
The Antidepressant Paroxetine Reduces the Cardiac Sodium Current



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
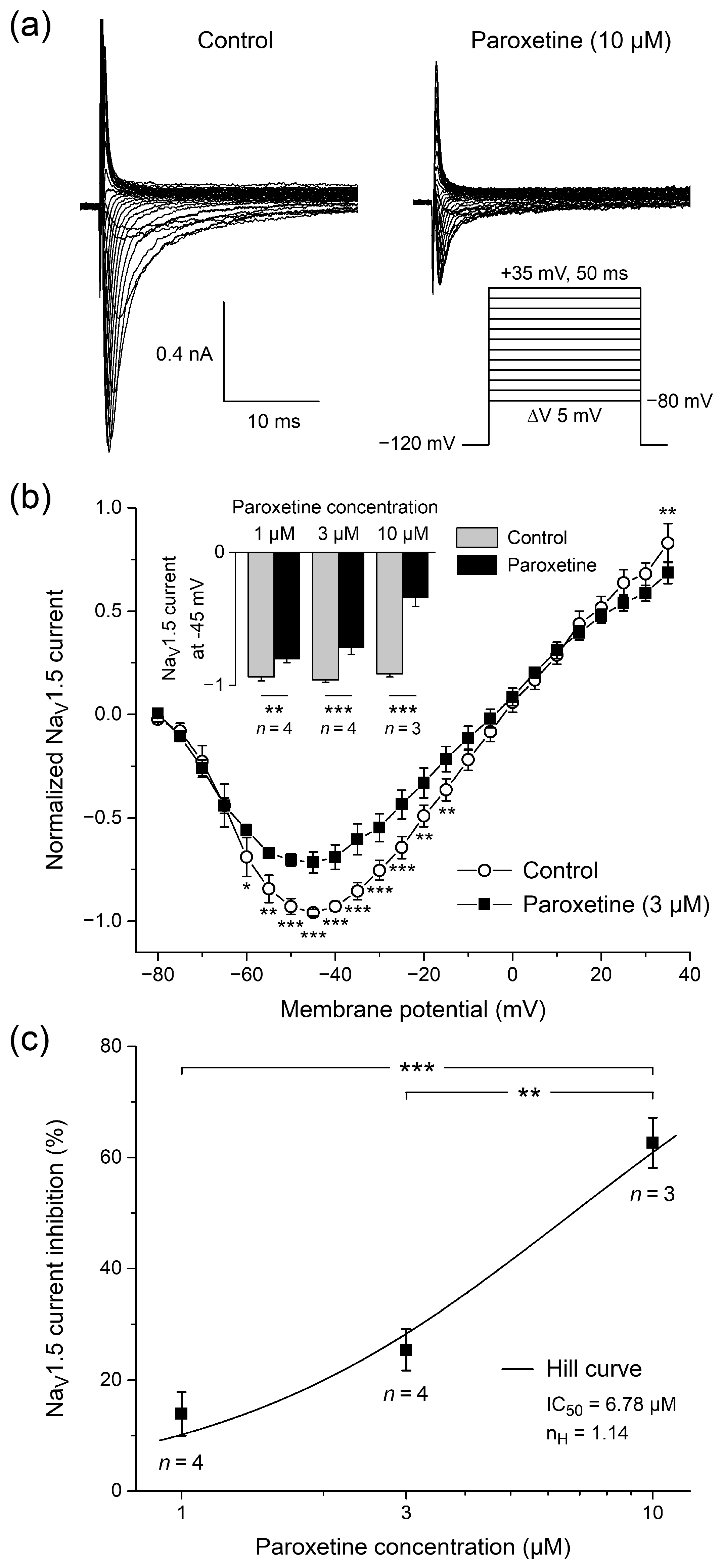
2.1. Effects of Paroxetine on NaV1.5 Current in HEK-293 Cells
2.1.1. Effects of Paroxetine on Peak NaV1.5 Current
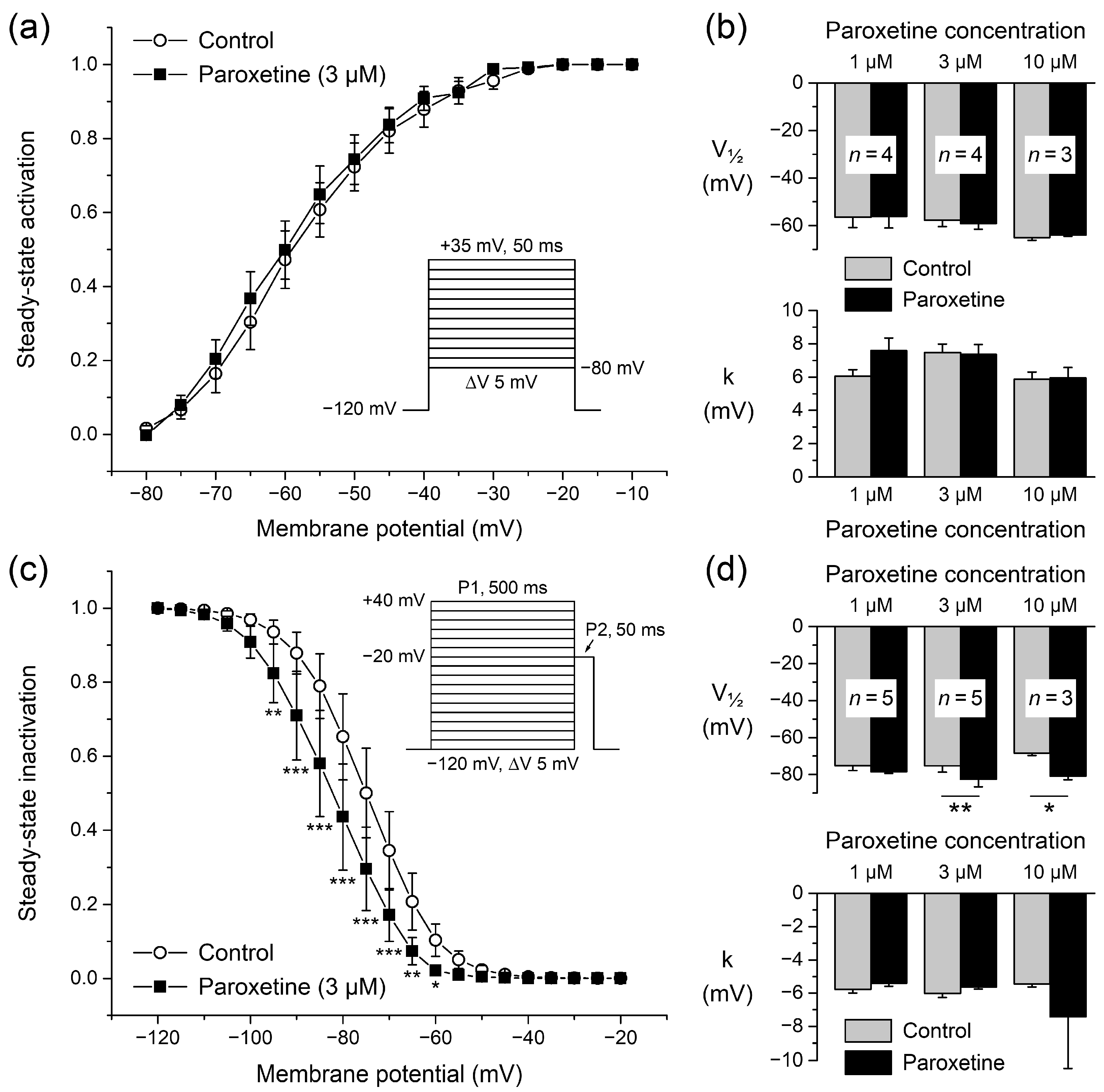
2.1.2. Effects of Paroxetine on Steady-State Activation
2.1.3. Effects of Paroxetine on Steady-State Inactivation
2.1.4. Effects of Paroxetine on Rates of Activation and Inactivation
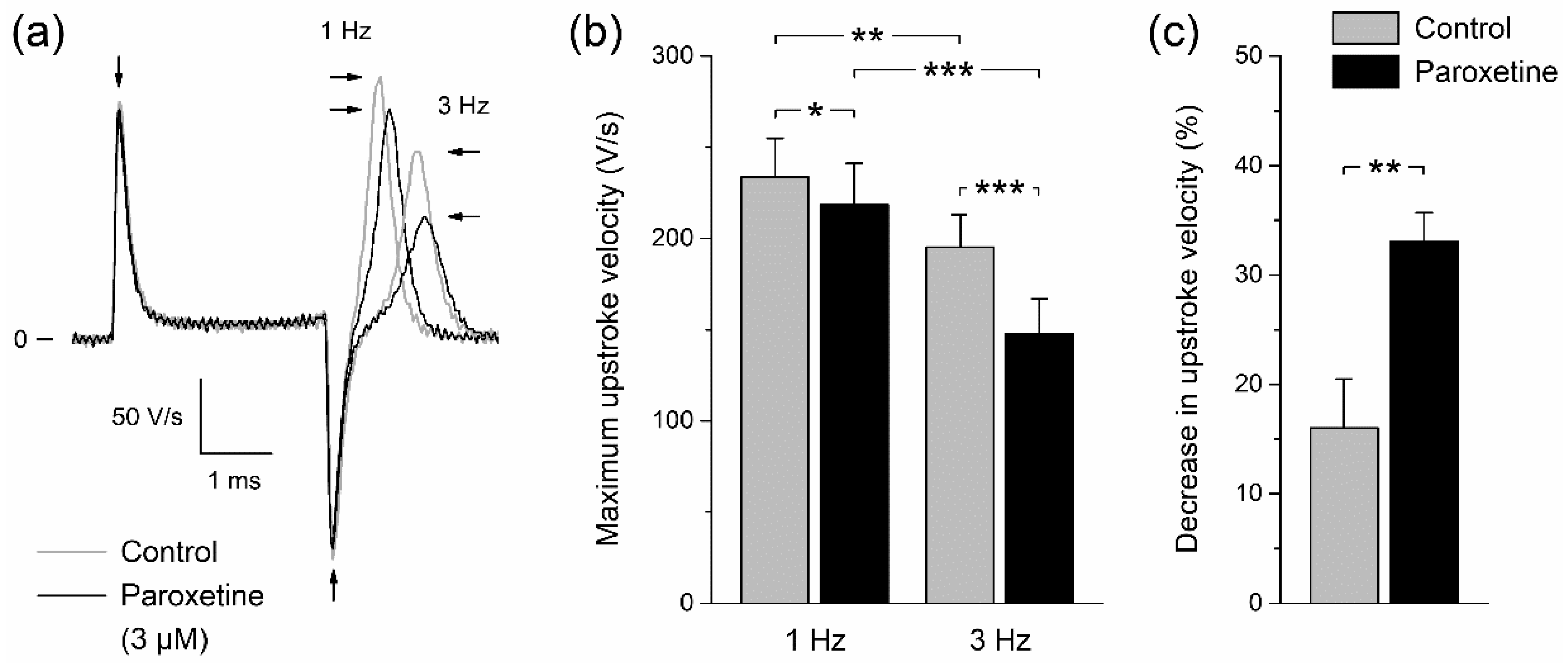
2.2. Effects of Paroxetine on Action Potentials of Rabbit Left Ventricular Cardiomyocytes
2.3. In Silico Experiments
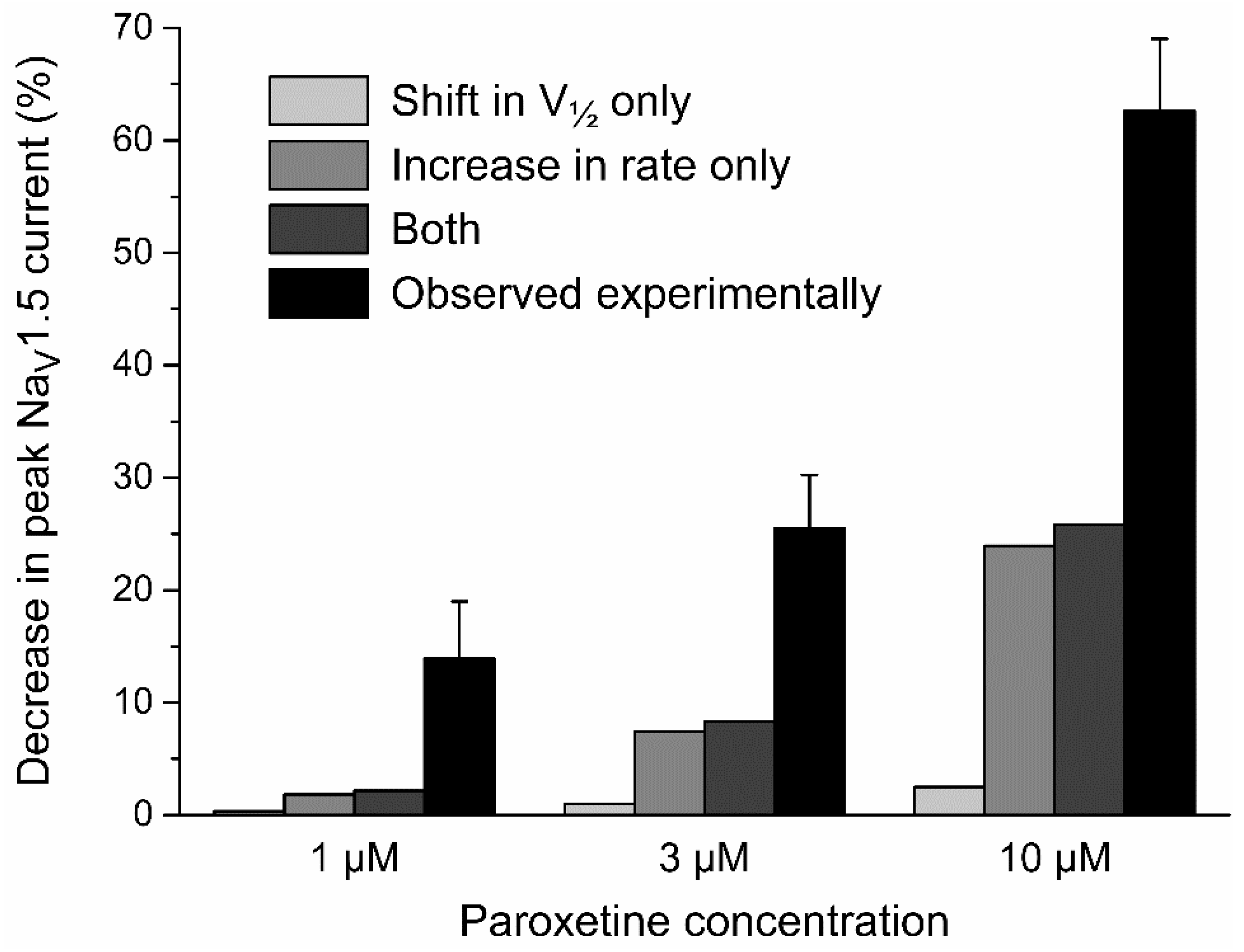
2.3.1. Effects of Paroxetine on Peak NaV1.5 Current
2.3.2. Effects of Paroxetine on Single Human Left Ventricular Cardiomyocytes
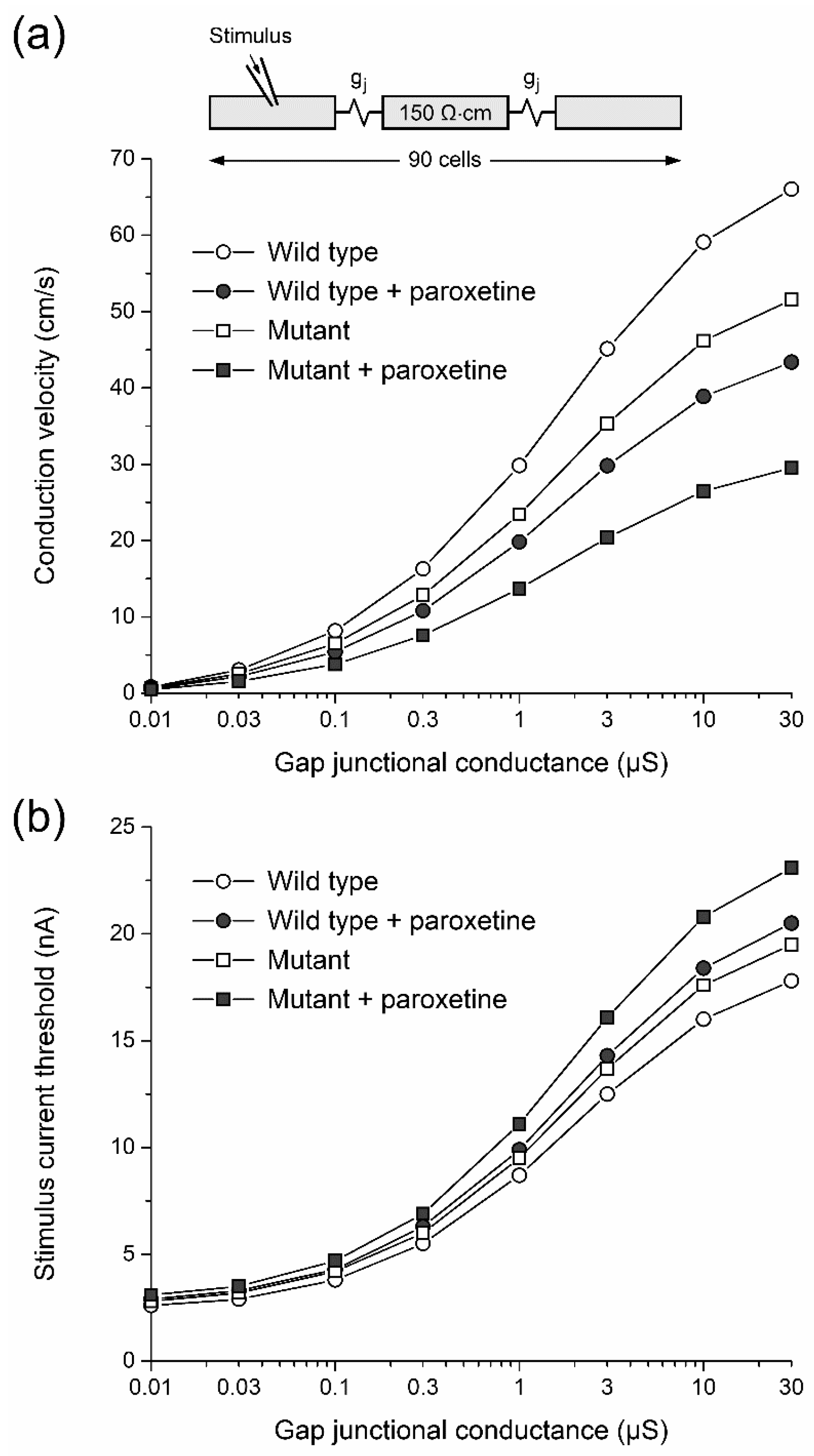
2.3.3. Effects of Paroxetine on Conduction Velocity and Excitability
3. Discussion
4. Materials and Methods
4.1. Cell Preparations
4.1.1. HEK-293 Cell Culture
4.1.2. Rabbit Ventricular Cardiomyocytes
4.2. Electrophysiology
4.2.1. Data Acquisition
4.2.2. Sodium Current Measurements
4.2.3. Action Potential Measurements
4.3. Computer Simulations
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kantor, E.D.; Rehm, C.D.; Haas, J.S.; Chan, A.T.; Giovannucci, E.L. Trends in prescription drug use among adults in the United States from 1999–2012. JAMA 2015, 314, 1818–1830. [Google Scholar] [CrossRef] [PubMed]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2016, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Berlin, J.A.; Glasser, S.C.; Ellenberg, S.S. Adverse event detection in drug development: Recommendations and obligations beyond phase 3. Am. J. Public Health 2008, 98, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, C.A. Proarrhythmia with non-antiarrhythmics: A review. Cardiology 2004, 102, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, J.E.; Chung, M.K.; Campbell, K.B.; Hammadah, M.; Joglar, J.A.; Leclerc, J.; Rajagopalan, B. Drug-induced arrhythmias: A scientific statement from the American Heart Association. Circulation 2020, 142, e214–e233. [Google Scholar] [CrossRef] [PubMed]
- Huikuri, H.V.; Castellanos, A.; Myerburg, R.J. Sudden death due to cardiac arrhythmias. N. Engl. J. Med. 2001, 345, 1473–1482. [Google Scholar] [CrossRef]
- Tse, G.; Chan, Y.W.F.; Keung, W.; Yan, B.P. Electrophysiological mechanisms of long and short QT syndromes. Int. J. Cardiol. Heart Vasc. 2017, 14, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, E.R.; Roden, D. Drug-induced arrhythmia: Pharmacogenomic prescribing? Eur. Heart J. 2012, 34, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, T.E.; Barcella, C.A.; Gerds, T.A.; Kessing, L.V.; Zylyftari, N.; Mohr, G.H.; Kragholm, K.; Polcwiartek, C.; Wissenberg, M.; Folke, F.; et al. Risk of out-of-hospital cardiac arrest in antidepressant drug users. Br. J. Clin. Pharmacol. 2022, 88, 3162–3171. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S. Clinical spectrum of SCN5A mutations: Long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Ackerman, M.J.; Antzelevitch, C.; Bezzina, C.R.; Borggrefe, M.; Cuneo, B.F.; Wilde, A.A.M. Inherited cardiac arrhythmias. Nat. Rev. Dis. Primers 2020, 6, 58. [Google Scholar] [CrossRef]
- Sawhney, V.; Thomas, G.; Webster, P.; Schilling, R. Resolution of Brugada-pattern ECG after withdrawal of the selective serotonin reuptake inhibitor paroxetine. Heart 2010, 96, 1165–1166. [Google Scholar] [CrossRef] [Green Version]
- Valuck, R. Selective serotonin reuptake inhibitors: A class review. Pharm. Ther. 2004, 29, 234–243. [Google Scholar]
- Dunner, D.L.; Lipschitz, A.; Pitts, C.D.; Davies, J.T. Efficacy and tolerability of controlled-release paroxetine in the treatment of severe depression: Post hoc analysis of pooled data from a subset of subjects in four double-blind clinical trials. Clin. Ther. 2005, 27, 1901–1911. [Google Scholar] [CrossRef]
- Reis, M.; Aamo, T.; Spigset, O.; Ahlner, J. Serum concentrations of antidepressant drugs in a naturalistic setting: Compilation based on a large therapeutic drug monitoring database. Ther. Drug Monit. 2009, 31, 42–56. [Google Scholar] [CrossRef]
- Sawamura, K.; Suzuki, Y.; Someya, T. Effects of dosage and CYP2D6-mutated allele on plasma concentration of paroxetine. Eur. J. Clin. Pharmacol. 2004, 60, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.G.; Goldie, A.; Papayanni-Papasthatis, S. Effect of paroxetine on the electrocardiogram. Psychopharmacology 1989, 97, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Funk, K.A.; Bostwick, J.R. A comparison of the risk of QT prolongation among SSRIs. Ann. Pharmacother. 2013, 47, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.S.; Jang, I.-J.; Kim, B.-H.; Kim, J.; Jeon, J.-Y.; Tae, Y.-M.; Yi, S.; Eum, S.; Cho, J.-Y.; Shin, S.-G.; et al. Changes in the QTc interval after administration of flecainide acetate, with and without coadministered paroxetine, in relation to cytochrome P450 2D6 genotype: Data from an open-label, two-period, single-sequence crossover study in healthy Korean male subjects. Clin. Ther. 2010, 32, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Krafte, D.S.; Davison, K.; Dugrenier, N.; Estep, K.; Josef, K.; Barchi, R.L.; Kallen, R.G.; Silver, P.J.; Ezrin, A.M. Pharmacological modulation of human cardiac Na+ channels. Eur. J. Pharmacol. 1994, 266, 245–254. [Google Scholar] [CrossRef]
- Brugada, R.; Brugada, J.; Antzelevitch, C.; Kirsch, G.E.; Potenza, D.; Towbin, J.A.; Brugada, P. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation 2000, 101, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Erfurth, A.; Loew, M.; Dobmeier, P.; Wendler, G. EKG-Veränderungen nach Paroxetin: Drei Fallberichte. Nervenarzt 1998, 69, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Dubnov-Raz, G.; Juurlink, D.N.; Fogelman, R.; Merlob, P.; Ito, S.; Koren, G.; Finkelstein, Y. Antenatal use of selective serotonin-reuptake inhibitors and QT interval prolongation in newborns. Pediatrics 2008, 122, e710–e715. [Google Scholar] [CrossRef] [PubMed]
- Leerssen, E.C.M.; Tak, R.O.; Breur, J.M.P.J. Severe transient neonatal long QT syndrome due to maternal paroxetine usage: A case report. Cardiol. Young 2019, 29, 1300–1301. [Google Scholar] [CrossRef] [PubMed]
- Wenzel-Seifert, K.; Wittmann, M.; Haen, E. Torsade de pointes episodes under treatment with selective serotonin reuptake inhibitors. Pharmacopsychiatry 2010, 43, 279–281. [Google Scholar] [CrossRef]
- Yokota, S.; Ishikura, Y.; Ono, H. Cardiovascular effects of paroxetine, a newly developed antidepressant, in anesthetized dogs in comparison with those of imipramine, amitriptyline and clomipramine. Jpn. J. Pharmacol. 1987, 45, 335–342. [Google Scholar] [CrossRef]
- Harmer, A.R.; Valentin, J.-P.; Pollard, C.E. On the relationship between block of the cardiac Na+ channel and drug-induced prolongation of the QRS complex. Br. J. Pharmacol. 2011, 164, 260–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BrugadaDrugs.org. Available online: https://www.brugadadrugs.org/ (accessed on 11 December 2022).
- Postema, P.G.; Wolpert, C.; Amin, A.S.; Probst, V.; Borggrefe, M.; Roden, D.M.; Priori, S.G.; Tan, H.L.; Hiraoka, M.; Brugada, J.; et al. Drugs and Brugada syndrome patients: Review of the literature, recommendations, and an up-to-date website (www.brugadadrugs.org). Heart Rhythm 2009, 6, 1335–1341. [Google Scholar] [CrossRef] [Green Version]
- CredibleMeds QTdrugs List. Available online: https://crediblemeds.org/ (accessed on 11 December 2022).
- Woosley, R.L.; Black, K.; Heise, C.W.; Romero, K. CredibleMeds.org: What does it offer? Trends Cardiovasc. Med. 2018, 28, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Dick, I.E.; Brochu, R.M.; Purohit, Y.; Kaczorowski, G.J.; Martin, W.J.; Priest, B.T. Sodium channel blockade may contribute to the analgesic efficacy of antidepressants. J. Pain 2007, 8, 315–324. [Google Scholar] [CrossRef]
- Wang, G.K.; Mitchell, J.; Wang, S.-Y. Block of persistent late Na+ currents by antidepressant sertraline and paroxetine. J. Membr. Biol. 2008, 222, 79–90. [Google Scholar] [CrossRef]
- Thériault, O.; Poulin, H.; Beaulieu, J.-M.; Chahine, M. Differential modulation of Nav1.7 and Nav1.8 channels by antidepressant drugs. Eur. J. Pharmacol. 2015, 764, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Berecki, G.; Wilders, R.; De Jonge, B.; Van Ginneken, A.C.G.; Verkerk, A.O. Re-evaluation of the action potential upstroke velocity as a measure of the Na+ current in cardiac myocytes at physiological conditions. PLoS ONE 2010, 5, e15772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.-R.; Ma, J.-H.; Zhang, P.-H.; Xing, J.-L. Blocking effect of methylflavonolamine on human NaV1.5 channels expressed in Xenopus laevis oocytes and on sodium currents in rabbit ventricular myocytes. Acta Pharmacol. Sin. 2010, 31, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, H.-F.; Zhang, H.; Wang, C.; Chen, Y.-F.; Ma, R.; Xiang, J.-Z.; Du, X.-L.; Tang, Q. Inhibitory effects of hesperetin on Nav1.5 channels stably expressed in HEK 293 cells and on the voltage-gated cardiac sodium current in human atrial myocytes. Acta Pharmacol. Sin. 2016, 37, 1563–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caves, R.E.; Carpenter, A.; Choisy, S.C.; Clennell, B.; Cheng, H.; McNiff, C.; Mann, B.; Milnes, J.T.; Hancox, J.C.; James, A.F. Inhibition of voltage-gated Na+ currents by eleclazine in rat atrial and ventricular myocytes. Heart Rhythm O2 2020, 1, 206–214. [Google Scholar] [CrossRef]
- Jia, L.; Eroglu, T.E.; Wilders, R.; Verkerk, A.O.; Tan, H.L. Carbamazepine increases the risk of sudden cardiac arrest by a reduction of the cardiac sodium current. Front. Cell Dev. Biol. 2022, 10, 891996. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Hong, L. Effects of mexiletine on a race-specific mutation in Nav1.5 associated with long QT syndrome. Front. Physiol. 2022, 13, 904664. [Google Scholar] [CrossRef]
- Föhr, K.J.; Rapp, M.; Fauler, M.; Zimmer, T.; Jungwirth, B.; Messerer, D.A.C. Block of voltage-gated sodium channels by aripiprazole in a state-dependent manner. Int. J. Mol. Sci. 2022, 23, 12890. [Google Scholar] [CrossRef]
- Ten Tusscher, K.H.W.J.; Noble, D.; Noble, P.J.; Panfilov, A.V. A model for human ventricular tissue. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, 1573–1589. [Google Scholar] [CrossRef]
- Ten Tusscher, K.H.W.J.; Panfilov, A.V. Cell model for efficient simulation of wave propagation in human ventricular tissue under normal and pathological conditions. Phys. Med. Biol. 2006, 51, 6141–6156. [Google Scholar] [CrossRef] [PubMed]
- Wilders, R. Arrhythmogenic right ventricular cardiomyopathy: Considerations from in silico experiments. Front. Physiol. 2012, 3, 168. [Google Scholar] [CrossRef] [Green Version]
- Sala, M.; Coppa, F.; Cappucciati, C.; Brambilla, P.; D’Allio, G.; Caverzasi, E.; Barale, F.; De Ferrari, G.M. Antidepressants: Their effects on cardiac channels, QT prolongation and Torsade de Pointes. Curr. Opin. Investig. Drugs 2006, 7, 256–263. [Google Scholar] [PubMed]
- Kléber, A.G.; Rudy, Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 2004, 84, 431–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, S.C.; Antzelevitch, C. Sodium channel block produces opposite electrophysiological effects in canine ventricular epicardium and endocardium. Circ. Res. 1991, 69, 277–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, P.; Schleimer, J.-H.; Schreiber, S. Modifications of sodium channel voltage dependence induce arrhythmia-favouring dynamics of cardiac action potentials. PLoS ONE 2020, 15, e0236949. [Google Scholar] [CrossRef]
- Remme, C.A.; Wilde, A.A.M.; Bezzina, C.R. Cardiac sodium channel overlap syndromes: Different faces of SCN5A mutations. Trends Cardiovasc. Med. 2008, 18, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.-H.; Lenkowski, P.W.; Lee, H.C.; Mounsey, P.; Patel, M.K. Modulation of Nav1.5 by β1- and β3-subunit co-expression in mammalian cells. Pflügers Arch. 2005, 449, 403–412. [Google Scholar] [CrossRef]
- O’Malley, H.A.; Isom, L.L. Sodium channel β subunits: Emerging targets in channelopathies. Annu. Rev. Physiol. 2015, 77, 481–504. [Google Scholar] [CrossRef] [Green Version]
- Amin, A.S.; Verkerk, A.O.; Bhuiyan, Z.A.; Wilde, A.A.M.; Tan, H.L. Novel Brugada syndrome-causing mutation in ion-conducting pore of cardiac Na+ channel does not affect ion selectivity properties. Acta Physiol. Scand. 2005, 185, 291–301. [Google Scholar] [CrossRef]
- Ghovanloo, M.-R.; Shuart, N.G.; Mezeyova, J.; Dean, R.A.; Ruben, P.C.; Goodchild, S.J. Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J. Biol. Chem. 2018, 293, 16546–16558. [Google Scholar] [CrossRef] [PubMed]
- Rapedius, M.; Obergrussberger, A.; Humphries, E.S.A.; Scholz, S.; Rinke-Weiss, I.; Goetze, T.A.; Brinkwirth, N.; Rotordam, M.G.; Strassmaier, T.; Randolph, A.; et al. There is no F in APC: Using physiological fluoride-free solutions for high throughput automated patch clamp experiments. Front. Mol. Neurosci. 2022, 15, 982316. [Google Scholar] [CrossRef] [PubMed]
- Sindrup, S.H.; Brøsen, K.; Gram, L.F. Pharmacokinetics of the selective serotonin reuptake inhibitor paroxetine: Nonlinearity and relation to the sparteine oxidation polymorphism. Clin. Pharmacol. Ther. 1992, 51, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Hiemke, C.; Bergemann, N.; Clement, H.W.; Conca, A.; Deckert, J.; Domschke, K.; Eckermann, G.; Egberts, K.; Gerlach, M.; Greiner, C.; et al. Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: Update 2017. Pharmacopsychiatry 2018, 51, 9–62. [Google Scholar] [CrossRef] [Green Version]
- Gilles, M.; Deuschle, M.; Kellner, S.; Shams, M.; Krumm, B.; Härtter, S.; Heuser, I.; Hiemke, C. Paroxetine serum concentrations in depressed patients and response to treatment. Pharmacopsychiatry 2005, 38, 118–121. [Google Scholar] [CrossRef]
- Duboff, E.A. Long-term treatment of major depressive disorder with paroxetine. J. Clin. Psychopharmacol. 1993, 13, 28S–33S. [Google Scholar] [CrossRef]
- Kaye, C.M.; Haddock, R.E.; Langley, P.F.; Mellows, G.; Tasker, T.C.G.; Zussman, B.D.; Greb, W.H. A review of the metabolism and pharmacokinetics of paroxetine in man. Acta Psychiatr. Scand. 1989, 80 (Suppl. S350), 60–75. [Google Scholar] [CrossRef] [PubMed]
- Gunasekara, N.S.; Noble, S.; Benfield, P. Paroxetine: An update of its pharmacology and therapeutic use in depression and a review of its use in other disorders. Drugs 1998, 55, 85–120. [Google Scholar] [CrossRef]
- Lewis, R.J.; Kemp, P.M.; Johnson, R.D. Paroxetine in postmortem fluids and tissues from nine aviation accident victims. J. Anal. Toxicol. 2015, 39, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.J.; Charlton, S.J. Assessing the minimum number of data points required for accurate IC50 determination. Assay Drug Dev. Technol. 2005, 3, 525–531. [Google Scholar] [CrossRef]
- Portero, V.; Wilders, R.; Casini, S.; Charpentier, F.; Verkerk, A.O.; Remme, C.A. KV4.3 expression modulates NaV1.5 sodium current. Front. Physiol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Veldkamp, M.W.; Geuzebroek, G.S.C.; Baartscheer, A.; Verkerk, A.O.; Schumacher, C.A.; Suarez, G.G.; Berger, W.R.; Casini, S.; Van Amersfoorth, S.C.M.; Scholman, K.T.; et al. Neurokinin-3 receptor activation selectively prolongs atrial refractoriness by inhibition of a background K+ channel. Nat. Commun. 2018, 9, 4357. [Google Scholar] [CrossRef] [Green Version]
- Barry, P.H.; Lynch, J.W. Liquid junction potentials and small cell effects in patch-clamp analysis. J. Membr. Biol. 1991, 121, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Rush, S.; Larsen, H. A practical algorithm for solving dynamic membrane equations. IEEE Trans. Biomed. Eng. 1978, 25, 389–392. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plijter, I.S.; Verkerk, A.O.; Wilders, R. The Antidepressant Paroxetine Reduces the Cardiac Sodium Current. Int. J. Mol. Sci. 2023, 24, 1904. https://doi.org/10.3390/ijms24031904
Plijter IS, Verkerk AO, Wilders R. The Antidepressant Paroxetine Reduces the Cardiac Sodium Current. International Journal of Molecular Sciences. 2023; 24(3):1904. https://doi.org/10.3390/ijms24031904
Chicago/Turabian StylePlijter, Ingmar S., Arie O. Verkerk, and Ronald Wilders. 2023. "The Antidepressant Paroxetine Reduces the Cardiac Sodium Current" International Journal of Molecular Sciences 24, no. 3: 1904. https://doi.org/10.3390/ijms24031904
APA StylePlijter, I. S., Verkerk, A. O., & Wilders, R. (2023). The Antidepressant Paroxetine Reduces the Cardiac Sodium Current. International Journal of Molecular Sciences, 24(3), 1904. https://doi.org/10.3390/ijms24031904