTP in the same manner as acute blockade of the ERK pathway. Hippocampus 2017, 27, 1239–1249. [Google Scholar] [CrossRef]
- Pucilowska, J.; Vithayathil, J.; Pagani, M.; Kelly, C.; Karlo, J.C.; Robol, C.; Morella, I.; Gozzi, A.; Brambilla, R.; Landreth, G.E. Pharmacological Inhibition of ERK Signaling Rescues Pathophysiology and Behavioral Phenotype Associated with 16p11.2 Chromosomal Deletion in Mice. J. Neurosci. 2018, 38, 6640–6652. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Larsen, R.S.; Bjorklund, G.R.; Li, X.; Wu, Y.; Philpot, B.D.; Snider, W.D.; Newbern, J.M. Layer specific and general requirements for ERK/MAPK signaling in the developing neocortex. Elife 2016, 5, e11123. [Google Scholar] [CrossRef]
- Papale, A.; Morella, I.M.; Indrigo, M.T.; Bernardi, R.E.; Marrone, L.; Marchisella, F.; Brancale, A.; Spanagel, R.; Brambilla, R.; Fasano, S. Impairment of cocaine-mediated behaviours in mice by clinically relevant Ras-ERK inhibitors. Elife 2016, 5, e17111. [Google Scholar] [CrossRef]
- Gabrielli, A.P.; Manzardo, A.M.; Butler, M.G. GeneAnalytics Pathways and Profiling of Shared Autism and Cancer Genes. Int. J. Mol. Sci. 2019, 20, 1166. [Google Scholar] [CrossRef] [Green Version]
- Vogel Ciernia, A.; LaSalle, J. The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat. Rev. Neurosci. 2016, 17, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Wang, G.; Luo, X.; Cheng, Y.; Song, Q.; Wan, J.; Moore, C.; Song, H.; Jin, P.; Qian, J.; et al. An all-to-all approach to the identification of sequence-specific readers for epigenetic DNA modifications on cytosine. Nat. Commun. 2021, 12, 795. [Google Scholar] [CrossRef]
- Kosaka, H.; Okamoto, Y.; Munesue, T.; Yamasue, H.; Inohara, K.; Fujioka, T.; Anme, T.; Orisaka, M.; Ishitobi, M.; Jung, M.; et al. Oxytocin efficacy is modulated by dosage and oxytocin receptor genotype in young adults with high-functioning autism: A 24-week randomized clinical trial. Transl. Psychiatry 2016, 6, e872. [Google Scholar] [CrossRef] [Green Version]
- Baribeau, D.A.; Dupuis, A.; Paton, T.A.; Scherer, S.W.; Schachar, R.J.; Arnold, P.D.; Szatmari, P.; Nicolson, R.; Georgiades, S.; Crosbie, J.; et al. Oxytocin Receptor Polymorphisms are Differentially Associated with Social Abilities across Neurodevelopmental Disorders. Sci. Rep. 2017, 7, 11618. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.J.; McDougle, C.J.; Hooker, J.M.; Zurcher, N.R. Epigenetics of Autism Spectrum Disorder: Histone Deacetylases. Biol. Psychiatry 2022, 91, 922–933. [Google Scholar] [CrossRef]
- Wilczynski, B.; Dabrowska, A.; Saczko, J.; Kulbacka, J. The Role of Chloride Channels in the Multidrug Resistance. Membranes 2021, 12, 38. [Google Scholar] [CrossRef]
- Nott, A.; Cheng, J.; Gao, F.; Lin, Y.T.; Gjoneska, E.; Ko, T.; Minhas, P.; Zamudio, A.V.; Meng, J.; Zhang, F.; et al. Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat. Neurosci. 2016, 19, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Rein, B. Mechanisms of synaptic transmission dysregulation in the prefrontal cortex: Pathophysiological implications. Mol. Psychiatry 2022, 27, 445–465. [Google Scholar] [CrossRef] [PubMed]
- Duclot, F.; Wang, H.; Youssef, C.; Liu, Y.; Wang, Z.; Kabbaj, M. Trichostatin A (TSA) facilitates formation of partner preference in male prairie voles (Microtus ochrogaster). Horm. Behav. 2016, 81, 68–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Wang, Q.; Yan, T.; Zhang, Y.; Xu, H.J.; Yu, H.P.; Tu, Z.; Guo, X.; Jiang, Y.H.; Li, X.J.; et al. Maternal valproic acid exposure leads to neurogenesis defects and autism-like behaviors in non-human primates. Transl. Psychiatry 2019, 9, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Servadio, M.; Manduca, A.; Melancia, F.; Leboffe, L.; Schiavi, S.; Campolongo, P.; Palmery, M.; Ascenzi, P.; di Masi, A.; Trezza, V. Impaired repair of DNA damage is associated with autistic-like traits in rats prenatally exposed to valproic acid. Eur. Neuropsychopharmacol. 2018, 28, 85–96. [Google Scholar] [CrossRef]
- Sun, W.; Poschmann, J.; Cruz-Herrera Del Rosario, R.; Parikshak, N.N.; Hajan, H.S.; Kumar, V.; Ramasamy, R.; Belgard, T.G.; Elanggovan, B.; Wong, C.C.Y.; et al. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell 2016, 167, 1385–1397.e11. [Google Scholar] [CrossRef] [Green Version]
- Bilbrough, T.; Piemontese, E.; Seitz, O. Dissecting the role of protein phosphorylation: A chemical biology toolbox. Chem. Soc. Rev. 2022, 51, 5691–5730. [Google Scholar] [CrossRef]
- Wang, L.; Pang, K.; Han, K.; Adamski, C.J.; Wang, W.; He, L.; Lai, J.K.; Bondar, V.V.; Duman, J.G.; Richman, R.; et al. An autism-linked missense mutation in SHANK3 reveals the modularity of Shank3 function. Mol. Psychiatry 2020, 25, 2534–2555. [Google Scholar] [CrossRef]
- Vien, T.N.; Ackley, M.A.; Doherty, J.J.; Moss, S.J.; Davies, P.A. Preventing Phosphorylation of the GABA (A) R beta3 Subunit Compromises the Behavioral Effects of Neuroactive Steroids. Front. Mol. Neurosci. 2022, 15, 817996. [Google Scholar] [CrossRef]
- McDonald, B.J.; Amato, A.; Connolly, C.N.; Benke, D.; Moss, S.J.; Smart, T.G. Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat. Neurosci. 1998, 1, 23–28. [Google Scholar] [CrossRef]
- Meyer Zu Reckendorf, S.; Moser, D.; Blechschmidt, A.; Joga, V.N.; Sinske, D.; Hegler, J.; Deininger, S.; Catanese, A.; Vettorazzi, S.; Antoniadis, G.; et al. Motoneuron-Specific PTEN Deletion in Mice Induces Neuronal Hypertrophy and Also Regeneration after Facial Nerve Injury. J. Neurosci. 2022, 42, 2474–2491. [Google Scholar] [CrossRef]
- Khlebodarova, T.M.; Kogai, V.V.; Trifonova, E.A.; Likhoshvai, V.A. Dynamic landscape of the local translation at activated synapses. Mol. Psychiatry 2018, 23, 107–114. [Google Scholar] [CrossRef]
- Chen, E.; Joseph, S. Fragile X mental retardation protein: A paradigm for translational control by RNA-binding proteins. Biochimie 2015, 114, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, T.; Liu, P.W. Regulation of the Stability and Localization of Post-synaptic Membrane Proteins by Liquid-Liquid Phase Separation. Front. Physiol. 2021, 12, 795757. [Google Scholar] [CrossRef]
- Lussier, M.P.; Nasu-Nishimura, Y.; Roche, K.W. Activity-dependent ubiquitination of the AMPA receptor subunit GluA2. J. Neurosci. 2011, 31, 3077–3081. [Google Scholar] [CrossRef] [Green Version]
- Kerrisk Campbell, M.; Sheng, M. USP8 Deubiquitinates SHANK3 to Control Synapse Density and SHANK3 Activity-Dependent Protein Levels. J. Neurosci. 2018, 38, 5289–5301. [Google Scholar] [CrossRef]
- Qiao, H.; Tian, Y.; Huo, Y.; Man, H.Y. Role of the DUB enzyme USP7 in dendritic arborization, neuronal migration, and autistic-like behaviors in mice. iScience 2022, 25, 104595. [Google Scholar] [CrossRef]
- Ambrozkiewicz, M.C.; Cuthill, K.J.; Harnett, D.; Kawabe, H.; Tarabykin, V. Molecular Evolution, Neurodevelopmental Roles and Clinical Significance of HECT-Type UBE3 E3 Ubiquitin Ligases. Cells 2020, 9, 2455. [Google Scholar] [CrossRef]
- Dai, W.; Xie, S.; Chen, C.; Choi, B.H. Ras sumoylation in cell signaling and transformation. Semin. Cancer Biol. 2021, 76, 301–309. [Google Scholar] [CrossRef]
- Schorova, L.; Martin, S. Sumoylation in Synaptic Function and Dysfunction. Front. Synaptic Neurosci. 2016, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Henley, J.M.; Craig, T.J.; Wilkinson, K.A. Neuronal SUMOylation: Mechanisms, physiology, and roles in neuronal dysfunction. Physiol. Rev. 2014, 94, 1249–1285. [Google Scholar] [CrossRef] [PubMed]
- Khayachi, A.; Gwizdek, C.; Poupon, G.; Alcor, D.; Chafai, M.; Casse, F.; Maurin, T.; Prieto, M.; Folci, A.; De Graeve, F.; et al. Sumoylation regulates FMRP-mediated dendritic spine elimination and maturation. Nat. Commun 2018, 9, 757. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Shi, Y.; Du, X.; Wang, J.; Zhang, Y.; Shan, S.; Yuan, Y.; Wang, R.; Zhou, C.; Liu, Y.; et al. SENP1 in the retrosplenial agranular cortex regulates core autistic-like symptoms in mice. Cell Rep. 2021, 37, 109939. [Google Scholar] [CrossRef] [PubMed]
- Agirman, G.; Hsiao, E.Y. SnapShot: The microbiota-gut-brain axis. Cell 2021, 184, 2524–2524.e1. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.W.; Pandya, J.D.; Shear, D.A. Gut Microbiota as a Therapeutic Target to Ameliorate the Biochemical, Neuroanatomical, and Behavioral Effects of Traumatic Brain Injuries. Front. Neurol. 2019, 10, 875. [Google Scholar] [CrossRef]
- Ghezzi, L.; Cantoni, C.; Rotondo, E.; Galimberti, D. The Gut Microbiome-Brain Crosstalk in Neurodegenerative Diseases. Biomedicines 2022, 10, 1486. [Google Scholar] [CrossRef]
- Dong, L.; Zheng, Q.; Cheng, Y.; Zhou, M.; Wang, M.; Xu, J.; Xu, Z.; Wu, G.; Yu, Y.; Ye, L.; et al. Gut Microbial Characteristics of Adult Patients with Epilepsy. Front. Neurosci. 2022, 16, 803538. [Google Scholar] [CrossRef]
- Tartaglione, A.M.; Villani, A.; Ajmone-Cat, M.A.; Minghetti, L.; Ricceri, L.; Pazienza, V.; De Simone, R.; Calamandrei, G. Maternal immune activation induces autism-like changes in behavior, neuroinflammatory profile and gut microbiota in mouse offspring of both sexes. Transl. Psychiatry 2022, 12, 384. [Google Scholar] [CrossRef]
- Xu, M.; Xu, X.; Li, J.; Li, F. Association Between Gut Microbiota and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Front. Psychiatry 2019, 10, 473. [Google Scholar] [CrossRef] [Green Version]
- Luna, R.A.; Oezguen, N.; Balderas, M.; Venkatachalam, A.; Runge, J.K.; Versalovic, J.; Veenstra-VanderWeele, J.; Anderson, G.M.; Savidge, T.; Williams, K.C. Distinct Microbiome-Neuroimmune Signatures Correlate with Functional Abdominal Pain in Children with Autism Spectrum Disorder. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 218–230. [Google Scholar] [CrossRef] [Green Version]
- Panisi, C.; Guerini, F.R.; Abruzzo, P.M.; Balzola, F.; Biava, P.M.; Bolotta, A.; Brunero, M.; Burgio, E.; Chiara, A.; Clerici, M.; et al. Autism Spectrum Disorder from the Womb to Adulthood: Suggestions for a Paradigm Shift. J. Pers. Med. 2021, 11, 70. [Google Scholar] [CrossRef]
- Tabouy, L.; Getselter, D.; Ziv, O.; Karpuj, M.; Tabouy, T.; Lukic, I.; Maayouf, R.; Werbner, N.; Ben-Amram, H.; Nuriel-Ohayon, M.; et al. Dysbiosis of microbiome and probiotic treatment in a genetic model of autism spectrum disorders. Brain Behav. Immun. 2018, 73, 310–319. [Google Scholar] [CrossRef]
- Liu, F.; Horton-Sparks, K.; Hull, V.; Li, R.W.; Martinez-Cerdeno, V. The valproic acid rat model of autism presents with gut bacterial dysbiosis similar to that in human autism. Mol. Autism 2018, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Sauer, A.K.; Bockmann, J.; Steinestel, K.; Boeckers, T.M.; Grabrucker, A.M. Altered Intestinal Morphology and Microbiota Composition in the Autism Spectrum Disorders Associated SHANK3 Mouse Model. Int. J. Mol. Sci. 2019, 20, 2134. [Google Scholar] [CrossRef] [Green Version]
- Mansur, F.; Teles, E.S.A.L.; Gomes, A.K.S.; Magdalon, J.; de Souza, J.S.; Griesi-Oliveira, K.; Passos-Bueno, M.R.; Sertie, A.L. Complement C4 Is Reduced in iPSC-Derived Astrocytes of Autism Spectrum Disorder Subjects. Int. J. Mol. Sci. 2021, 22, 7579. [Google Scholar] [CrossRef]
- Li, T.; Chen, X.; Zhang, C.; Zhang, Y.; Yao, W. An update on reactive astrocytes in chronic pain. J. Neuroinflamm. 2019, 16, 140. [Google Scholar] [CrossRef]
- Liu, X.X.; Yang, L.; Shao, L.X.; He, Y.; Wu, G.; Bao, Y.H.; Lu, N.N.; Gong, D.M.; Lu, Y.P.; Cui, T.T.; et al. Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis. J. Exp. Med. 2020, 217, e20180992. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhu, H.; Ni, Z.; Xin, Q.; Zhou, T.; Wu, R.; Gao, G.; Gao, Z.; Ma, H.; Li, H.; et al. Dynamics of a disinhibitory prefrontal microcircuit in controlling social competition. Neuron 2022, 110, 516–531.e6. [Google Scholar] [CrossRef]
- Yu, X.; Taylor, A.M.W.; Nagai, J.; Golshani, P.; Evans, C.J.; Coppola, G.; Khakh, B.S. Reducing Astrocyte Calcium Signaling In Vivo Alters Striatal Microcircuits and Causes Repetitive Behavior. Neuron 2018, 99, 1170–1187.e9. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.C.; Trask, S.; Rosenkranz, J.A. Maturation of amygdala inputs regulate shifts in social and fear behaviors: A substrate for developmental effects of stress. Neurosci. Biobehav. Rev. 2021, 125, 11–25. [Google Scholar] [CrossRef]
- Liu, D.; Nanclares, C.; Simbriger, K.; Fang, K.; Lorsung, E.; Le, N.; Amorim, I.S.; Chalkiadaki, K.; Pathak, S.S.; Li, J.; et al. Autistic-like behavior and cerebellar dysfunction in Bmal1 mutant mice ameliorated by mTORC1 inhibition. Mol. Psychiatry 2022. [Google Scholar] [CrossRef] [PubMed]
- Karalis, V.; Bateup, H.S. Current Approaches and Future Directions for the Treatment of mTORopathies. Dev. Neurosci. 2021, 43, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Meng, F.; Fujita, H.; Morgado, F.; Kazemi, Y.; Rice, L.C.; Ren, C.; Escamilla, C.O.; Gibson, J.M.; Sajadi, S.; et al. Regulation of autism-relevant behaviors by cerebellar-prefrontal cortical circuits. Nat. Neurosci. 2020, 23, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.R.; Gonda, X.; Tarazi, F.I. Autism Spectrum Disorder: Classification, diagnosis and therapy. Pharmacol. Ther. 2018, 190, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Samuel, N.; Vetkas, A.; Pancholi, A.; Sarica, C.; Loh, A.; Germann, J.; Harmsen, I.E.; Tasserie, J.; Milano, V.; Yamamoto, K.; et al. A Network-Based Approach to Glioma Surgery: Insights from Functional Neurosurgery. Cancers 2021, 13, 6127. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, S.; Lugo-Marin, J.; Setien-Ramos, I.; Gisbert-Gustemps, L.; Arteaga-Henriquez, G.; Diez-Villoria, E.; Ramos-Quiroga, J.A. Transcranial direct current stimulation in Autism Spectrum Disorder: A systematic review and meta-analysis. Eur. Neuropsychopharmacol. 2021, 48, 89–109. [Google Scholar] [CrossRef]
- Ameis, S.H.; Blumberger, D.M.; Croarkin, P.E.; Mabbott, D.J.; Lai, M.C.; Desarkar, P.; Szatmari, P.; Daskalakis, Z.J. Treatment of Executive Function Deficits in autism spectrum disorder with repetitive transcranial magnetic stimulation: A double-blind, sham-controlled, pilot trial. Brain Stimul. 2020, 13, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Li, Y.M.; Xiang, D.X. Supplement intervention associated with nutritional deficiencies in autism spectrum disorders: A systematic review. Eur. J. Nutr. 2018, 57, 2571–2582. [Google Scholar] [CrossRef]
- Cannell, J.J. Vitamin D and autism, what’s new? Rev. Endocr. Metab. Disord. 2017, 18, 183–193. [Google Scholar] [CrossRef]
- Agostoni, C.; Nobile, M.; Ciappolino, V.; Delvecchio, G.; Tesei, A.; Turolo, S.; Crippa, A.; Mazzocchi, A.; Altamura, C.A.; Brambilla, P. The Role of Omega-3 Fatty Acids in Developmental Psychopathology: A Systematic Review on Early Psychosis, Autism, and ADHD. Int. J. Mol. Sci. 2017, 18, 2608. [Google Scholar] [CrossRef] [Green Version]
- Fattorusso, A.; Di Genova, L.; Dell’Isola, G.B.; Mencaroni, E.; Esposito, S. Autism Spectrum Disorders and the Gut Microbiota. Nutrients 2019, 11, 521. [Google Scholar] [CrossRef]
- Gogou, M.; Kolios, G. Are therapeutic diets an emerging additional choice in autism spectrum disorder management? World J. Pediatr. 2018, 14, 215–223. [Google Scholar] [CrossRef]
- Sanchack, K.E.; Thomas, C.A. Autism Spectrum Disorder: Primary Care Principles. Am. Fam. Physician 2016, 94, 972–979. [Google Scholar]
- Goel, R.; Hong, J.S.; Findling, R.L.; Ji, N.Y. An update on pharmacotherapy of autism spectrum disorder in children and adolescents. Int. Rev. Psychiatry 2018, 30, 78–95. [Google Scholar] [CrossRef]
- Maneeton, N.; Maneeton, B.; Putthisri, S.; Woottiluk, P.; Narkpongphun, A.; Srisurapanont, M. Risperidone for children and adolescents with autism spectrum disorder: A systematic review. Neuropsychiatr. Dis. Treat. 2018, 14, 1811–1820. [Google Scholar] [CrossRef] [Green Version]
- Hendriksen, J.G.; Klinkenberg, S.; Collin, P.; Wong, B.; Niks, E.H.; Vles, J.S. Diagnosis and treatment of obsessive compulsive behavior in a boy with Duchenne muscular dystrophy and autism spectrum disorder: A case report. Neuromuscul. Disord. 2016, 26, 659–661. [Google Scholar] [CrossRef]
- AlOlaby, R.R.; Sweha, S.R.; Silva, M.; Durbin-Johnson, B.; Yrigollen, C.M.; Pretto, D.; Hagerman, R.J.; Tassone, F. Molecular biomarkers predictive of sertraline treatment response in young children with fragile X syndrome. Brain Dev. 2017, 39, 483–492. [Google Scholar] [CrossRef]
- Politte, L.C.; Scahill, L.; Figueroa, J.; McCracken, J.T.; King, B.; McDougle, C.J. A randomized, placebo-controlled trial of extended-release guanfacine in children with autism spectrum disorder and ADHD symptoms: An analysis of secondary outcome measures. Neuropsychopharmacology 2018, 43, 1772–1778. [Google Scholar] [CrossRef] [Green Version]
- Schroder, C.M.; Malow, B.A.; Maras, A.; Melmed, R.D.; Findling, R.L.; Breddy, J.; Nir, T.; Shahmoon, S.; Zisapel, N.; Gringras, P. Pediatric Prolonged-Release Melatonin for Sleep in Children with Autism Spectrum Disorder: Impact on Child Behavior and Caregiver’s Quality of Life. J. Autism Dev. Disord. 2019, 49, 3218–3230. [Google Scholar] [CrossRef] [Green Version]
- Maras, A.; Schroder, C.M.; Malow, B.A.; Findling, R.L.; Breddy, J.; Nir, T.; Shahmoon, S.; Zisapel, N.; Gringras, P. Long-Term Efficacy and Safety of Pediatric Prolonged-Release Melatonin for Insomnia in Children with Autism Spectrum Disorder. J. Child Adolesc. Psychopharmacol. 2018, 28, 699–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.M.; Kurtzberg, J. Cell therapy for diverse central nervous system disorders: Inherited metabolic diseases and autism. Pediatr. Res. 2018, 83, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Gao, X.; Yang, L. Repetitive Restricted Behaviors in Autism Spectrum Disorder: From Mechanism to Development of Therapeutics. Front. Neurosci. 2022, 16, 780407. [Google Scholar] [CrossRef] [PubMed]
- Murias, M.; Major, S.; Compton, S.; Buttinger, J.; Sun, J.M.; Kurtzberg, J.; Dawson, G. Electrophysiological Biomarkers Predict Clinical Improvement in an Open-Label Trial Assessing Efficacy of Autologous Umbilical Cord Blood for Treatment of Autism. Stem Cells Transl. Med. 2018, 7, 783–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, G.; Sun, J.M.; Davlantis, K.S.; Murias, M.; Franz, L.; Troy, J.; Simmons, R.; Sabatos-DeVito, M.; Durham, R.; Kurtzberg, J. Autologous Cord Blood Infusions Are Safe and Feasible in Young Children with Autism Spectrum Disorder: Results of a Single-Center Phase I Open-Label Trial. Stem Cells Transl. Med. 2017, 6, 1332–1339. [Google Scholar] [CrossRef]
- De la Torre-Ubieta, L.; Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 22, 345–361. [Google Scholar] [CrossRef] [Green Version]
- Vicidomini, C.; Ponzoni, L.; Lim, D.; Schmeisser, M.J.; Reim, D.; Morello, N.; Orellana, D.; Tozzi, A.; Durante, V.; Scalmani, P.; et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 2017, 22, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Caneja, C.M.; State, M.W.; Hagerman, R.J.; Jacquemont, S.; Marin, O.; Bagni, C.; Umbricht, D.; Simonoff, E.; de Andres-Trelles, F.; Kaale, A.; et al. A white paper on a neurodevelopmental framework for drug discovery in autism and other neurodevelopmental disorders. Eur. Neuropsychopharmacol. 2021, 48, 49–88. [Google Scholar] [CrossRef]
- Lee, E.; Lee, S.; Shin, J.J.; Choi, W.; Chung, C.; Lee, S.; Kim, J.; Ha, S.; Kim, R.; Yoo, T.; et al. Excitatory synapses and gap junctions cooperate to improve Pv neuronal burst firing and cortical social cognition in Shank2-mutant mice. Nat. Commun. 2021, 12, 5116. [Google Scholar] [CrossRef]
- Nisar, S.; Bhat, A.A.; Masoodi, T.; Hashem, S.; Akhtar, S.; Ali, T.A.; Amjad, S.; Chawla, S.; Bagga, P.; Frenneaux, M.P.; et al. Genetics of glutamate and its receptors in autism spectrum disorder. Mol. Psychiatry 2022, 27, 2380–2392. [Google Scholar] [CrossRef]
- Wink, L.K.; Reisinger, D.L.; Horn, P.; Shaffer, R.C.; O’Brien, K.; Schmitt, L.; Dominick, K.R.; Pedapati, E.V.; Erickson, C.A. Brief Report: Intranasal Ketamine in Adolescents and Young Adults with Autism Spectrum Disorder-Initial Results of a Randomized, Controlled, Crossover, Pilot Study. J. Autism Dev. Disord. 2021, 51, 1392–1399. [Google Scholar] [CrossRef]
- Wink, L.K.; Adams, R.; Horn, P.S.; Tessier, C.R.; Bantel, A.P.; Hong, M.; Shaffer, R.C.; Pedapati, E.V.; Erickson, C.A. A Randomized Placebo-Controlled Cross-Over Pilot Study of Riluzole for Drug-Refractory Irritability in Autism Spectrum Disorder. J. Autism Dev. Disord. 2018, 48, 3051–3060. [Google Scholar] [CrossRef]
- Sundberg, M.; Tochitsky, I.; Buchholz, D.E.; Winden, K.; Kujala, V.; Kapur, K.; Cataltepe, D.; Turner, D.; Han, M.J.; Woolf, C.J.; et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol. Psychiatry 2018, 23, 2167–2183. [Google Scholar] [CrossRef]
- Chaki, S.; Fukumoto, K. Role of Serotonergic System in the Antidepressant Actions of mGlu2/3 Receptor Antagonists: Similarity to Ketamine. Int. J. Mol. Sci. 2019, 20, 1270. [Google Scholar] [CrossRef] [Green Version]
- Veenstra-VanderWeele, J.; Cook, E.H.; King, B.H.; Zarevics, P.; Cherubini, M.; Walton-Bowen, K.; Bear, M.F.; Wang, P.P.; Carpenter, R.L. Arbaclofen in Children and Adolescents with Autism Spectrum Disorder: A Randomized, Controlled, Phase 2 Trial. Neuropsychopharmacology 2017, 42, 1390–1398. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, C.C.; Dai, Y.; Luo, Q.; Ji, Y.; Wang, K.; Deng, S.; Yu, J.; Xu, M.; Du, X.; et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl. Psychiatry 2020, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Yui, K.; Imataka, G.; Sasaki, H.; Kawasaki, Y.; Okanshi, T.; Shiroki, R.; Yoshihara, S. Improvement in Impaired Social Cognition but Not Seizures by Everolimus in a Child with Tuberous Sclerosis-Associated Autism through Increased Serum Antioxidant Proteins and Oxidant/Antioxidant Status. Case Rep. Pediatr. 2019, 2019, 2070619. [Google Scholar] [CrossRef]
- Zhang, F.; Rein, B.; Zhong, P.; Shwani, T.; Conrow-Graham, M.; Wang, Z.J.; Yan, Z. Synergistic inhibition of histone modifiers produces therapeutic effects in adult Shank3-deficient mice. Transl. Psychiatry 2021, 11, 99. [Google Scholar] [CrossRef]
- Wang, Z.J.; Zhong, P.; Ma, K.; Seo, J.S.; Yang, F.; Hu, Z.; Zhang, F.; Lin, L.; Wang, J.; Liu, T.; et al. Amelioration of autism-like social deficits by targeting histone methyltransferases EHMT1/2 in Shank3-deficient mice. Mol. Psychiatry 2020, 25, 2517–2533. [Google Scholar] [CrossRef]
- Rapanelli, M.; Williams, J.B.; Ma, K.; Yang, F.; Zhong, P.; Patel, R.; Kumar, M.; Qin, L.; Rein, B.; Wang, Z.J.; et al. Targeting histone demethylase LSD1 for treatment of deficits in autism mouse models. Mol. Psychiatry 2022, 27, 3355–3366. [Google Scholar] [CrossRef]
- Valtcheva, S.; Froemke, R.C. Neuromodulation of maternal circuits by oxytocin. Cell Tissue Res. 2019, 375, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Marotta, R.; Risoleo, M.C.; Messina, G.; Parisi, L.; Carotenuto, M.; Vetri, L.; Roccella, M. The Neurochemistry of Autism. Brain Sci. 2020, 10, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, S.; Harony-Nicolas, H. Oxytocin and Animal Models for Autism Spectrum Disorder. Curr. Top. Behav. Neurosci. 2018, 35, 213–237. [Google Scholar] [PubMed]
- Parker, K.J.; Oztan, O.; Libove, R.A.; Mohsin, N.; Karhson, D.S.; Sumiyoshi, R.D.; Summers, J.E.; Hinman, K.E.; Motonaga, K.S.; Phillips, J.M.; et al. A randomized placebo-controlled pilot trial shows that intranasal vasopressin improves social deficits in children with autism. Sci. Transl. Med. 2019, 11, eaau7356. [Google Scholar] [CrossRef]
- Baribeau, D.; Anagnostou, E. Novel treatments for autism spectrum disorder based on genomics and systems biology. Pharmacol. Ther. 2022, 230, 107939. [Google Scholar] [CrossRef]
- Capano, L.; Dupuis, A.; Brian, J.; Mankad, D.; Genore, L.; Hastie Adams, R.; Smile, S.; Lui, T.; Odrobina, D.; Foster, J.A.; et al. A pilot dose finding study of pioglitazone in autistic children. Mol. Autism 2018, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Wykes, R.C.; Lignani, G. Gene therapy and editing: Novel potential treatments for neuronal channelopathies. Neuropharmacology 2018, 132, 108–117. [Google Scholar] [CrossRef]
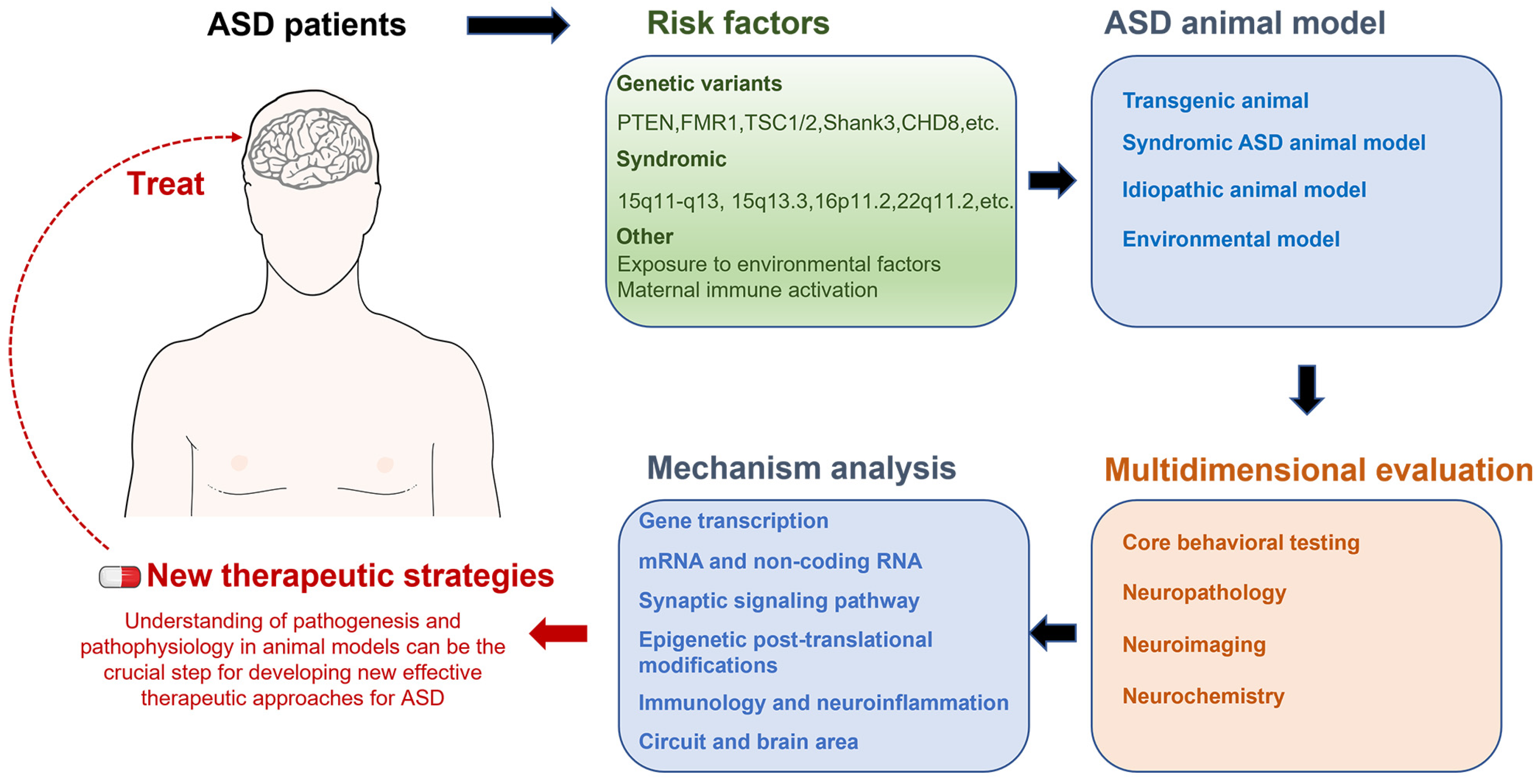
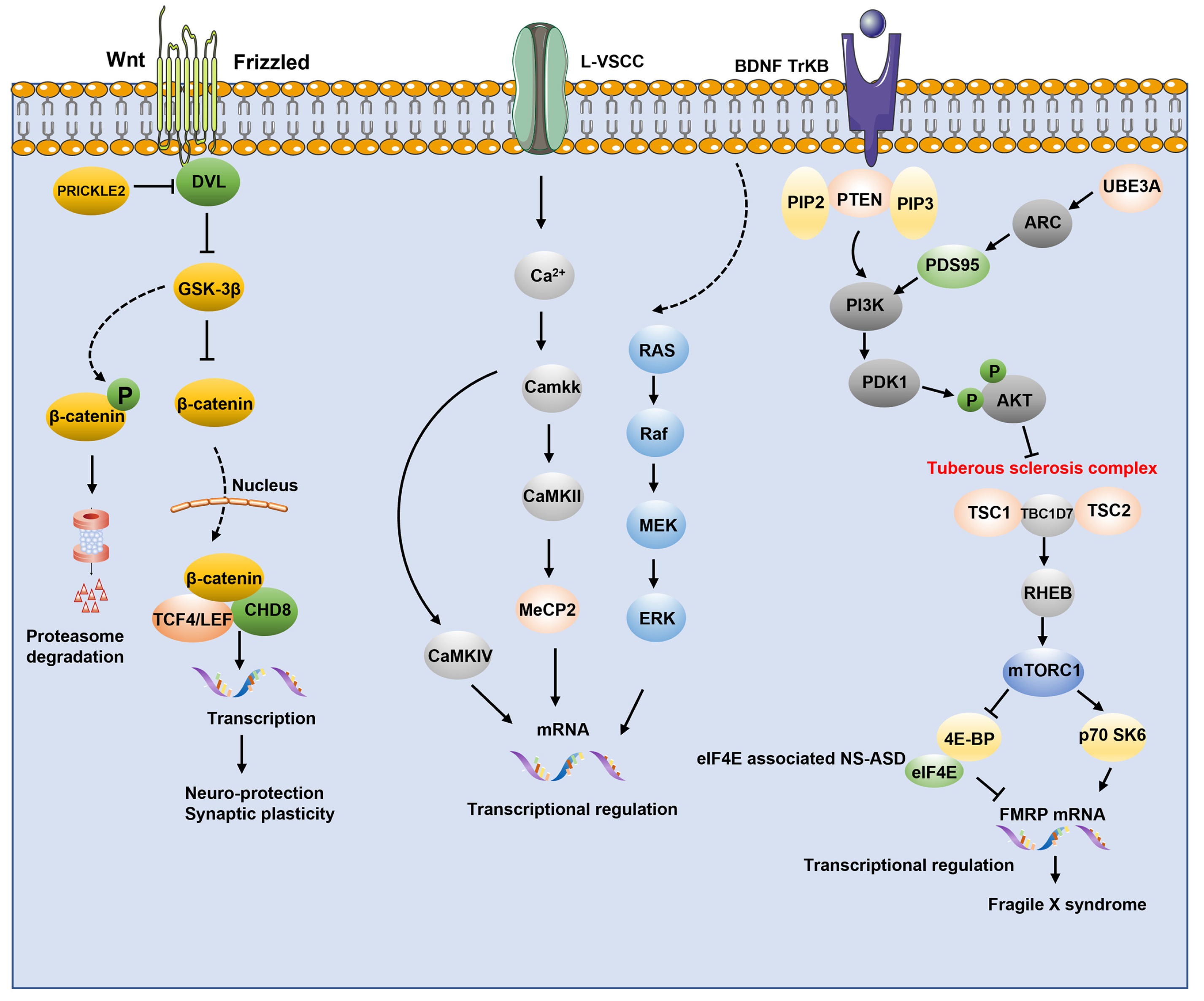




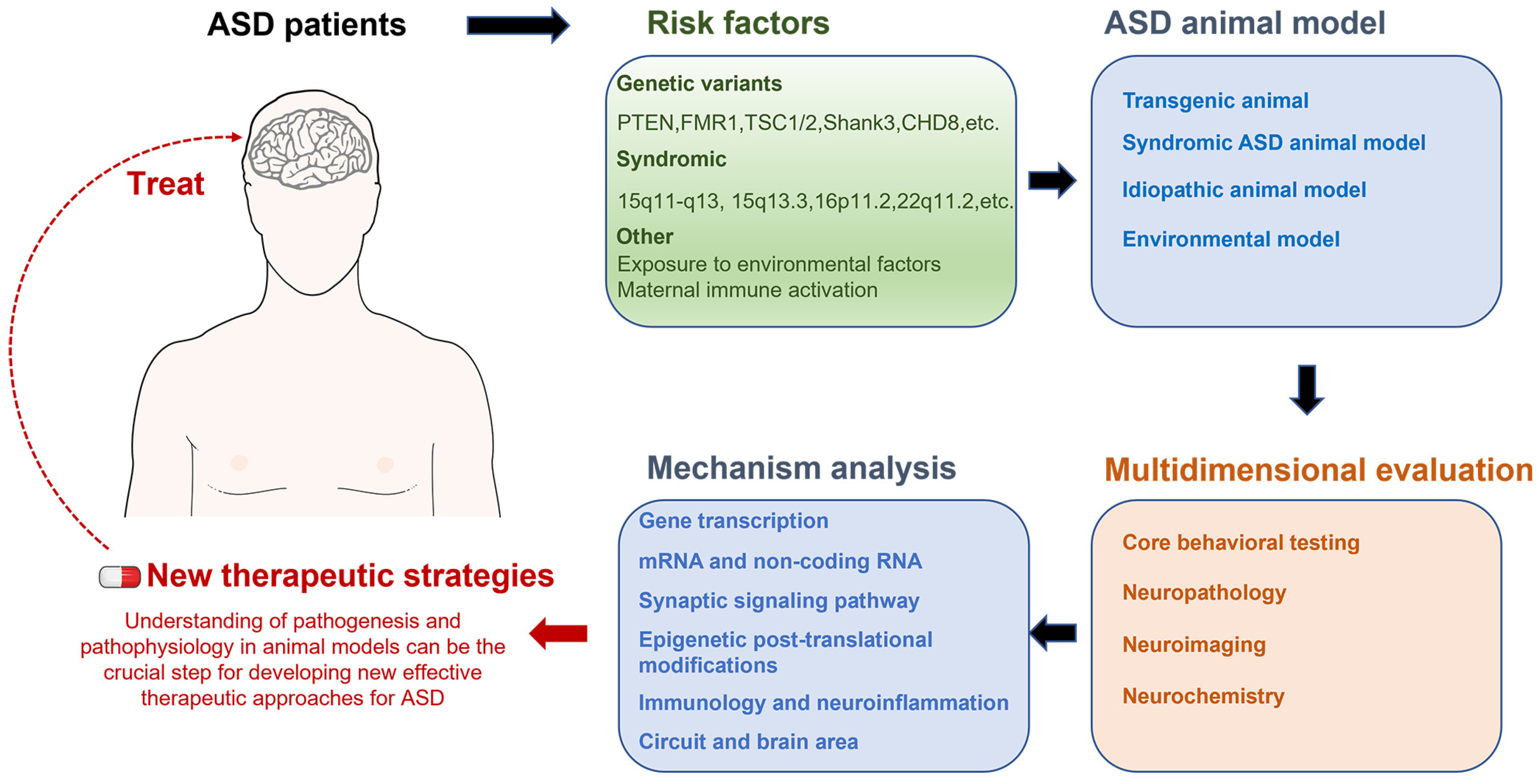{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene Symbol | Molecular Function/Copy Number | Molecular, Cellular and Circuit Phenotypes | Refs |
|---|---|---|---|---|
| Genetic animal models | NRXNs | Synaptic adhesion molecule | ↓Glutamatergic trans. and synaptic density | [7,8,9,10] |
| NLGNs | Synaptic adhesion molecule | Context-dependent impaired glutamatergic and GABAergic trans; ↓Brain volume, cerebellar deficit; ↓GABAergic trans. in D1-MSN in NAc; ↓Brain volume | [11,12,13,14] | |
| SHANK3 | Synaptic scaffolding molecule | Striatal dysfunction; ↓Activity-dependent AMPAR distribution and LTP; ↓Glutamatergic trans. by presynaptic mechanism, ↓LTP; ↓NMDAR function, Rac1, PAK, cofilin signaling defects, F-actin dysregulation in PFC | [15,16,17,18,19] | |
| TSC1/2 | Translational regulator | Cerebellar deficits; Brain enlargement, hyperactive mTOR signaling, autophagy deficiency | [20,21,22] | |
| FMR1 | Translational regulator | ↑mGluR function, immature protrusion; PI3K signaling, ↑spine density, impaired AMPAR-mediated synaptic plasticity; Hypersensitivity to ERK1/2 pathway activation, ↑protein synthesis; ↑Fetal or early postnatal GABA and Cl–, abnormal EEG | [23,24,25] | |
| MECP2 | Translational regulator | ↓Dopamine transporter (DAT) and tyrosine hydroxylase (TH) in the striatum; Altered cortical and cerebellar volumes; Cortical LTP deficit; ↓cortical BDNF levels; Impaired PI3K/AKT/mTOR pathway; ↓CB1 and CB2 receptor levels; Hippocampal circuit dysfunction | [26,27,28,29] | |
| CHD8 | Translational regulator | CHD8 regulates different sets of genes associated with ASD by direct and indirect mechanisms | [30,31,32] | |
| SCN1A | Na+ channel | ↓GABAergic interneuron firing | [33] | |
| SYNGAP1 | Alternative splicing | Major constituent of the PSD essential for postsynaptic signaling; SYNGAP1 regulates the postmitotic maturation of human neurons made from hiPSCs, which influences how activity develops within nascent neural networks | [34,35] | |
| ADNP | Translational regulator | Potential transcription factor; mediate some of the neuroprotective peptide VIP-associated effects involving normal growth | [36] | |
| ANK2 | Protein transport | endocytosis and intracellular protein transport; continuous directional cell migration | [37] | |
| CUL3 | Transport | Mediate ubiquitination of target proteins; releases the GATOR1 complex-mediated inhibition of the TORC1 pathway | [38] | |
| PTEN | Apoptosis | Neuron positioning, dendritic development and synapse formation | [39] | |
| TBR1 | Transcription | Neuronal migration, laminar and areal identity, and axonal projection; blocks the formation of the corticospinal (CS) tract from layer 6 projection neurons | [40] | |
| SCN2A | Ion transport | Mediates the voltage-dependent sodium ion permeability of excitable membranes | [41] | |
| TRIP12 | DNA repair | Ubiquitin fusion degradation (UFD) pathway and | [42] | |
| regulation of DNA repair | ||||
| RELN | Cell adhesion | Regulates microtubule function in neurons and neuronal migration; affects migration of sympathetic preganglionic neurons in the spinal cord | [43] | |
| UBE3A | Proteolysis | Acts as a regulator of synaptic development by mediating ubiquitination and degradation of ARC; Synergizes with WBP2 in enhancing PGR activity | [37] | |
| Cntnap2 | Cell adhesion | Plays a role in the formation of functionally distinct domains critical for saltatory conduction of nerve impulses in myelinated nerve fibers | [44] | |
| Grin2b | Ion transport | Irreversible neuronal death; neural pattern formation; long-term depression (LTD) of hippocampus membrane currents and in synaptic plasticity | [45,46] | |
| Copy number variation | 15q11-q13 | Deletion | ↑excitatory synaptic event frequency amplitude, density of dendritic protrusions,↓inhibitory synaptic transmission; Impaired activity-dependent synaptic plasticity and homoeostatic synaptic scaling | [47,48] |
| 15q13.3 | Microdeletion | ↑endoplasmic reticulum stress; Dysregulated neuronal gene expression; ↑cholinergic activity; ↑homomeric CHRNA7 channel activity | [49,50] | |
| 16p11.2 | Deletion | ↑soma size and dendrite length in 16pdel neurons; ↓neuronal size and dendrite length in 16pdup neurons; ↓synaptic density | [51,52] | |
| 22q11.2 | Duplication | ↓spontaneous neuronal activity and calcium signalling; ↓expression of miR-1290 | [53] | |
| Idiopathic animal models | BTBR-T+ tfl/J | ↓GABAergic inhibitory transmission; ↓5HT2A receptor density and activity; ↑glutamatergic transmission in cortico-striatal circuitry; Impaired dopamine D2 receptor function; ↓BDNF expression in hippocampus and cortex; Absence of corpus callosum, lack of hippocampal commissure; ↓cortical thickness; ↓cerebral white and gray matter; Impaired cortico-thalamic function; Altered volumes of cerebellum, brainstem, striatum, and hippocampus | [54,55,56,57,58,59] | |
| BALB/cByJ | Interleukin change; neuroinflammation | [60] | ||
| Environmental models | VPA | ↑glutamatergic excitatory signaling; Hyperexcitable local connectivity;↓parvalbumin-positive inhibitory interneurons;↑brain serotonin levels; Apical dendritic arborization complexity;↓PTEN expression and↑p-AKT protein levels in hippocampus and cortex | [61,62,63,64] | |
| Maternal immune activation | Changes of various interleukins in brain | [65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Wang, B.; Wu, C.; Wang, J.; Sun, M. Autism Spectrum Disorder: Neurodevelopmental Risk Factors, Biological Mechanism, and Precision Therapy. Int. J. Mol. Sci. 2023, 24, 1819. https://doi.org/10.3390/ijms24031819
Wang L, Wang B, Wu C, Wang J, Sun M. Autism Spectrum Disorder: Neurodevelopmental Risk Factors, Biological Mechanism, and Precision Therapy. International Journal of Molecular Sciences. 2023; 24(3):1819. https://doi.org/10.3390/ijms24031819
Chicago/Turabian StyleWang, Ling, Binquan Wang, Chunyan Wu, Jie Wang, and Mingkuan Sun. 2023. "Autism Spectrum Disorder: Neurodevelopmental Risk Factors, Biological Mechanism, and Precision Therapy" International Journal of Molecular Sciences 24, no. 3: 1819. https://doi.org/10.3390/ijms24031819
APA StyleWang, L., Wang, B., Wu, C., Wang, J., & Sun, M. (2023). Autism Spectrum Disorder: Neurodevelopmental Risk Factors, Biological Mechanism, and Precision Therapy. International Journal of Molecular Sciences, 24(3), 1819. https://doi.org/10.3390/ijms24031819