
Gut Microbiota-Derived TMAO: A Causal Factor Promoting Atherosclerotic Cardiovascular Disease?
 , , , and
, , , and
Abstract
:1. Introduction
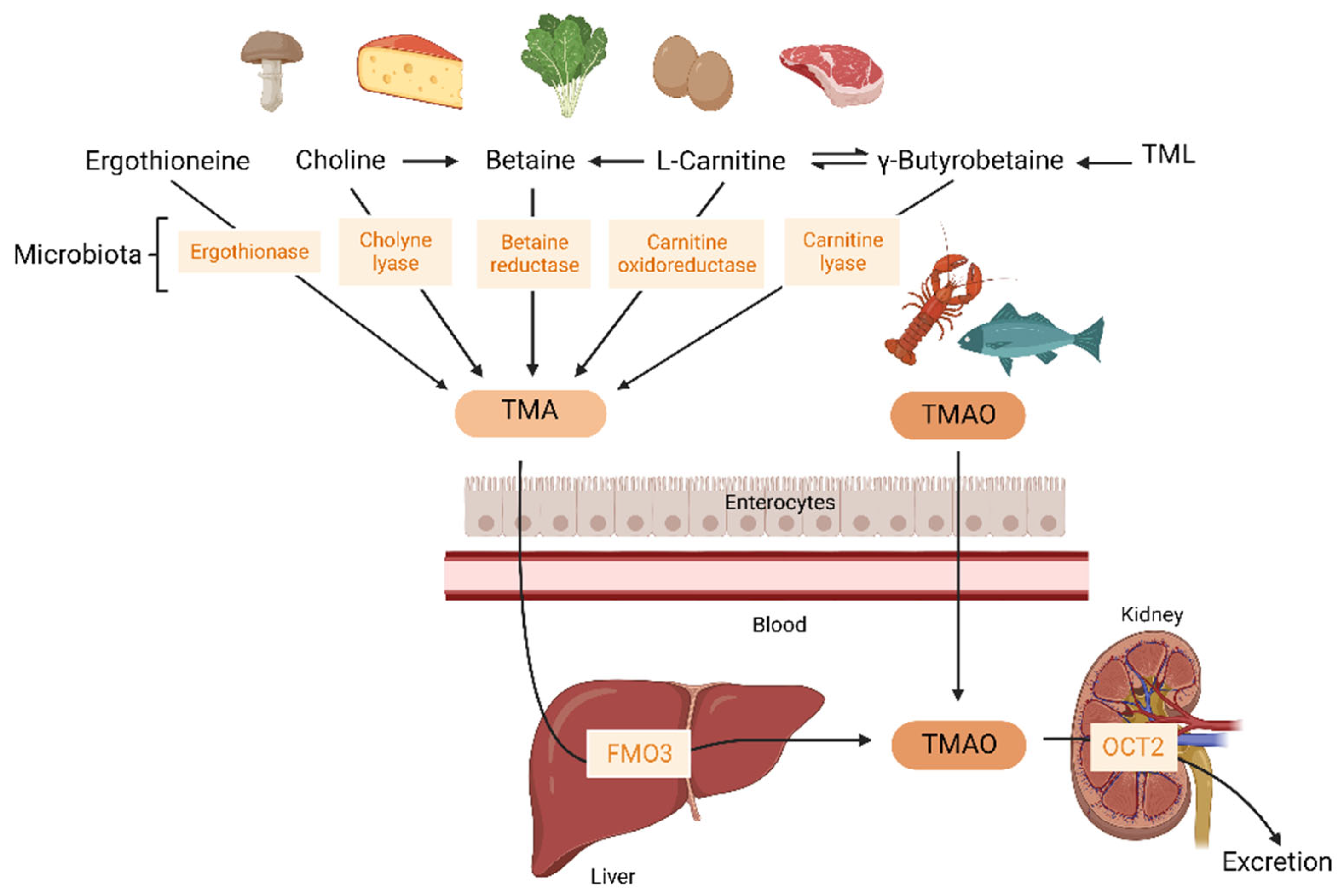
2. Trimethylamine-N-Oxide (TMAO)
2.1. Microbial Pathways Leading to TMA Production in the Gut
2.2. Dietary Precursors of TMA and TMAO
2.3. TMAO Metabolism
3. TMAO in Prospective Studies

{kind=link}
{kind=link}
| Year | N | Follow-Up | Outcomes | Covariates in the Adjusted Models | Hazard Ratio (95% CI) and p Value | Ref. |
|---|---|---|---|---|---|---|
| Individuals free of known CVD: community-based population | ||||||
| 2013 | 4007 | 3 years | MACE | 1: age, sex, smoking status, SBP, LDLc, HDLc, diabetes, CRP 2: model 1 + myeloperoxidase level, eGFR, total white-cell count, BMI, medication 3: model 2 + extent of the disease | UA: 2.54 (1.96–3.28) p < 0.001 1: 1.88 (1.44–2.44) p < 0.001 2: 1.49 (1.10–2.03) p = 0.01 3: 1.43 (1.05–1.94) p = 0.02 | [45] |
| 2021 | 4131 | 15 years | ASCVD | Age, sex, race, study site, education, income, health status, smoking status, alcohol intake, physical activity, BMI, WC, lipid-lowering medication, antihypertensive medication, antibiotics, T2D, HDL-C, LDL-C, TG, CRP, SBP, DBP, diet, eGFR | Adj: 1.07 (0.90–1.27) p = 0.516 Adj: 1.37 (0.98–1.90) p = 0.084 (individuals with eGFR < 60 mL/min 1.73 m2) Adj: 0.98 (0.80–1.20) p = 0.903 (individuals with eGFR ≥ 60 mL/min 1.73 m2) | [34] |
| 2022 | 5333 | 13.2 years | All-cause mortality Cardiovascular mortality Non-cardiovascular mortality | 1: age, sex, race, and enrollment site 2: model 1 + education, household income, smoking, BMI, physical activity, treated hypertension, instrumental activities of daily living, self-reported health status, systolic blood pressure, HDL cholesterol, prevalent atrial fibrillation, prevalent coronary heart disease, myocardial infarction, prevalent diabetes, prevalent chronic obstructive pulmonary disease, and reported daily intake of eggs, fish, liver, non-processed red meat, processed meat, and total calories 3: model 2 + eGFR | 1: 1.36 (1.24–1.51) p < 0.001 2: 1.30 (1.17–1.44) p < 0.001 3: 1.07 (0.96–1.19) p = 0.51 1: 1.50 (1.27–1.77) p < 0.0001 2: 1.35 (1.14–1.60) p < 0.0001 3: 1.09 (0.91–1.30) p = 0.65 1: 1.29 (1.14–1.46) p < 0.0001 2: 1.27 (1.12–1.44) p = 0.0003 3: 1.06 (0.93–1.21) p = 0.61 | [32] |
| 2022 | 6393 | 10.5 years | All-cause mortality Cardiovascular mortality Non-cardiovascular mortality | 1: age and sex 2: model 1 + HT, diabetes mellitus, total cholesterol, smoking, BMI, and eGFR | UA: 3.07 (2.48–3.80) p < 0.001 1: 1.09 (0.88–1.37) p = 0.43 2: 1.11 (0.88–1.40) p = 0.38 UA: 3.75 (2.56–5.49) p < 0.001 1: 1.13 (0.77–1.70) p = 0.54 2: 0.97 (0.65–1.46) p = 0.88 UA: 2.77 (2.14–3.60) p < 0.001 1: 1.07 (0.82–1.40) p = 0.64 2: 1.17 (0.88–1.54) p = 0.28 | [33] |
| Patients with CVD or CAD | ||||||
| 2015 | 339 | 8 years | MACE | eGFR | UA: 1.01 (0.98–1.04) p = 0.493 Adj: 0.991 (0.98–1.00) p = 0.062 | [26] |
| 2016 | 2235 | 5 years | All-cause mortality | 1: age, sex, SBP, LDL-C, HDL-C, smoking, and diabetes mellitus 2: model 1 + hsCRP and eGFR 3: model 2 + medications (angiotensin-converting enzyme inhibitor, angiotensin-receptor blocker, β-blocker, aspirin, or statin), number of stenotic vessels, myeloperoxidase, and BNP | UA: 3.90 (2.78–5.48) p < 0.0001 1: 2.61 (1.82–3.76) p < 0.0001 2: 1.95 (1.33–2.86) p = 0.003 3: 1.71 (1.11–2.61) p = 0.0138 | [36] |
| 2021 | 1287 | 15 years | ASCVD | Age, sex, race, study site, education, income, health status, smoking status, alcohol intake, physical activity, BMI, WC, lipid-lowering medication, antihypertensive medication, antibiotics, T2D, HDL-C, LDL-C, TG, CRP, SBP, DBP, diet, eGFR | 1.10 (0.87–1.39) p = 0.179 | [34] |
| 2021 | 449 | 9 years | All-cause mortality | 1: BMI, diabetes status, eGFR 2: model 1 + age, sex, smoking status, SBP, DBP, hsCRP, HDL-C, LDL-C, white blood cell count, and in LIFE-CAD additionally for presence of CAD | UA: 1.61 (1.36–1.92) p < 0.001 1: 1.30 (1.07–1.58) p = 0.009 2: 1.24 (1.01–1.51) p = 0.040 | [35] |
| 2022 | 1726 | 5 years | All-cause mortality Cardiovascular mortality AMI | 1: age, gender, and history of CAD 2: pre-defined patient characteristics, cardiovascular risk factors, and medical history, including age, gender, body mass index, smoking history, positive cardiovascular family history, hypertension, hypercholesterolemia, history of diabetes, history of stroke, history of CAD, previous AMI, history of heart failure and adjudicated functionally relevant coronary artery disease* Both models also adjusted adding Cystatin-C | UA: 1.42 (1.29–1.58) p < 0.001 1: 1.28 (1.13–1.43) p < 0.001 1 *: 1.16 (1.03–1.30) p = 0.013 2: 1.23 (1.09–1.38) p < 0.001 2 *: 1.11 (0.99–1.26) p = 0.084 UA: 1.60 (1.39–1.84) p < 0.001 1: 1.44 (1.4–1.68) p < 0.001 1 *: 1.28 (1.10–1.48) p < 0.001 2: 1.36 (1.16–1.58) p < 0.001 2 *: 1.19 (1.01–1.40) p = 0.032 UA: 1.38 (1.17–1.65) p < 0.001 1: 1.33 (1.11–1.60) p = 0.002 1 *: 1.22 (1.01–1.48) p = 0.043 2: 1.32 (1.08–1.60) p = 0.006 2 *: 1.17 (0.95–1.45) p = 0.146 | [46] |
| 2022 | 4132 | 9.8 years | All-cause mortality Cardiovascular mortality Non-cardiovascular mortality | 1: age and sex 2: model 1 + HT, diabetes mellitus, total cholesterol, smoking, BMI, and eGFR | UA: 2.29 (1.88–2.79) p < 0.001 1: 1.32 (1.08–1.62) p = 0.01 2: 1.07 (0.86–1.32) p = 0.56 UA: 2.73 (2.00–3.72) p < 0.001 1: 1.52 (1.11–2.09) p = 0.01 2: 1.16 (0.83–1.62) p = 0.38 UA: 2.02 (1.57–2.61) p < 0.001 1: 1.20 (0.92–1.56) p = 0.18 2: 1.02 (0.77–1.34) p = 0.90 | [33] |
| ACS patients | ||||||
| 2017 | 1079 | 2 years | All-cause mortality Death/MI | Age, sex, SBP, heart rate, past histories of MI/angina, increased BP and diabetes, Killip score, STEMI class, revascularization, and medication | UA: 1.21 (0.98–1.48) p = 0.074 UA: 1.4 (1.26–1.55) p < 0.0005 Adj: 1.21 (1.03–1.439) p = 0.023 | [37] |
| 2017 | 530 | 30 days 6 months 7 years | MACE (30 days and 6 months) All-cause mortality (7 years) | Age, gender, HDL-C, LDL-C, smoking, presence or absence of a history of diabetes mellitus, HT, hyperlipidemia, revascularization or CAD, CRP level, eGFR, initial cTnT level, diagnosis of either STEMI, nonSTEMI, or unstable angina | UA: 2.29 (1.39–3.79) p < 0.01 Adj: 6.3 (1.89–21.00) p < 0.01 UA: 2.48 (1.51–4.09) p < 0.001 Adj: 5.65 (1.91–16.74) p < 0.01 UA: 3.72 (2.23–6.20) p < 0.001 Adj: 1.81 (1.04–3.15) p < 0.05 | [38] |
| 2021 | 326 | 9 years | All-cause mortality | 1: BMI, diabetes status, eGFR 2: model 1 + age, sex, smoking status, SBP, DBP, hsCRP, HDL-C, LDL-C, white blood cell count, and in LIFE-CAD additionally for presence of CAD | UA: 1.39 (0.98–1.97) p = 0.066 1: 1.10 (0.74–1.65) p = 0.638 2: 1.07 (0.70–1.63) p = 0.771 | [35] |
| 2021 | 292 | 7 years | Cardiovascular mortality | Prior MI, diabetes, age, gender, diabetes, hemoglobin and creatinine levels at admission | UA: 21.33 (4.7–96.5) p < 0.001 Adj: 11.62 (2.3–59.7) p = 0.003 | [47] |
| 2022 | 309 | 6.7 years | MACE | 1: age, sex, BMI, smoking, HT, dyslipidemia, type 2 diabetes, unstable angina, STEMI, nonSTEMI, statin medication, beta-blockers, oral antidiabetic medication, insulin medication, diuretics, aspirin 2: model 1 + eGFR | UA: 2.63 (1.69–4.08) p < 0.001 1: 1.83 (1.08–3.09) p = 0.043 2: 1.66 (0.98–2.82) p = 0.119 | [42] |
| Stable CKD patients | ||||||
| 2015 | 521 | 5 years | All-cause mortality | Age, sex, systolic blood pressure, LDL-C, HDL-C, smoking, diabetes mellitus, hsCRP, and eGFR | UA: 2.76 (1.74–4.37) p < 0.001 Adj: 1.93 (1.13–3.29) p < 0.05 | [41] |
| 2016 | 2529 | 3 years | Ischemic events | 1: age, sex, race, diabetes and cardiovascular comorbidities at baseline 2: model 1 + traditional cardiovascular risk factors 3: model 2 + chronic kidney disease-specific risk factors 4: Adjusted for all covariates with statically significant association with ischemic events | UA: 1.45 (1.28–1.64) 1: 1.38 (1.21–1.57) p < 0.0001 2: 1.39 (1.22–1.59) p = 0.0055 3: 1.24 (1.07–1.43) p = 0.065 4: 1.23 (1.06–1.42) p = 0.0059 | [39] |
| 2016 | 339 | 3.3 years | All-cause mortality | 1: age, race, sex 2: model 1 + SBP, LDL-C, HDL-C, smoking, CRP, eGFR | 1: 2.45 (1.09–5.50) p = 0.02 2: 1.25 (0.48–3.28) p = 0.37 | [40] |
4. TMAO in Genetic Studies
5. Mechanisms Related to TMAO Atherogenic Effect
5.1. TMAO, Cholesterol Metabolism Disruption, and Atherosclerotic Plaque Formation
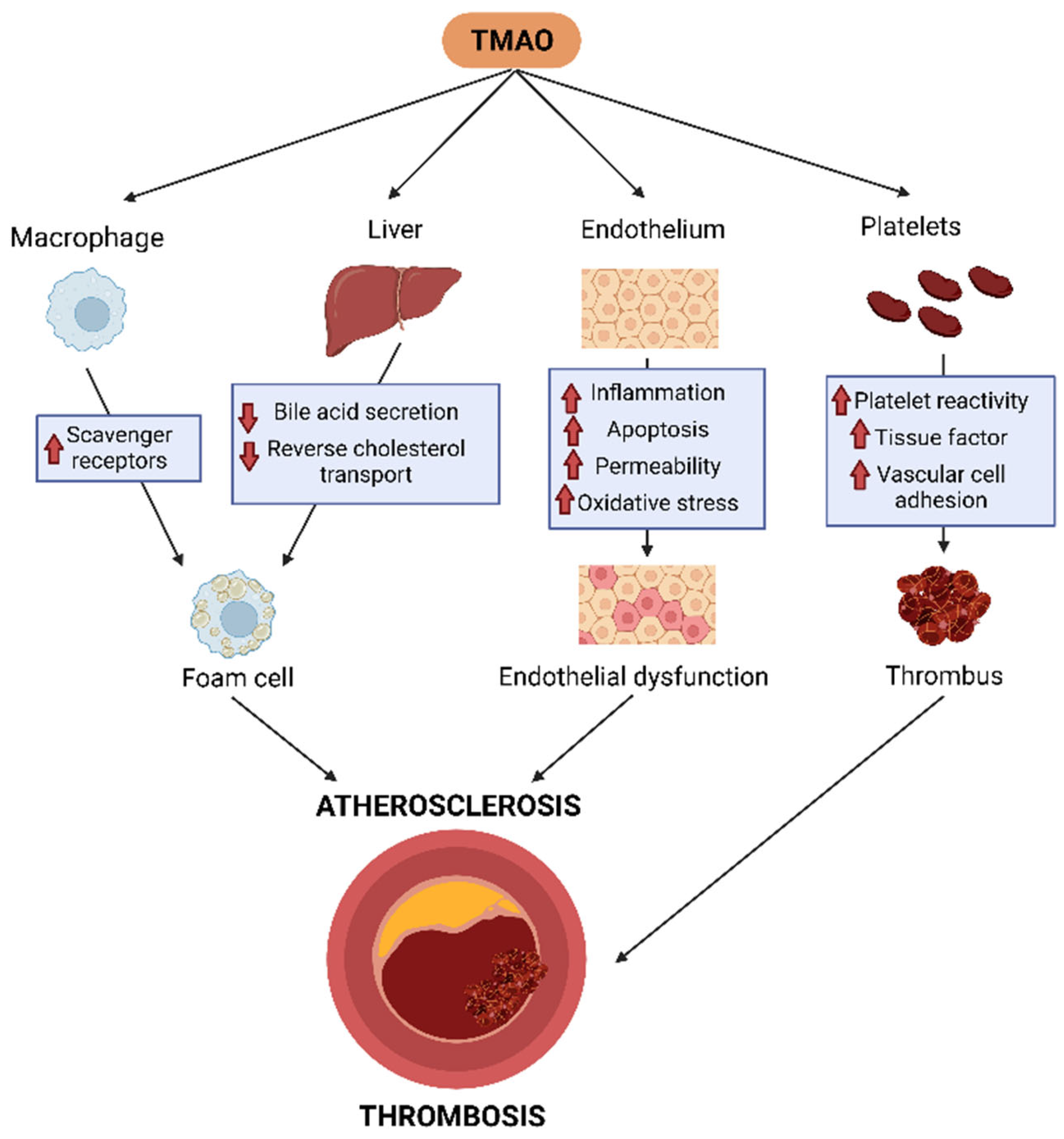
5.2. TMAO, Inflammation, and Endothelial Dysfunction
5.3. TMAO, Platelet Activation, and Thrombosis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, C.; Moré, M.; Bellamine, A. Trimethylamine N-Oxide in Relation to Cardiometabolic Health—Cause or Effect? Nutrients 2020, 12, 1330. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Levison, B.S.; Culley, M.K.; Buffa, J.A.; Wang, Z.; Gregory, J.C.; Org, E.; Wu, Y.; Li, L.; Smith, J.D.; et al. γ-Butyrobetaine Is a Proatherogenic Intermediate in Gut Microbial Metabolism of L-Carnitine to TMAO. Cell Metab. 2014, 20, 799–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Heianza, Y.; Ma, W.; Manson, J.E.; Rexrode, K.M.; Qi, L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J. Am. Heart Assoc. 2017, 6, e004947. [Google Scholar] [CrossRef]
- Qi, J.; You, T.; Li, J.; Pan, T.; Xiang, L.; Han, Y.; Zhu, L. Circulating Trimethylamine N-oxide and the Risk of Cardiovascular Diseases: A Systematic Review and Meta-analysis of 11 Prospective Cohort Studies. J. Cell. Mol. Med. 2018, 22, 185–194. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [Green Version]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the Trimethylamine-Producing Bacteria of the Human Gut Microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- Craciun, S.; Balskus, E.P. Microbial Conversion of Choline to Trimethylamine Requires a Glycyl Radical Enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 21307–21312. [Google Scholar] [CrossRef]
- Zhu, Y.; Jameson, E.; Crosatti, M.; Schäfer, H.; Rajakumar, K.; Bugg, T.D.H.; Chen, Y. Carnitine Metabolism to Trimethylamine by an Unusual Rieske-Type Oxygenase from Human Microbiota. Proc. Natl. Acad. Sci. USA 2014, 111, 4268–4273. [Google Scholar] [CrossRef] [Green Version]
- Falony, G.; Vieira-Silva, S.; Raes, J. Microbiology Meets Big Data: The Case of Gut Microbiota–Derived Trimethylamine. Annu. Rev. Microbiol. 2015, 69, 305–321. [Google Scholar] [CrossRef]
- Cho, C.E.; Taesuwan, S.; Malysheva, O.V.; Bender, E.; Tulchinsky, N.F.; Yan, J.; Sutter, J.L.; Caudill, M.A. Trimethylamine-N-Oxide (TMAO) Response to Animal Source Foods Varies among Healthy Young Men and Is Influenced by Their Gut Microbiota Composition: A Randomized Controlled Trial. Mol. Nutr. Food Res. 2017, 61, 1600324. [Google Scholar] [CrossRef]
- Cho, C.E.; Caudill, M.A. Trimethylamine-N-Oxide: Friend, Foe, or Simply Caught in the Cross-Fire? Trends Endocrinol. Metab. 2017, 28, 121–130. [Google Scholar] [CrossRef]
- Seline, K.-G.; Johein, H. The Determination of L-Carnitine in Several Food Samples. Food Chem. 2007, 105, 793–804. [Google Scholar] [CrossRef]
- Patterson, K.Y.; Bhagwat, S.A.; Williams, J.R.; Howe, J.C.; Holden, J.M.; Zeisel, S.H.; Dacosta, K.A.; Mar, M.-H. USDA Database for the Choline Content of Common Foods, Release 2 (2008); Nutrient Data Laboratory, Beltsville Human Nutrition Research Center, ARS, USDA: Beltsville, MD, USA, 2015. [Google Scholar]
- Servillo, L.; Giovane, A.; Cautela, D.; Castaldo, D.; Balestrieri, M.L. Where Does Nε-Trimethyllysine for the Carnitine Biosynthesis in Mammals Come From? PLoS ONE 2014, 9, e84589. [Google Scholar] [CrossRef] [Green Version]
- Stremmel, W.; Schmidt, K.V.; Schuhmann, V.; Kratzer, F.; Garbade, S.F.; Langhans, C.-D.; Fricker, G.; Okun, J.G. Blood Trimethylamine-N-Oxide Originates from Microbiota Mediated Breakdown of Phosphatidylcholine and Absorption from Small Intestine. PLoS ONE 2017, 12, e0170742. [Google Scholar] [CrossRef]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-Oxide, a Metabolite Associated with Atherosclerosis, Exhibits Complex Genetic and Dietary Regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Schugar, R.C.; Brown, J.M. Emerging Roles of Flavin Monooxygenase 3 in Cholesterol Metabolism and Atherosclerosis. Curr. Opin. Lipidol. 2015, 26, 426–431. [Google Scholar] [CrossRef] [Green Version]
- Esposito, T.; Varriale, B.; D’Angelo, R.; Amato, A.; Sidoti, A. Regulation of Flavin-Containing Mono-Oxygenase (Fmo3) Gene Expression by Steroids in Mice and Humans. Horm. Mol. Biol. Clin. Investig. 2014, 20, 99–109. [Google Scholar] [CrossRef]
- Rohrmann, S.; Linseisen, J.; Allenspach, M.; von Eckardstein, A.; Müller, D. Plasma Concentrations of Trimethylamine-N-Oxide Are Directly Associated with Dairy Food Consumption and Low-Grade Inflammation in a German Adult Population. J. Nutr. 2016, 146, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühn, T.; Rohrmann, S.; Sookthai, D.; Johnson, T.; Katzke, V.; Kaaks, R.; von Eckardstein, A.; Müller, D. Intra-Individual Variation of Plasma Trimethylamine-N-Oxide (TMAO), Betaine and Choline over 1 Year. Clin. Chem. Lab. Med. 2017, 55, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, R.; Merz, B.; Rist, M.J.; Ferrario, P.G.; Bub, A.; Kulling, S.E.; Watzl, B. Associations of Current Diet with Plasma and Urine TMAO in the KarMeN Study: Direct and Indirect Contributions. Mol. Nutr. Food Res. 2017, 61, 1700363. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Awwad, H.M.; Kirsch, S.H.; Waldura, C.; Herrmann, W.; Graeber, S.; Geisel, J. Plasma Trimethylamine-N-Oxide Following Supplementation with Vitamin D or D plus B Vitamins. Mol. Nutr. Food Res. 2017, 61, 1600358. [Google Scholar] [CrossRef]
- Mueller, D.M.; Allenspach, M.; Othman, A.; Saely, C.H.; Muendlein, A.; Vonbank, A.; Drexel, H.; von Eckardstein, A. Plasma Levels of Trimethylamine-N-Oxide Are Confounded by Impaired Kidney Function and Poor Metabolic Control. Atherosclerosis 2015, 243, 638–644. [Google Scholar] [CrossRef]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide Is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. JASN 2016, 27, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Taesuwan, S.; Cho, C.E.; Malysheva, O.V.; Bender, E.; King, J.H.; Yan, J.; Thalacker-Mercer, A.E.; Caudill, M.A. The Metabolic Fate of Isotopically Labeled Trimethylamine-N-Oxide (TMAO) in Humans. J. Nutr. Biochem. 2017, 45, 77–82. [Google Scholar] [CrossRef]
- Miyake, T.; Mizuno, T.; Mochizuki, T.; Kimura, M.; Matsuki, S.; Irie, S.; Ieiri, I.; Maeda, K.; Kusuhara, H. Involvement of Organic Cation Transporters in the Kinetics of Trimethylamine N-Oxide. J. Pharm. Sci. 2017, 106, 2542–2550. [Google Scholar] [CrossRef] [Green Version]
- Teft, W.A.; Morse, B.L.; Leake, B.F.; Wilson, A.; Mansell, S.E.; Hegele, R.A.; Ho, R.H.; Kim, R.B. Identification and Characterization of Trimethylamine-N-Oxide Uptake and Efflux Transporters. Mol. Pharm. 2017, 14, 310–318. [Google Scholar] [CrossRef]
- Sanchez-Gimenez, R.; Ahmed-Khodja, W.; Molina, Y.; Peiró, O.M.; Bonet, G.; Carrasquer, A.; Fragkiadakis, G.A.; Bulló, M.; Bardaji, A.; Papandreou, C. Gut Microbiota-Derived Metabolites and Cardiovascular Disease Risk: A Systematic Review of Prospective Cohort Studies. Nutrients 2022, 14, 2654. [Google Scholar] [CrossRef]
- Fretts, A.M.; Hazen, S.L.; Jensen, P.; Budoff, M.; Sitlani, C.M.; Wang, M.; de Oliveira Otto, M.C.; DiDonato, J.A.; Lee, Y.; Psaty, B.M.; et al. Association of Trimethylamine N-Oxide and Metabolites With Mortality in Older Adults. JAMA Netw. Open 2022, 5, e2213242. [Google Scholar] [CrossRef]
- Bjørnestad, E.Ø.; Dhar, I.; Svingen, G.F.T.; Pedersen, E.R.; Ørn, S.; Svenningsson, M.M.; Tell, G.S.; Ueland, P.M.; Sulo, G.; Laaksonen, R.; et al. Circulating Trimethylamine N-Oxide Levels Do Not Predict 10-Year Survival in Patients with or without Coronary Heart Disease. J. Intern. Med. 2022, 292, 915–924. [Google Scholar] [CrossRef]
- Lee, Y.; Nemet, I.; Wang, Z.; Lai, H.T.M.; de Oliveira Otto, M.C.; Lemaitre, R.N.; Fretts, A.M.; Sotoodehnia, N.; Budoff, M.; DiDonato, J.A.; et al. Longitudinal Plasma Measures of Trimethylamine N-Oxide and Risk of Atherosclerotic Cardiovascular Disease Events in Community-Based Older Adults. J. Am. Heart Assoc. 2021, 10, e020646. [Google Scholar] [CrossRef]
- Ringel, C.; Dittrich, J.; Gaudl, A.; Schellong, P.; Beuchel, C.F.; Baber, R.; Beutner, F.; Teren, A.; Engel, C.; Wirkner, K.; et al. Association of Plasma Trimethylamine N-Oxide Levels with Atherosclerotic Cardiovascular Disease and Factors of the Metabolic Syndrome. Atherosclerosis 2021, 335, 62–67. [Google Scholar] [CrossRef]
- Senthong, V.; Wang, Z.; Li, X.S.; Fan, Y.; Wu, Y.; Tang, W.H.W.; Hazen, S.L. Intestinal Microbiota-Generated Metabolite Trimethylamine-N-Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J. Am. Heart Assoc. 2016, 5, e002816. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Heaney, L.M.; Jones, D.J.L.; Ng, L.L. Trimethylamine N-Oxide and Risk Stratification after Acute Myocardial Infarction. Clin. Chem. 2017, 63, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Li, X.S.; Obeid, S.; Klingenberg, R.; Gencer, B.; Mach, F.; Räber, L.; Windecker, S.; Rodondi, N.; Nanchen, D.; Muller, O.; et al. Gut Microbiota-Dependent Trimethylamine N-Oxide in Acute Coronary Syndromes: A Prognostic Marker for Incident Cardiovascular Events beyond Traditional Risk Factors. Eur. Heart J. 2017, 38, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.B.; Morse, B.L.; Djurdjev, O.; Tang, M.; Muirhead, N.; Barrett, B.; Holmes, D.T.; Madore, F.; Clase, C.M.; Rigatto, C.; et al. Advanced Chronic Kidney Disease Populations Have Elevated Trimethylamine N-Oxide Levels Associated with Increased Cardiovascular Events. Kidney Int. 2016, 89, 1144–1152. [Google Scholar] [CrossRef]
- Robinson-Cohen, C.; Newitt, R.; Shen, D.D.; Rettie, A.E.; Kestenbaum, B.R.; Himmelfarb, J.; Yeung, C.K. Association of FMO3 Variants and Trimethylamine N-Oxide Concentration, Disease Progression, and Mortality in CKD Patients. PLoS ONE 2016, 11, e0161074. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut Microbiota-Dependent Trimethylamine N-Oxide (TMAO) Pathway Contributes to Both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney Disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef]
- Sanchez-Gimenez, R.; Peiró, Ó.M.; Bonet, G.; Carrasquer, A.; Fragkiadakis, G.A.; Bulló, M.; Papandreou, C.; Bardaji, A. Plasma Trimethylamine-N-Oxide, Its Precursors and Risk of Cardiovascular Events in Patients with Acute Coronary Syndrome: Mediating Effects of Renal Function. Front. Cardiovasc. Med. 2022, 9, 1000815. [Google Scholar] [CrossRef]
- Zhang, W.; Miikeda, A.; Zuckerman, J.; Jia, X.; Charugundla, S.; Zhou, Z.; Kaczor-Urbanowicz, K.E.; Magyar, C.; Guo, F.; Wang, Z.; et al. Inhibition of Microbiota-Dependent TMAO Production Attenuates Chronic Kidney Disease in Mice. Sci. Rep. 2021, 11, 518. [Google Scholar] [CrossRef]
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Amrein, M.; Li, X.S.; Walter, J.; Wang, Z.; Zimmermann, T.; Strebel, I.; Honegger, U.; Leu, K.; Schäfer, I.; Twerenbold, R.; et al. Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide (TMAO) and Cardiovascular Risk in Patients with Suspected Functionally Relevant Coronary Artery Disease (FCAD). Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2022, 111, 692–704. [Google Scholar] [CrossRef]
- Eyileten, C.; Jarosz-Popek, J.; Jakubik, D.; Gasecka, A.; Wolska, M.; Ufnal, M.; Postula, M.; Toma, A.; Lang, I.M.; Siller-Matula, J.M. Plasma Trimethylamine-N-Oxide Is an Independent Predictor of Long-Term Cardiovascular Mortality in Patients Undergoing Percutaneous Coronary Intervention for Acute Coronary Syndrome. Front. Cardiovasc. Med. 2021, 8, 728724. [Google Scholar] [CrossRef]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.C.; Timpson, N.; Davey Smith, G. Mendelian Randomization: Using Genes as Instruments for Making Causal Inferences in Epidemiology. Stat. Med. 2008, 27, 1133–1163. [Google Scholar] [CrossRef]
- Jia, J.; Dou, P.; Gao, M.; Kong, X.; Li, C.; Liu, Z.; Huang, T. Assessment of Causal Direction Between Gut Microbiota-Dependent Metabolites and Cardiometabolic Health: A Bidirectional Mendelian Randomization Analysis. Diabetes 2019, 68, 1747–1755. [Google Scholar] [CrossRef]
- Zhuang, Z.; Gao, M.; Yang, R.; Liu, Z.; Cao, W.; Huang, T. Causal Relationships between Gut Metabolites and Alzheimer’s Disease: A Bidirectional Mendelian Randomization Study. Neurobiol. Aging 2021, 100, 119.e15–119.e18. [Google Scholar] [CrossRef]
- Wang, H.; Luo, Q.; Ding, X.; Chen, L.; Zhang, Z. Trimethylamine N-Oxide and Its Precursors in Relation to Blood Pressure: A Mendelian Randomization Study. Front. Cardiovasc. Med. 2022, 9, 922441. [Google Scholar] [CrossRef]
- Ge, X.; Zheng, L.; Zhuang, R.; Yu, P.; Xu, Z.; Liu, G.; Xi, X.; Zhou, X.; Fan, H. The Gut Microbial Metabolite Trimethylamine N-Oxide and Hypertension Risk: A Systematic Review and Dose-Response Meta-Analysis. Adv. Nutr. 2020, 11, 66–76. [Google Scholar] [CrossRef]
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-Oxide Promotes Atherosclerosis via CD36-Dependent MAPK/JNK Pathway. Biomed. Pharmacother. 2018, 97, 941–947. [Google Scholar] [CrossRef]
- Mohammadi, A.; Vahabzadeh, Z.; Jamalzadeh, S.; Khalili, T. Trimethylamine-N-Oxide, as a Risk Factor for Atherosclerosis, Induces Stress in J774A.1 Murine Macrophages. Adv. Med. Sci. 2018, 63, 57–63. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Wang, Z.; Fan, Y.; Levison, B.; Hazen, J.E.; Donahue, L.M.; Wu, Y.; Hazen, S.L. Prognostic Value of Elevated Levels of Intestinal Microbe-Generated Metabolite Trimethylamine-N-Oxide in Patients with Heart Failure: Refining the Gut Hypothesis. J. Am. Coll. Cardiol. 2014, 64, 1908–1914. [Google Scholar] [CrossRef] [Green Version]
- Manor, O.; Zubair, N.; Conomos, M.P.; Xu, X.; Rohwer, J.E.; Krafft, C.E.; Lovejoy, J.C.; Magis, A.T. A Multi-Omic Association Study of Trimethylamine N-Oxide. Cell Rep. 2018, 24, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal Microbiota Metabolism of L-Carnitine, a Nutrient in Red Meat, Promotes Atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Collins, H.L.; Drazul-Schrader, D.; Sulpizio, A.C.; Koster, P.D.; Williamson, Y.; Adelman, S.J.; Owen, K.; Sanli, T.; Bellamine, A. L-Carnitine Intake and High Trimethylamine N-Oxide Plasma Levels Correlate with Low Aortic Lesions in ApoE(-/-) Transgenic Mice Expressing CETP. Atherosclerosis 2016, 244, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, A.; Najar, A.G.; Yaghoobi, M.M.; Jahani, Y.; Vahabzadeh, Z. Trimethylamine-N-Oxide Treatment Induces Changes in the ATP-Binding Cassette Transporter A1 and Scavenger Receptor A1 in Murine Macrophage J774A.1 Cells. Inflammation 2016, 39, 393–404. [Google Scholar] [CrossRef]
- Mohammadi, A.; Gholamhoseyniannajar, A.; Yaghoobi, M.M.; Jahani, Y.; Vahabzadeh, Z. Expression Levels of Heat Shock Protein 60 and Glucose-Regulated Protein 78 in Response to Trimethylamine-N-Oxide Treatment in Murine Macrophage J774A.1 Cell Line. Cell. Mol. Biol. 2015, 61, 94–100. [Google Scholar]
- Canyelles, M.; García-Osuna, Á.; Junza, A.; Yanes, O.; Puig, N.; Ordóñez-Llanos, J.; Sionis, A.; Sans-Roselló, J.; Alquézar-Arbé, A.; Santos, D.; et al. The Capacity of APOB-Depleted Plasma in Inducing ATP-Binding Cassette A1/G1-Mediated Macrophage Cholesterol Efflux—But Not Gut Microbial-Derived Metabolites—Is Independently Associated with Mortality in Patients with ST-Segment Elevation Myocardial Infarction. Biomedicines 2021, 9, 1336. [Google Scholar] [CrossRef]
- Mistry, R.H.; Verkade, H.J.; Tietge, U.J.F. Reverse Cholesterol Transport Is Increased in Germ-Free Mice—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temel, R.E.; Brown, J.M. Biliary and Nonbiliary Contributions to Reverse Cholesterol Transport. Curr. Opin. Lipidol. 2012, 23, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, D.M.; Wang, Z.; Lee, R.; Meng, Y.; Che, N.; Charugundla, S.; Qi, H.; Wu, J.; Pan, C.; Brown, J.M.; et al. Flavin Containing Monooxygenase 3 Exerts Broad Effects on Glucose and Lipid Metabolism and Atherosclerosis. J. Lipid Res. 2015, 56, 22–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulanger, C.M. Endothelium. Arterioscler. Thromb. Vasc. Biol. 2016, 36, e26–e31. [Google Scholar] [CrossRef] [Green Version]
- Chou, R.-H.; Chen, C.-Y.; Chen, I.-C.; Huang, H.-L.; Lu, Y.-W.; Kuo, C.-S.; Chang, C.-C.; Huang, P.-H.; Chen, J.-W.; Lin, S.-J. Trimethylamine N-Oxide, Circulating Endothelial Progenitor Cells, and Endothelial Function in Patients with Stable Angina. Sci. Rep. 2019, 9, 4249. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-Oxide in Atherogenesis: Impairing Endothelial Self-Repair Capacity and Enhancing Monocyte Adhesion. Biosci. Rep. 2017, 37, BSR20160244. [Google Scholar] [CrossRef] [Green Version]
- Brunt, V.E.; Gioscia-Ryan, R.A.; Casso, A.G.; VanDongen, N.S.; Ziemba, B.P.; Sapinsley, Z.J.; Richey, J.J.; Zigler, M.C.; Neilson, A.P.; Davy, K.P.; et al. Trimethylamine-N-Oxide Promotes Age-Related Vascular Oxidative Stress and Endothelial Dysfunction in Mice and Healthy Humans. Hypertension 2020, 76, 101–112. [Google Scholar] [CrossRef]
- Li, T.; Chen, Y.; Gua, C.; Li, X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front. Physiol. 2017, 8, 350. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-Oxide Induces Inflammation and Endothelial Dysfunction in Human Umbilical Vein Endothelial Cells via Activating ROS-TXNIP-NLRP3 Inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef]
- Boini, K.M.; Hussain, T.; Li, P.-L.; Koka, S.S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell. Physiol. Biochem. 2017, 44, 152–162. [Google Scholar] [CrossRef]
- Wu, P.; Chen, J.; Chen, J.; Tao, J.; Wu, S.; Xu, G.; Wang, Z.; Wei, D.; Yin, W. Trimethylamine N-Oxide Promotes ApoE−/− Mice Atherosclerosis by Inducing Vascular Endothelial Cell Pyroptosis via the SDHB/ROS Pathway. J. Cell. Physiol. 2020, 235, 6582–6591. [Google Scholar] [CrossRef]
- Singh, G.B.; Zhang, Y.; Boini, K.M.; Koka, S. High Mobility Group Box 1 Mediates TMAO-Induced Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 20, e3570. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Wang, Z.; Tang, W.H.W.; Hazen, S.L. Gut Microbe-Generated Trimethylamine N-Oxide from Dietary Choline Is Prothrombotic in Subjects. Circulation 2017, 135, 1671–1673. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-Lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a Gut Microbe–Targeted Nonlethal Therapeutic to Inhibit Thrombosis Potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef]
- Skye, S.M.; Zhu, W.; Romano, K.A.; Guo, C.-J.; Wang, Z.; Jia, X.; Kirsop, J.; Haag, B.; Lang, J.M.; DiDonato, J.A.; et al. Microbial Transplantation with Human Gut Commensals Containing CutC Is Sufficient to Transmit Enhanced Platelet Reactivity and Thrombosis Potential. Circ. Res. 2018, 123, 1164–1176. [Google Scholar] [CrossRef]
- Zhu, W.; Buffa, J.A.; Wang, Z.; Warrier, M.; Schugar, R.; Shih, D.M.; Gupta, N.; Gregory, J.C.; Org, E.; Fu, X.; et al. Flavin Monooxygenase 3, the Host Hepatic Enzyme in the Metaorganismal Trimethylamine N-Oxide-Generating Pathway, Modulates Platelet Responsiveness and Thrombosis Risk. J. Thromb. Haemost. JTH 2018, 16, 1857–1872. [Google Scholar] [CrossRef] [Green Version]
- Shih, D.M.; Zhu, W.; Schugar, R.C.; Meng, Y.; Jia, X.; Miikeda, A.; Wang, Z.; Zieger, M.; Lee, R.; Graham, M.; et al. Genetic Deficiency of Flavin-Containing Monooxygenase 3 ( Fmo3) Protects against Thrombosis but Has only a Minor Effect on Plasma Lipid Levels-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1045–1054. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Qiu, X.; Liu, Y.; Yuan, C.; Yang, X. Trimethylamine N-Oxide Promotes Tissue Factor Expression and Activity in Vascular Endothelial Cells: A New Link between Trimethylamine N-Oxide and Atherosclerotic Thrombosis. Thromb. Res. 2019, 177, 110–116. [Google Scholar] [CrossRef]
- Witkowski, M.; Witkowski, M.; Friebel, J.; Buffa, J.A.; Li, X.S.; Wang, Z.; Sangwan, N.; Li, L.; DiDonato, J.A.; Tizian, C.; et al. Vascular Endothelial Tissue Factor Contributes to Trimethylamine N-Oxide-Enhanced Arterial Thrombosis. Cardiovasc. Res. 2022, 118, 2367–2384. [Google Scholar] [CrossRef] [PubMed]
- Trøseid, M.; Ueland, T.; Hov, J.R.; Svardal, A.; Gregersen, I.; Dahl, C.P.; Aakhus, S.; Gude, E.; Bjørndal, B.; Halvorsen, B.; et al. Microbiota-Dependent Metabolite Trimethylamine-N-Oxide Is Associated with Disease Severity and Survival of Patients with Chronic Heart Failure. J. Intern. Med. 2015, 277, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Shrestha, K.; Borowski, A.G.; Wu, Y.; Troughton, R.W.; Klein, A.L.; Hazen, S.L. Intestinal Microbiota-Dependent Phosphatidylcholine Metabolites, Diastolic Dysfunction, and Adverse Clinical Outcomes in Chronic Systolic Heart Failure. J. Card. Fail. 2015, 21, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Yazaki, Y.; Voors, A.A.; Jones, D.J.L.; Chan, D.C.S.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.; Hillege, H.L.; et al. Association with Outcomes and Response to Treatment of Trimethylamine N-Oxide in Heart Failure: Results from BIOSTAT-CHF. Eur. J. Heart Fail. 2019, 21, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israr, M.Z.; Bernieh, D.; Salzano, A.; Cassambai, S.; Yazaki, Y.; Heaney, L.M.; Jones, D.J.L.; Ng, L.L.; Suzuki, T. Association of Gut-Related Metabolites with Outcome in Acute Heart Failure. Am. Heart J. 2021, 234, 71–80. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Wang, Z.; Li, X.S.; Fan, Y.; Li, D.S.; Wu, Y.; Hazen, S.L. Increased Trimethylamine N-Oxide Portends High Mortality Risk Independent of Glycemic Control in Patients with Type 2 Diabetes Mellitus. Clin. Chem. 2017, 63, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Croyal, M.; Saulnier, P.-J.; Aguesse, A.; Gand, E.; Ragot, S.; Roussel, R.; Halimi, J.-M.; Ducrocq, G.; Cariou, B.; Montaigne, D.; et al. Plasma Trimethylamine N-Oxide and Risk of Cardiovascular Events in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2020, 105, dgaa188. [Google Scholar] [CrossRef]
- Winther, S.A.; Øllgaard, J.C.; Hansen, T.W.; von Scholten, B.J.; Reinhard, H.; Ahluwalia, T.S.; Wang, Z.; Gæde, P.; Parving, H.-H.; Hazen, S.; et al. Plasma Trimethylamine N-Oxide and Its Metabolic Precursors and Risk of Mortality, Cardiovascular and Renal Disease in Individuals with Type 2-Diabetes and Albuminuria. PLoS ONE 2021, 16, e0244402. [Google Scholar] [CrossRef]
- Wargny, M.; Croyal, M.; Ragot, S.; Gand, E.; Jacobi, D.; Trochu, J.-N.; Prieur, X.; Le May, C.; Goronflot, T.; Cariou, B.; et al. Nutritional Biomarkers and Heart Failure Requiring Hospitalization in Patients with Type 2 Diabetes: The SURDIAGENE Cohort. Cardiovasc. Diabetol. 2022, 21, 101. [Google Scholar] [CrossRef]
- Senthong, V.; Wang, Z.; Fan, Y.; Wu, Y.; Hazen, S.L.; Tang, W.H.W. Trimethylamine N-Oxide and Mortality Risk in Patients With Peripheral Artery Disease. J. Am. Heart Assoc. 2016, 5, e004237. [Google Scholar] [CrossRef]
- Roncal, C.; Martínez-Aguilar, E.; Orbe, J.; Ravassa, S.; Fernandez-Montero, A.; Saenz-Pipaon, G.; Ugarte, A.; Estella-Hermoso de Mendoza, A.; Rodriguez, J.A.; Fernández-Alonso, S.; et al. Trimethylamine-N-Oxide (TMAO) Predicts Cardiovascular Mortality in Peripheral Artery Disease. Sci. Rep. 2019, 9, 15580. [Google Scholar] [CrossRef]
- Skagen, K.; Trøseid, M.; Ueland, T.; Holm, S.; Abbas, A.; Gregersen, I.; Kummen, M.; Bjerkeli, V.; Reier-Nilsen, F.; Russell, D.; et al. The Carnitine-Butyrobetaine-Trimethylamine-N-Oxide Pathway and Its Association with Cardiovascular Mortality in Patients with Carotid Atherosclerosis. Atherosclerosis 2016, 247, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Kaysen, G.A.; Johansen, K.L.; Chertow, G.M.; Dalrymple, L.S.; Kornak, J.; Grimes, B.; Dwyer, T.; Chassy, A.W.; Fiehn, O. Associations of Trimethylamine N-Oxide With Nutritional and Inflammatory Biomarkers and Cardiovascular Outcomes in Patients New to Dialysis. J. Ren. Nutr. 2015, 25, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Zou, J.-Z.; Chen, J.; Tan, X.; Xiang, F.-F.; Shen, B.; Hu, J.-C.; Wang, J.-L.; Wang, Y.-Q.; Yu, J.-B.; et al. Association of Trimethylamine N-Oxide with Cardiovascular and All-Cause Mortality in Hemodialysis Patients. Ren. Fail. 2020, 42, 1004–1014. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canyelles, M.; Borràs, C.; Rotllan, N.; Tondo, M.; Escolà-Gil, J.C.; Blanco-Vaca, F. Gut Microbiota-Derived TMAO: A Causal Factor Promoting Atherosclerotic Cardiovascular Disease? Int. J. Mol. Sci. 2023, 24, 1940. https://doi.org/10.3390/ijms24031940
Canyelles M, Borràs C, Rotllan N, Tondo M, Escolà-Gil JC, Blanco-Vaca F. Gut Microbiota-Derived TMAO: A Causal Factor Promoting Atherosclerotic Cardiovascular Disease? International Journal of Molecular Sciences. 2023; 24(3):1940. https://doi.org/10.3390/ijms24031940
Chicago/Turabian StyleCanyelles, Marina, Carla Borràs, Noemí Rotllan, Mireia Tondo, Joan Carles Escolà-Gil, and Francisco Blanco-Vaca. 2023. "Gut Microbiota-Derived TMAO: A Causal Factor Promoting Atherosclerotic Cardiovascular Disease?" International Journal of Molecular Sciences 24, no. 3: 1940. https://doi.org/10.3390/ijms24031940