
Cembrane Diterpenes Possessing Nonaromatic Oxacycles from the Hainan Soft Coral Sarcophyton mililatensis
 and
and
Abstract
:1. Introduction
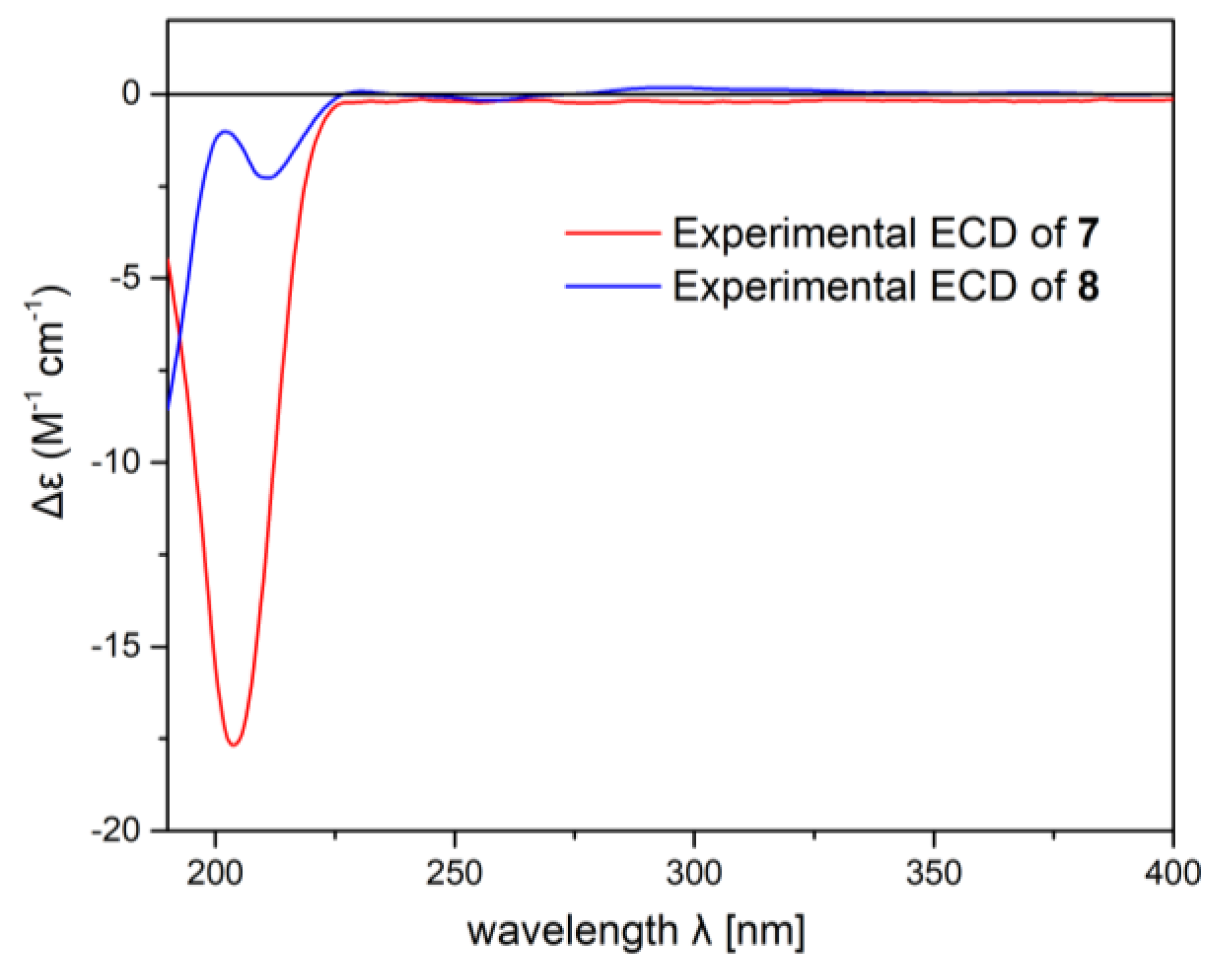
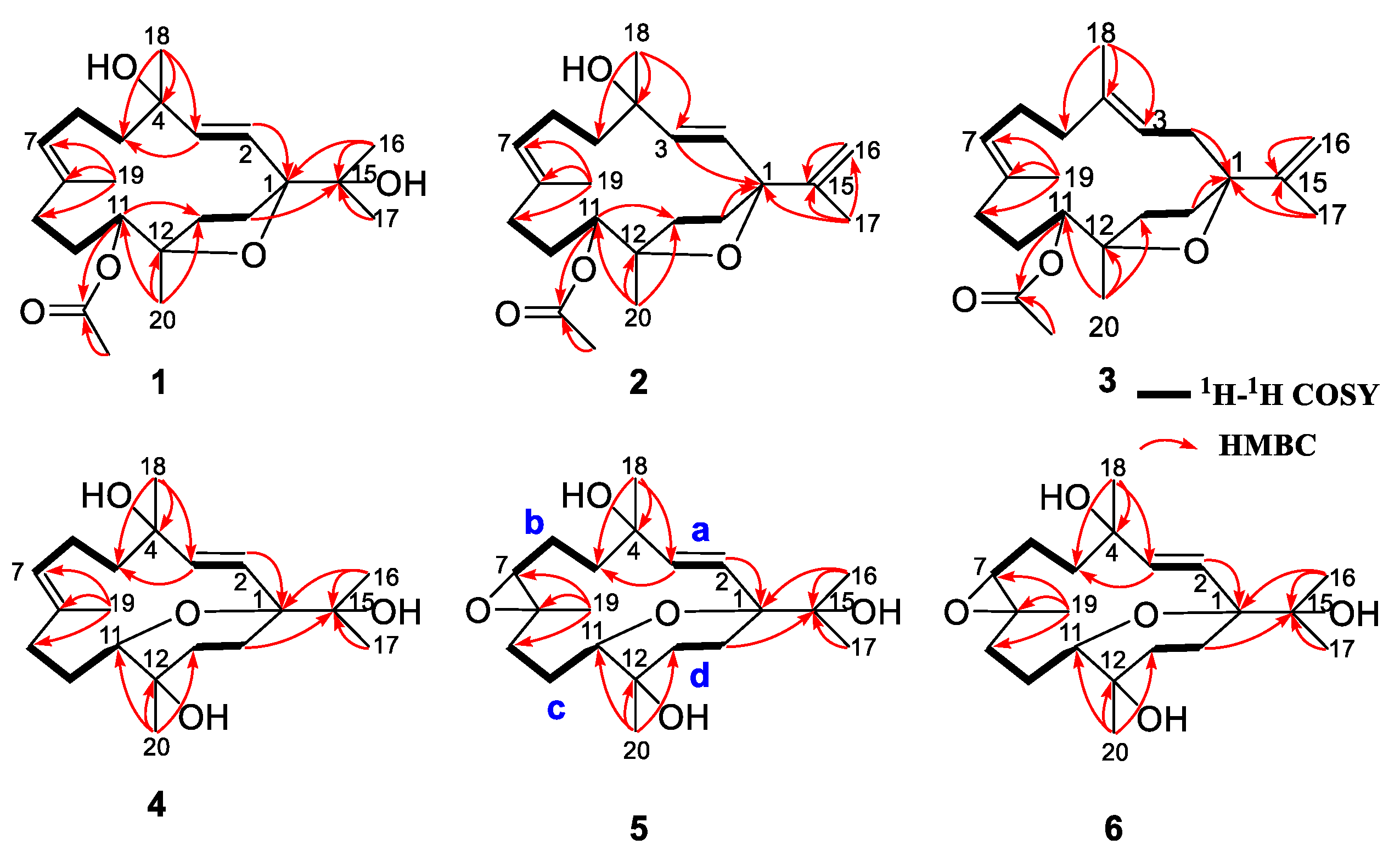
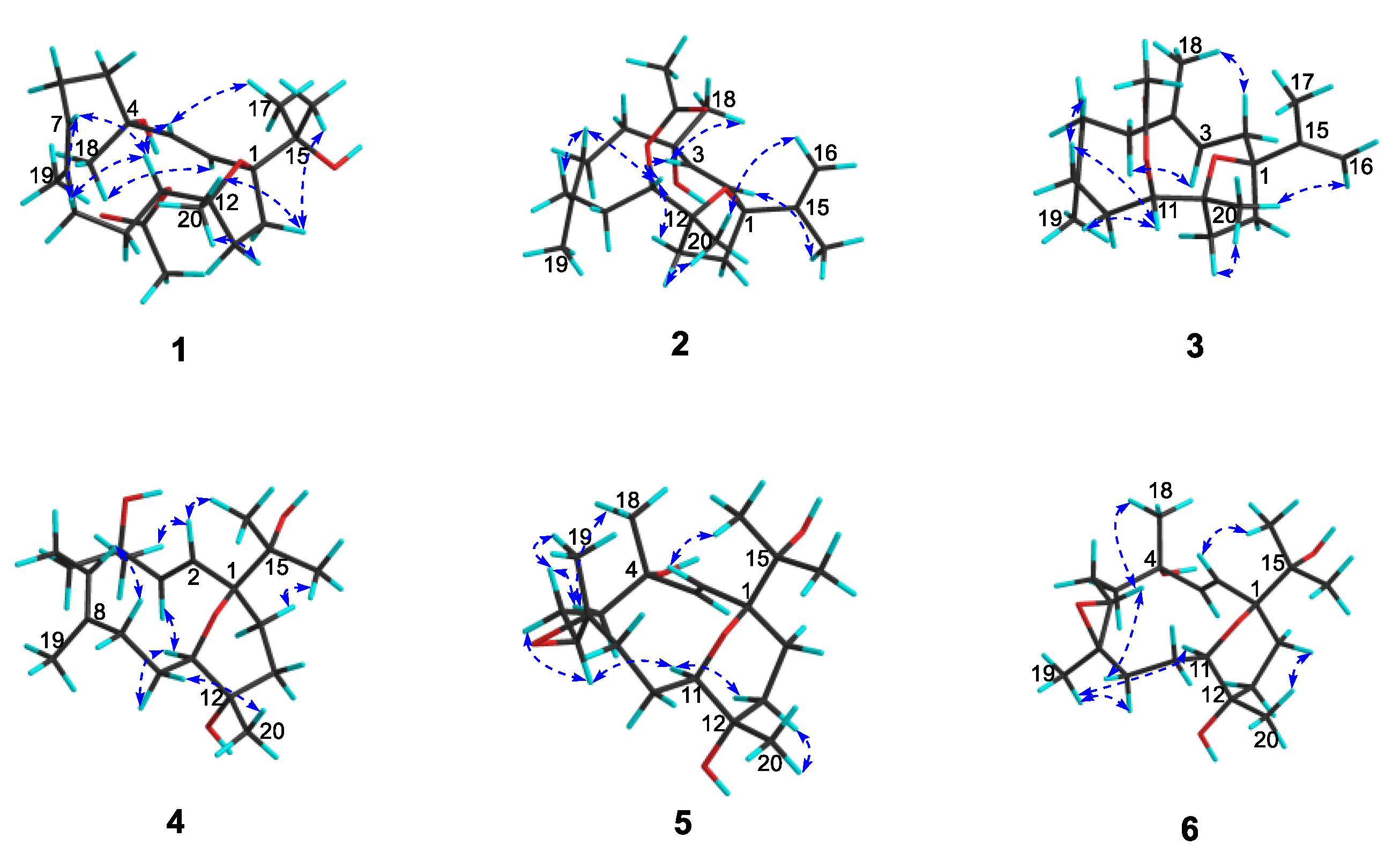
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Extraction and Isolation
3.4. Spectroscopic Data of Compounds
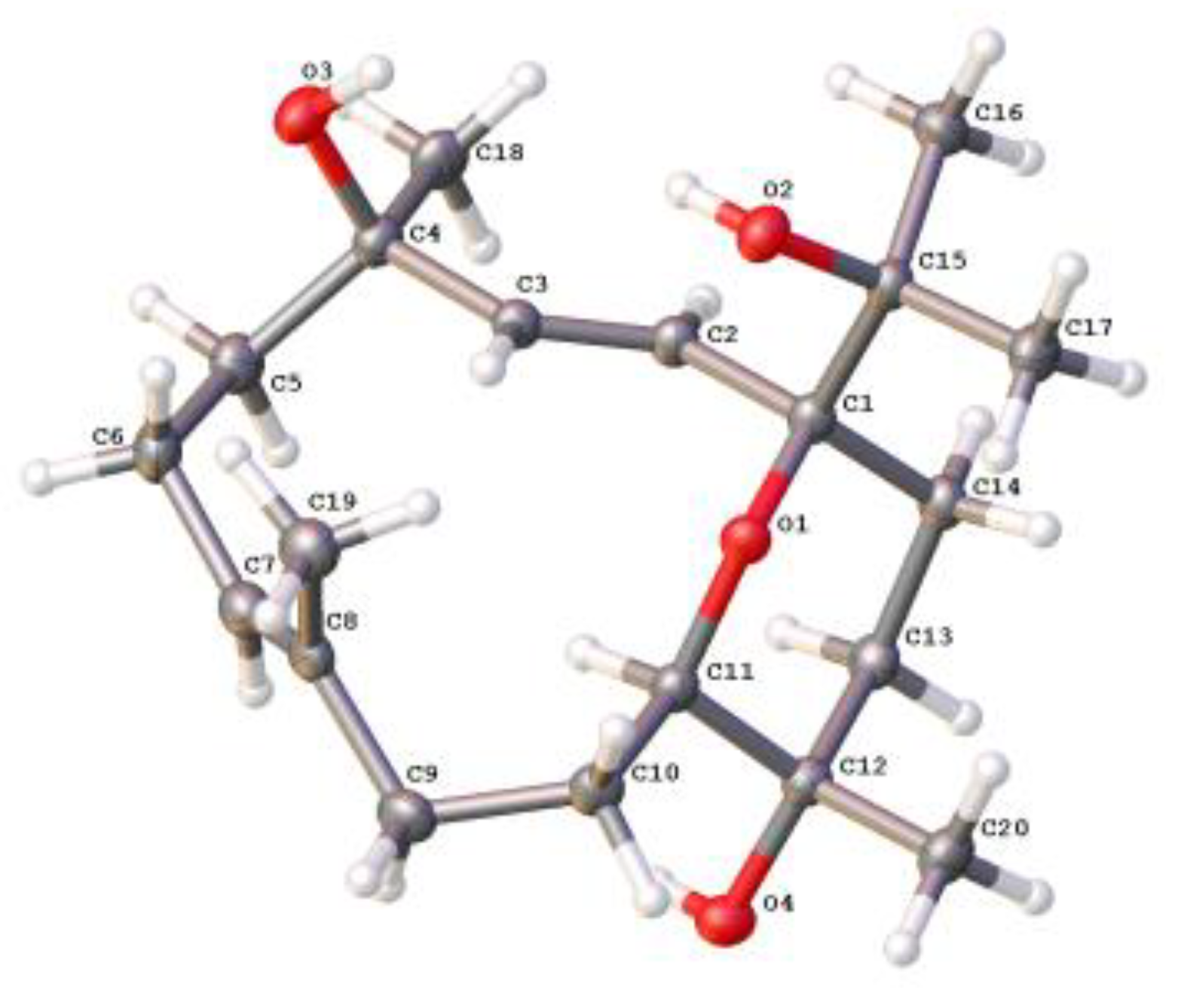
3.5. Crystal Structure Analysis of 4
3.6. TNF-α Inhibitory Activity Bioassay
3.7. ECD Computational Protocol
3.8. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haque, N.; Parveen, S.; Tang, T.; Wei, J.; Huang, Z. Marine natural products in clinical use. Mar. Drugs 2022, 20, 528. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, C. Marine natural products in medicinal chemistry. ACS Med. Chem. Lett. 2018, 9, 959–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Dickschat, J.S.; Guo, Y.-W. Diving into the world of marine 2,11-cyclized cembranoids: A summary of new compounds and their biological activities. Nat. Prod. Rep. 2020, 37, 1367–1383. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhou, X.-F.; Lin, X.-P.; Liu, J.; Peng, Y.; Yang, X.-W.; Liu, Y. Cembrane diterpenes chemistry and biological properties. Curr. Org. Chem. 2012, 16, 1512–1539. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2022, 39, 1122–1171. [Google Scholar] [CrossRef]
- Lai, D.; Geng, Z.; Deng, Z.; van Ofwegen, L.; Proksch, P.; Lin, W. Cembranoids from the soft coral Sinularia rigida with antifouling activities. J. Agric. Food Chem. 2013, 61, 4585–4592. [Google Scholar] [CrossRef]
- Liang, L.-F.; Chen, W.-T.; Li, X.-W.; Wang, H.-Y.; Guo, Y.-W. New bicyclic cembranoids from the South China Sea soft coral Sarcophyton trocheliophorum. Sci. Rep. 2017, 7, 46584. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-F.; Zeng, Y.-B.; Li, W.-S.; Luo, H.; Zhang, H.-Y.; Guo, Y.-W. Xishaglaucumins A–J, new cembranoids with anti-inflammatory activities from the South China Sea soft coral Sarcophyton glaucum. Chin. J. Chem. 2022, 40, 79–90. [Google Scholar] [CrossRef]
- Rao, C.B.; Satyanarayana, C.; Rao, D.S.; Rao, D.V.; Fahy, E.; Faulkner, D.J. Metabolites of the soft coral Sinularia ovispiculata from the Indian Ocean. J. Nat. Prod. 1993, 56, 2003–2007. [Google Scholar] [CrossRef]
- Yin, F.-Z.; Yao, L.-G.; Zhang, Z.-Y.; Wang, J.-R.; Wang, H.; Guo, Y.-W. Polyoxygenated cembranoids from the Hainan soft coral Lobophytum crassum. Tetrahedron 2021, 90, 132204. [Google Scholar] [CrossRef]
- Hegazy, M.E.F.; Su, J.-H.; Sung, P.-J.; Sheu, J.-H. Cembranoids with 3,14-ether linkage and a secocembrane with bistetrahydrofuran from the Dongsha Atoll soft coral Lobophytum sp. Mar. Drugs 2011, 9, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.; Chen, Y.-W.; Huang, C.-Y.; Tseng, Y.-J.; Lin, C.-C.; Dai, C.-F.; Wu, Y.-C.; Sheu, J.-H. Isolation and structure elucidation of cembranoids from a Dongsha Atoll soft coral Sarcophyton stellatum. Mar. Drugs 2018, 16, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattorusso, E.; Romano, A.; Taglialatela-Scafati, O.; Irace, C.; Maffettone, C.; Bavestrello, G.; Cerrano, C. Oxygenated cembranoids of the decaryiol type from the Indonesian soft coral Lobophytum sp. Tetrahedron 2009, 65, 2898–2904. [Google Scholar] [CrossRef]
- Li, S.-W.; Cuadrado, C.; Huan, X.-J.; Yao, L.-G.; Miao, Z.-H.; Daranas, A.H.; Guo, Y.-W. Rare new bicyclic cembranoid ethers and a novel trihydroxy prenylated guaiane from the Xisha soft coral Lobophytum sp. Bioorg. Chem. 2020, 103, 104223. [Google Scholar] [CrossRef]
- Li, S.-W.; Cuadrado, C.; Yao, L.-G.; Daranas, A.H.; Guo, Y.-W. Quantum mechanical–NMR-aided configuration and conformation of two unreported macrocycles isolated from the soft coral Lobophytum sp.: Energy calculations versus coupling constants. Org. Lett. 2020, 22, 4093–4096. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. The use of X-ray crystallography to determine absolute configuration. Chirality 2008, 20, 681–690. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. Assignment of the absolute configuration of polyfunctional compounds by NMR using chiral derivatizing agents. Chem. Rev. 2012, 112, 4603–4641. [Google Scholar] [CrossRef]
- Mándi, A.; Kurtán, T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef]
- Liang, L.-F.; Kurtán, T.; Mándi, A.; Yao, L.-G.; Li, J.; Lan, L.-F.; Guo, Y.-W. Structural, stereochemical, and bioactive studies of cembranoids from Chinese soft coral Sarcophyton trocheliophorum. Tetrahedron 2018, 74, 1933–1941. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-L.; Jin, T.-Y.; Liu, X.-H.; Zhang, J.-R.; Shi, X.; Wang, M.-F.; Huang, R.; Zhang, Y.; Liu, K.-C.; Li, G.-Q. Sinudenoids A–E, C19-norcembranoid diterpenes with unusual scaffolds from the soft coral Sinularia densa. Org. Lett. 2022, 24, 9007–9011. [Google Scholar] [CrossRef]
- Chao, C.-H.; Lin, K.-H.; Huang, C.-Y.; Hwang, T.-L.; Dai, C.-F.; Huang, H.-C.; Sheu, J.-H. Computationally assisted structural elucidation of cembranoids from the soft coral Sarcophyton tortuosum. Mar. Drugs 2022, 20, 297. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Kurtán, T.; Mándi, A.; Yan, X.-H.; Zhang, W.; Guo, Y.-W. Biscembranoids formed from an α,β-unsaturated γ-lactone ring as a dienophile: Structure revision and establishment of their absolute configurations using theoretical calculations of electronic circulardichroism spectra. J. Org. Chem. 2013, 78, 3113–3119. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Cai, F.-Y.; Lauro, G.; Tang, H.; Su, L.; Wang, H.-L.; Li, H.H.; Mándi, A.; Kurtán, T.; Riccio, R.; et al. Immunomodulatory biscembranoids and assignment of their relative and absolute configurations: Data set modulation in the density functional theory/nuclear magnetic resonance approach. J. Nat. Prod. 2019, 82, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liu, J.; Huang, J.; Wang, Y.; Leng, X.; Li, T.; Ouyang, H.; Yan, X.; He, S. Bistrochelides H–L: Biscembranoids from the South China Sea soft coral Sarcophyton serenei. Phytochemistry 2022, 204, 113438. [Google Scholar] [CrossRef]
- Liu, J.; Gu, Y.-C.; Su, M.-Z.; Guo, Y.-W. Chemistry and bioactivity of secondary metabolites from South China Sea marine fauna and flora: Recent research advances and perspective. Acta Pharmacol. Sin. 2022, 43, 3062–3079. [Google Scholar] [CrossRef]
- Bu, Q.; Yang, M.; Yan, X.-Y.; Yao, L.-G.; Guo, Y.-W.; Liang, L.-F. New flexible cembrane-type macrocyclic diterpenes as TNF-α inhibitors from the South China Sea soft coral Sarcophyton mililatensis. Int. J. Biol. Macromol. 2022, 222, 880–886. [Google Scholar] [CrossRef]
- Bu, Q.; Yang, M.; Yan, X.-Y.; Li, S.-W.; Ge, Z.-Y.; Zhang, L.; Yao, L.-G.; Guo, Y.-W.; Liang, L.-F. Mililatensols A–C, new records of sarsolenane and capnosane diterpenes from soft coral Sarcophyton mililatensis. Mar. Drugs 2022, 20, 566. [Google Scholar] [CrossRef]
- Yang, M.; Li, X.-L.; Wang, J.-R.; Lei, X.; Tang, W.; Li, X.-W.; Sun, H.; Guo, Y.-W. Sarcomililate A, an unusual diterpenoid with tricyclo[11.3.0.02,16]hexadecane carbon skeleton, and its potential biogenetic precursors from the Hainan soft coral Sarcophyton mililatensis. J. Org. Chem. 2019, 84, 2568–2576. [Google Scholar] [CrossRef]
- Li, S.; Ye, F.; Zhu, Z.; Huang, H.; Mao, S.; Guo, Y. Cembrane-type diterpenoids from the South China Sea soft coral Sarcophyton mililatensis. Acta Pharm. Sin. B 2018, 8, 944–955. [Google Scholar] [CrossRef]
- Cuong, N.X.; Tuan, T.A.; Kiem, P.V.; Minh, C.V.; Choi, E.M.; Kim, Y.H. New cembranoid diterpenes from the Vietnamese soft coral Sarcophyton mililatensis stimulate osteoblastic differentiation in MC3T3-E1 cells. Chem. Pharm. Bull. 2008, 56, 988–992. [Google Scholar] [CrossRef]
- Minh, C.V.; Cuong, N.X.; Tuan, T.A.; Choi, E.M.; Kim, Y.H.; Van Kiem, P. A new 9,11-secosterol from the Vietnamese sea soft coral, Sarcophyton mililatensis, increases the function of osteoblastic MC3T3-E1 cells. Nat. Prod. Commun. 2007, 2, 1095–1100. [Google Scholar] [CrossRef]
- Bowden, B.F.; Coll, J.C.; Mitchell, S.J. Studies of Australian soft corals. XVIII. Further cembranoid diterpenes from soft corals of the genus Sarcophyton. Aust. J. Chem. 1980, 33, 879–884. [Google Scholar] [CrossRef]
- Blackman, A.J.; Bowden, B.F.; Coll, J.C.; Frick, B.; Mahendran, M.; Mitchell, S.J. Studies of Australian soft corals. XXIX. Several new cembranoid diterpenes from Nephthea brassica and related diterpenes from a Sarcophyton species. Aust. J. Chem. 1982, 35, 1873–1880. [Google Scholar] [CrossRef]
- Xu, X.-H.; Kong, C.-H.; Lin, C.-J.; Wang, X.; Lu, J.-H. Isolation and identification of a novel cembrane-type diterpenoid from the soft coral Sarcophyton crassocaule. Chem. J. Chin. Univ. 2003, 24, 1023–1025. [Google Scholar]
- Huang, H.-C.; Chao, C.-H.; Kuo, Y.-H.; Sheu, J.-H. Crassocolides G-M, cembranoids from the Formosan soft coral Sarcophyton crassocaule. Chem. Biodivers. 2009, 6, 1232–1242. [Google Scholar] [CrossRef]
- Shaker, K.H.; Müller, M.; Ghani, M.A.; Dahse, H.-M.; Seifert, K. Terpenes from the soft corals Litophyton arboreum and Sarcophyton ehrenbergi. Chem. Biodivers. 2010, 7, 2007–2015. [Google Scholar] [CrossRef]
- He, M.M.; Smith, A.S.; Oslob, J.D.; Flanagan, W.M.; Braisted, A.C.; Whitty, A.; Cancilla, M.T.; Wang, J.; Lugovskoy, A.A.; Yoburn, J.C.; et al. Small-molecule inhibition of TNF-α. Science 2005, 310, 1022–1025. [Google Scholar] [CrossRef]
- MacroModel, Schrödinger LLC: Portland, OR, USA, 2012.
- Gaussian 09, Revision B.01, Gaussian, Inc.: Wallingford, CT, USA, 2010.
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 b | |||
|---|---|---|---|---|---|---|
| δH mult. (J, Hz) | δC, type | δH mult. (J, Hz) | δC, Type | δH mult. (J, Hz) | δC, Type | |
| 1 | 91.4, qC | 88.1, qC | 89.6, qC | |||
| 2 | 5.55 d (15.8) | 129.8, CH | 5.68 d (15.5) | 130.4, CH | 2.14 m; 2.35 m | 35.7, CH2 |
| 3 | 6.11 d (15.8) | 137.4, CH | 5.92 d (15.5) | 136.4, CH | 5.12 t (6.0) | 119.9, CH |
| 4 | 72.4, qC | 74.1, qC | 136.6, qC | |||
| 5 | 1.69 m; 1.86 m | 41.6, CH2 | 1.77 m; 1.82 m | 43.5, CH2 | 2.21 m | 38.0, CH2 |
| 6 | 2.01 m; 2.63 m | 22.9, CH2 | 2.20 m | 24.3, CH2 | 2.22 m | 24.9, CH2 |
| 7 | 5.42 dd (4.3, 10.5) | 130.1, CH | 5.37 dd (6.2, 8.8) | 128.3, CH | 5.18 t (6.0) | 126.2, CH |
| 8 | 132.1, qC | 132.6, qC | 132.9, qC | |||
| 9 | 1.86 m; 2.00 m | 34.8, CH2 | 1.76 m; 2.00 m | 34.2, CH2 | 1.67 m; 2.02 m | 33.8, CH2 |
| 10 | 1.54 m; 1.89 m | 27.3, CH2 | 1.56 m; 1.96 m | 27.3, CH2 | 1.53 m; 1.85 m | 27.5, CH2 |
| 11 | 5.07 d (9.4) | 77.1, CH | 5.01 d (9.7) | 76.5, CH | 4.88 d (10.2) | 75.7, CH |
| 12 | 84.5, qC | 84.2, qC | 83.5, qC | |||
| 13 | 1.58 m; 1.74 m | 35.8, CH2 | 1.59 m; 1.78 m | 35.3, CH2 | 1.55 m; 1.77 m | 35.3, CH2 |
| 14 | 1.68 m; 2.31 m | 30.8, CH2 | 2.03 m | 35.3, CH2 | 1.78 m; 1.93 m | 32.0, CH2 |
| 15 | 72.9, qC | 149.5, qC | 150.2, qC | |||
| 16 | 1.12 s | 24.6, CH3 | 4.71 s; 4.96 s | 109.4, CH2 | 4.80 s; 5.10 s | 110.1, CH2 |
| 17 | 1.10 s | 26.0, CH3 | 1.71 s | 19.2, CH3 | 1.75 s | 20.1, CH3 |
| 18 | 1.34 s | 29.0, CH3 | 1.28 s | 28.6, CH3 | 1.56 s | 16.8, CH3 |
| 19 | 1.70 s | 16.3, CH3 | 1.66 s | 17.1, CH3 | 1.63 s | 17.6, CH3 |
| 20 | 1.16 s | 20.7, CH3 | 1.18 s | 21.0, CH3 | 1.18 s | 21.8, CH3 |
| OAc | 171.2, qC | 171.2, qC | 171.3, qC | |||
| 2.00 s | 21.4, CH3 | 2.05 s | 21.4, CH3 | 2.06 s | 21.5, CH3 | |
| No. | 4 a | 5 b | 6 b | |||
|---|---|---|---|---|---|---|
| δH mult. (J, Hz) | δC, Type | δH mult. (J, Hz) | δC, type | δH mult. (J, Hz) | δC, Type | |
| 1 | 81.1, qC | 81.8, qC | 80.3, qC | |||
| 2 | 5.20 d (16.4) | 125.1, CH | 5.44 d (15.9) | 124.1, CH | 5.53 d (16.0) | 124.4, CH |
| 3 | 6.11 d (16.4) | 141.5, CH | 5.75 d (15.9) | 140.7, CH | 6.26 d (16.0) | 142.8, CH |
| 4 | 72.7, qC | 74.0, qC | 73.6, qC | |||
| 5 | 1.55 m; 1.93 m | 43.3, CH2 | 1.76 m; 2.03 m | 42.4, CH2 | 1.86 m | 38.0, CH2 |
| 6 | 2.06 m; 2.58 m | 22.9, CH2 | 2.08 m; 2.30 m | 24.2, CH2 | 1.59 m; 2. 02 m | 25.0, CH2 |
| 7 | 5.23 dd (5.1, 10.2) | 131.0, CH | 2.89 d (9.7) | 66.8, CH | 2.95 dd (4.2, 9.0) | 67.0, CH |
| 8 | 132.6, qC | 61.9, qC | 60.3, qC | |||
| 9 | 1.51 m; 1.70 m | 39.0, CH2 | 1.41 m; 2.15 m | 35.5, CH2 | 1.41 m; 2.03 m | 33.3, CH2 |
| 10 | 1.42 m; 1.88 m | 26.6, CH2 | 1.42 m; 1.87 m | 25.0, CH2 | 1.46 m; 1.90 m | 24.4, CH2 |
| 11 | 3.35 t (4.1) | 75.6, CH | 3.51 t (5.1) | 75.0, CH | 3.49 t (10.8) | 74.1, CH |
| 12 | 70.9, qC | 71.2, qC | 70.3, qC | |||
| 13 | 2.01 m; 2.21 m | 37.3, CH2 | 1.53 m; 1.70 m | 37.3, CH2 | 1.71 m | 36.7, CH2 |
| 14 | 1.54 m; 2.16 m | 28.5, CH2 | 1.60 m; 2.22 m | 28.2, CH2 | 1.61 m; 2.12 m | 28.2, CH2 |
| 15 | 75.1, qC | 74.6, qC | 75.0, qC | |||
| 16 | 1.16 s | 24.7, CH3 | 1.16 s | 24.8, CH3 | 1.17 s | 24.7, CH3 |
| 17 | 1.09 s | 24.1, CH3 | 1.11 s | 24.0, CH3 | 1.14 s | 24.4, CH3 |
| 18 | 1.37 s | 28.0, CH3 | 1.28 s | 27.7, CH3 | 1.39 s | 27.2, CH3 |
| 19 | 1.70 s | 15.2, CH3 | 1.38 s | 17.8, CH3 | 1.27 s | 15.5, CH3 |
| 20 | 1.18 s | 19.6, CH3 | 1.17 s | 19.2, CH3 | 1.16 s | 19.7, CH3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Yang, M.; Chen, Z.-H.; Ge, Z.-Y.; Li, S.-W.; Yan, X.-Y.; Yao, L.-G.; Liang, L.-F.; Guo, Y.-W. Cembrane Diterpenes Possessing Nonaromatic Oxacycles from the Hainan Soft Coral Sarcophyton mililatensis. Int. J. Mol. Sci. 2023, 24, 1979. https://doi.org/10.3390/ijms24031979
Zhang L, Yang M, Chen Z-H, Ge Z-Y, Li S-W, Yan X-Y, Yao L-G, Liang L-F, Guo Y-W. Cembrane Diterpenes Possessing Nonaromatic Oxacycles from the Hainan Soft Coral Sarcophyton mililatensis. International Journal of Molecular Sciences. 2023; 24(3):1979. https://doi.org/10.3390/ijms24031979
Chicago/Turabian StyleZhang, Ling, Min Yang, Zi-Hui Chen, Zeng-Yue Ge, Song-Wei Li, Xian-Yun Yan, Li-Gong Yao, Lin-Fu Liang, and Yue-Wei Guo. 2023. "Cembrane Diterpenes Possessing Nonaromatic Oxacycles from the Hainan Soft Coral Sarcophyton mililatensis" International Journal of Molecular Sciences 24, no. 3: 1979. https://doi.org/10.3390/ijms24031979