
Biological Activity of Novel Organotin Compounds with a Schiff Base Containing an Antioxidant Fragment


 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
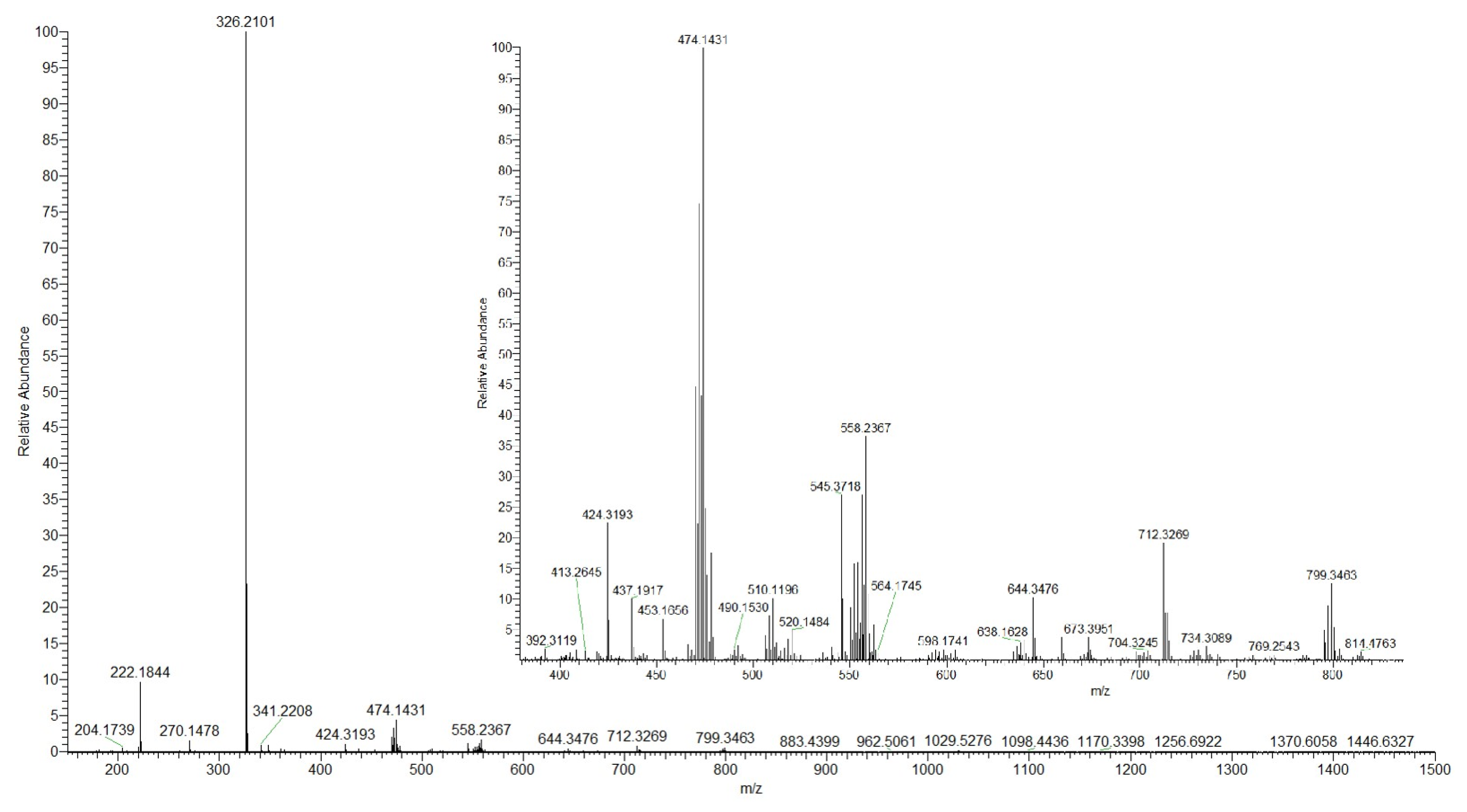
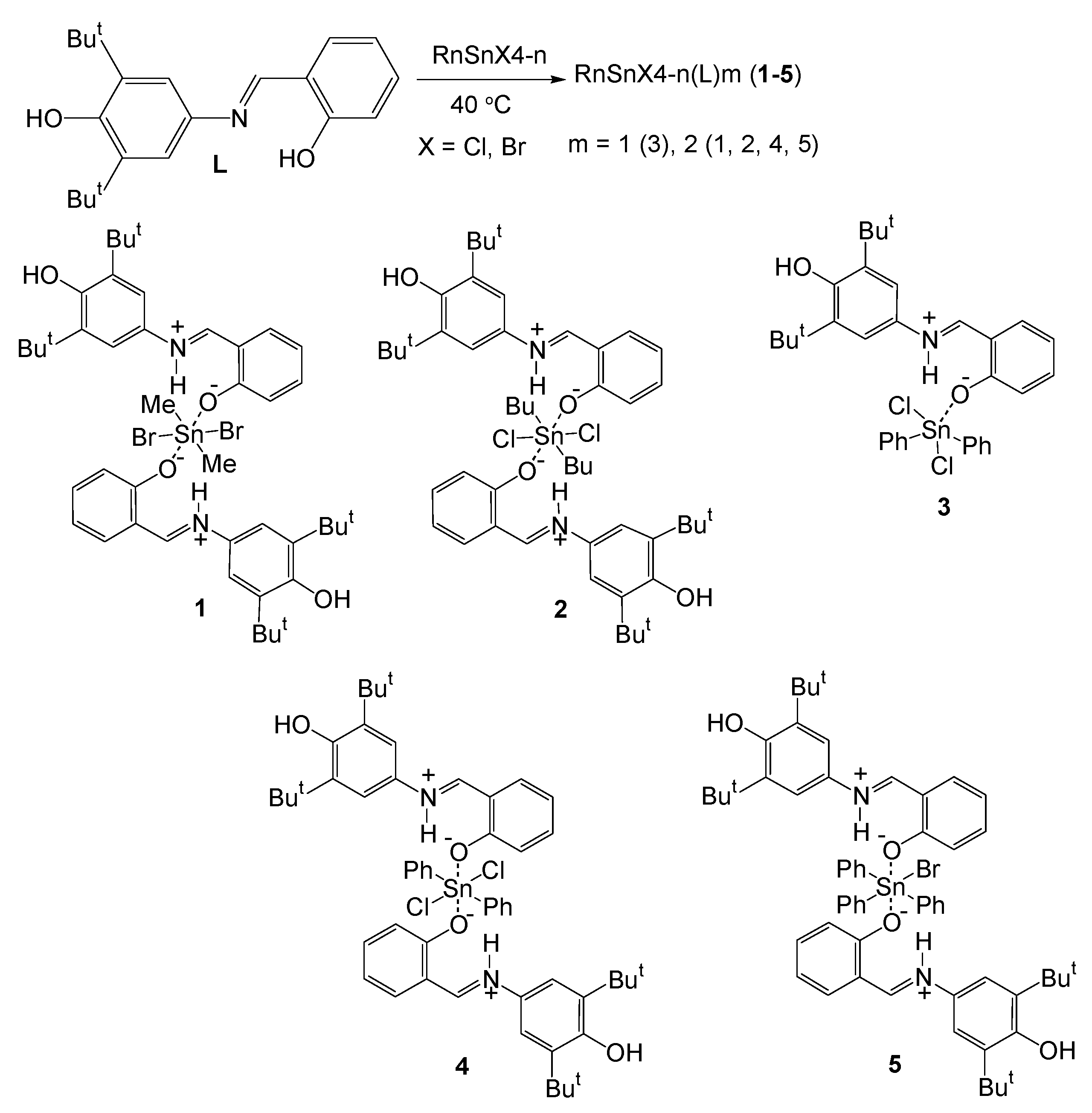
2.1. Synthesis and Characterization
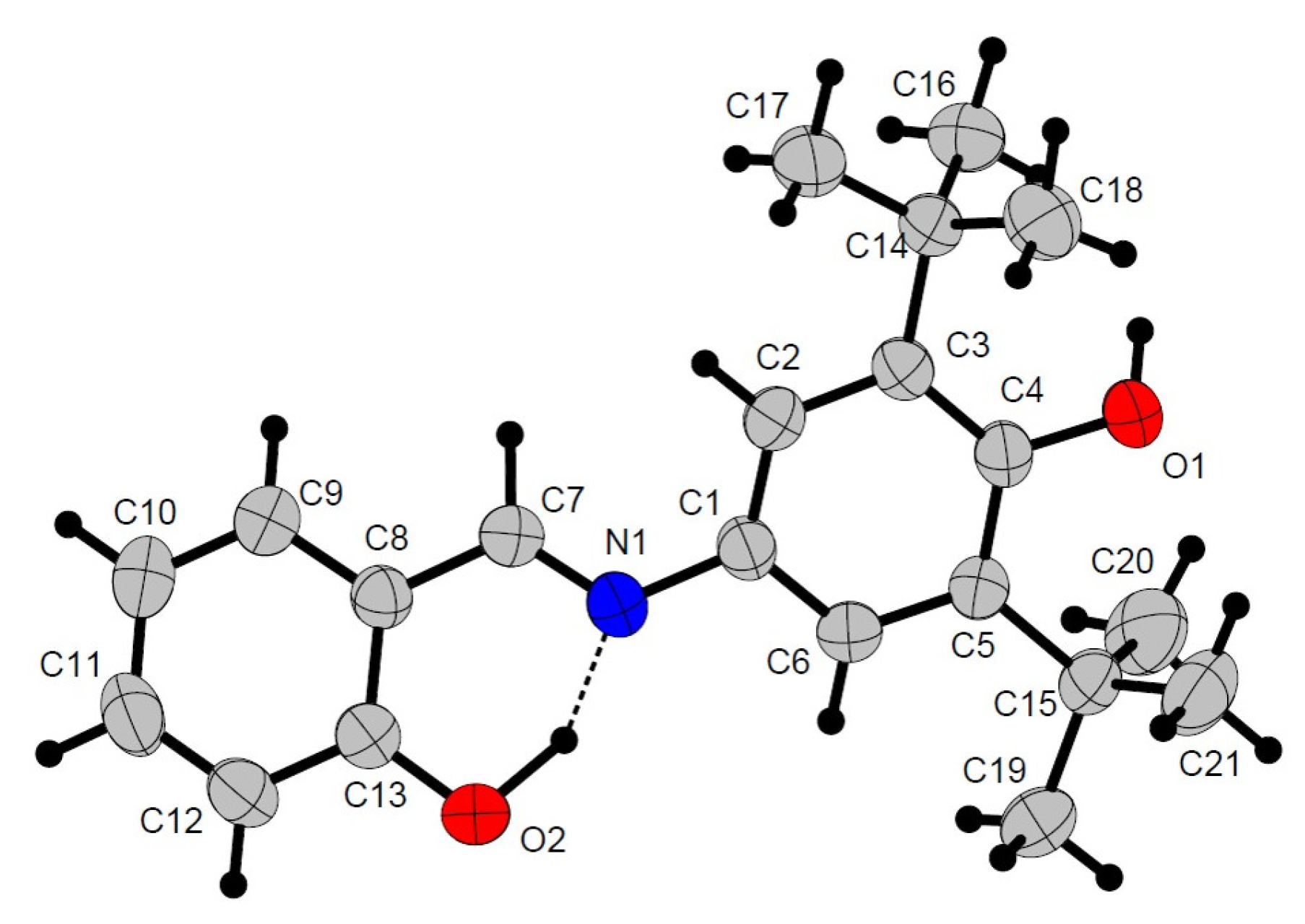
2.2. Crystal Structures
2.3. CUPRAC Assay (Cu2+ Reducing)
2.4. DPPH Radical Scavenging Activity
2.5. Lipoxygenase Inhibition Assessment
2.6. Antiproliferative Activity
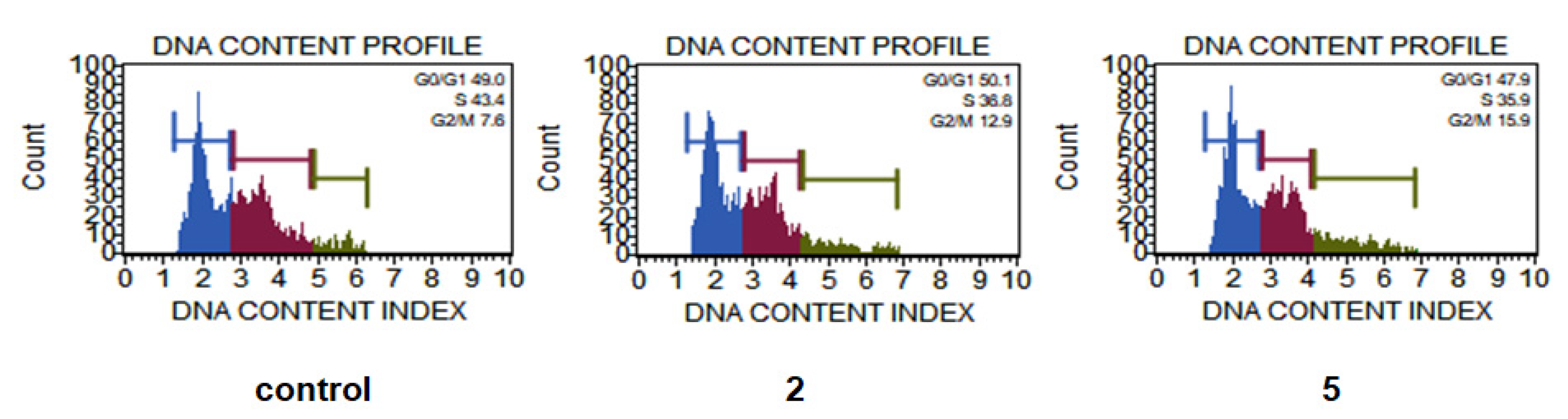
2.7. Cell Death and Cell Cycle Analysis
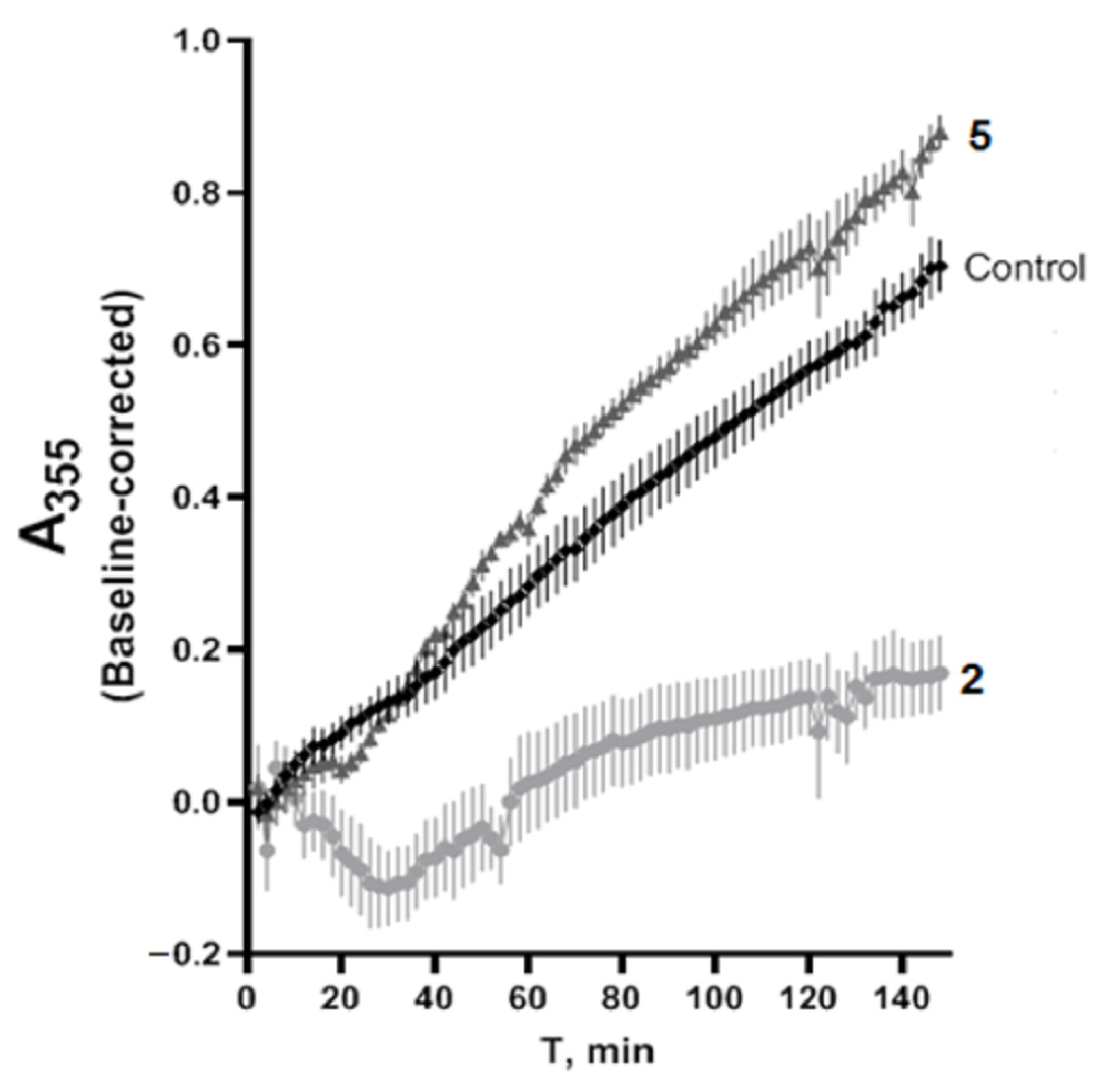
2.8. Tubulin Polymerization
3. Materials and Methods
3.1. Reagents and Materials
3.2. Synthesis
3.2.1. Synthesis and Characterization of Me2SnBr2(L)2 (1)
3.2.2. Synthesis and Characterization of Bu2SnCl2(L)2 (2)
3.2.3. Synthesis and Characterization of Ph2SnCl2(L) (3)
3.2.4. Synthesis and Characterization of Ph2SnCl2(L)2 (4)
3.2.5. Synthesis and Characterization of Ph3SnBr(L)2 (5)
3.3. Crystallographic Data Collection and Structure Determination
3.4. CUPRAC Assay (Cu2+ Reducing)
3.5. DPPH Radical Scavenging Activity
3.6. Lipoxygenase Inhibition Assessment
3.7. Antiproliferative Activity [28]
3.8. Cell Death and Cell Cycle Analysis
3.9. Tubulin Polymerization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Milaeva, E.R.; Tyurin, V.Y. Hybrid metal complexes with opposed biological modes of action—Promising selective drug candidates. Pure Appl. Chem. 2017, 89, 1065–1088. [Google Scholar] [CrossRef]
- Milaeva, E.R.; Shpakovsky, D.B.; Gracheva, Y.A.; Antonenko, T.A.; Ksenofontova, T.D.; Nikitin, E.A.; Berseneva, D.A. Novel selective anticancer agents based on Sn and Au complexes. Mini-review. Pure Appl. Chem. 2020, 92, 1201–1216. [Google Scholar] [CrossRef]
- Antonenko, T.A.; Gracheva, Y.A.; Shpakovsky, D.B.; Vorobyev, M.A.; Tafeenko, V.A.; Mazur, D.M.; Milaeva, E.R. Cytotoxic activity of organotin compounds containing non-steroidal anti-inflammatory drugs. J. Organomet. Chem. 2022, 960, 122191. [Google Scholar] [CrossRef]
- Adeyemi, J.O.; Onwudiwe, D.C. Organotin(IV) Dithiocarbamate Complexes: Chemistry and Biological Activity. Molecules 2018, 23, 2571. [Google Scholar] [CrossRef]
- Xanthopoulou, M.N.; Hadjikakou, S.K.; Hadjiliadis, N.; Milaeva, E.R.; Gracheva, J.A.; Tyurin, V.Y.; Kourkoumelis, N.; Christoforidis, K.C.; Metsios, A.K.; Karkabounas, S.; et al. Biological studies of new organotin(IV) complexes of thioamide ligands. Eur. J. Med. Chem. 2008, 43, 327–335. [Google Scholar] [CrossRef]
- Dave, S.; Bansal, N. Analgesic And Anti-Inflammatory Activities Of Schif F Base Metal Complexes—A Review. Int. J. Basic App. Chem. Sci. 2013, 3, 31–40. [Google Scholar]
- Elshafie, H.S.; Sadeek, S.A.; Camele, I.; Mohamed, A.A. Biochemical Characterization of New Gemifloxacin Schiff Base (GMFX-o-phdn) Metal Complexes and Evaluation of Their Antimicrobial Activity against Some Phyto- or Human Pathogens. Int. J. Mol. Sci. 2022, 23, 2110. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Ahmed, F.M.; Zordok, W.A.; El-Shwiniy, W.H.; Sadeek, S.A.; Elshafie, H.S. Novel Enrofloxacin Schiff Base Metal Complexes: Synthesis, Spectroscopic Characterization, Computational Simulation and Antimicrobial Investigation against Some Food and Phyto-Pathogens. Inorganics 2022, 10, 177. [Google Scholar] [CrossRef]
- El-Attar, M.S.; Ahmed, F.M.; Sadeek, S.A.; Mohamed, S.F.; Zordok, W.A.; El-Shwiniy, W.H. Characterization, DFT, and antimicrobial evaluation of some new N2O2 tetradentate Schiff base metal complexes. Appl. Organomet. Chem. 2022, 36, e6826. [Google Scholar] [CrossRef]
- Gielen, M.; Tiekink, E.R.T. (Eds.) Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine; Wiley: Chichester, UK, 2008; p. 656. [Google Scholar]
- Shpakovsky, D.B.; Banti, C.N.; Mukhatova, E.M.; Gracheva, Y.A.; Osipova, V.P.; Berberova, N.T.; Albov, D.V.; Antonenko, T.A.; Aslanov, L.A.; Milaeva, E.R.; et al. Synthesis, antiradical activity and in vitro cytotoxicity of novel organotin complexes based on 2,6-di-tert-butyl-4-mercaptophenol. Dalton Trans. 2014, 43, 6880–6890. [Google Scholar] [CrossRef]
- Kolyada, M.N.; Osipova, V.P.; Berberova, N.T.; Shpakovsky, D.B.; Milaeva, E.R. Porphyrins with Phenolic Fragments at the Periphery of the Macrocycle as Perspective Antioxidants, Cytoprotectors and Heavy Metal Scavengers. Chem. Heterocycl. Compd. 2021, 57, 875–888. [Google Scholar] [CrossRef]
- Antonenko, T.A.; Shpakovsky, D.B.; Vorobyov, M.A.; Gracheva, Y.A.; Kharitonashvili, E.V.; Dubova, L.G.; Shevtsova, E.F.; Tafeenko, V.A.; Aslanov, L.A.; Iksanova, A.G.; et al. Antioxidative vs cytotoxic activities of organotin complexes bearing 2,6-di-tert-butylphenol moieties. Appl. Organomet. Chem. 2018, 32, e4381. [Google Scholar] [CrossRef]
- Apak, R.; Güçlü, K.; Özyürek, M.; Karademir, S.E. Novel Total Antioxidant Capacity Index for Dietary Polyphenols and Vitamins C and E, Using Their Cupric Ion Reducing Capability in the Presence of Neocuproine: CUPRAC Method. J. Agric. Food. Chem. 2004, 52, 7970–7981. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Ozturk, I.; Filimonova, S.; Hadjikakou, S.K.; Kourkoumelis, N.; Dokorou, V.; Manos, M.J.; Tasiopoulos, A.J.; Barsan, M.M.; Butler, I.S.; Milaeva, E.R.; et al. Structural Motifs and Biological Studies of New Antimony(III) Iodide Complexes with Thiones. Inorg. Chem. 2010, 49, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Nikš, M.; Otto, M. Towards an optimized MTT assay. J. Immunol. Methods 1990, 130, 149–151. [Google Scholar] [CrossRef]
- Hadjikakou, S.K.; Hadjiliadis, N. Antiproliferative and anti-tumor activity of organotin compounds. Coord. Chem. Rev. 2009, 253, 235–249. [Google Scholar] [CrossRef]
- Banti, C.N.; Hadjikakou, S.K.; Sismanoglu, T.; Hadjiliadis, N. Anti-proliferative and antitumor activity of organotin(IV) compounds. An overview of the last decade and future perspectives. J. Inorg. Biochem. 2019, 194, 114–152. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- Milaeva, E.R.; Shpakovsky, D.B.; Gracheva, Y.A.; Antonenko, T.A.; Osolodkin, D.I.; Palyulin, V.A.; Shevtsov, P.N.; Neganova, M.E.; Vinogradova, D.V.; Shevtsova, E.F. Some insight into the mode of cytotoxic action of organotin compounds with protective 2,6-di-tert-butylphenol fragments. J. Organomet. Chem. 2015, 782, 96–102. [Google Scholar] [CrossRef]
- Cheng, Z.; Lu, X.; Feng, B. A review of research progress of antitumor drugs based on tubulin targets. Transl. Cancer Res. 2020, 9, 4020–4027. [Google Scholar] [CrossRef] [PubMed]
- Medzhidov, A.A.; Kasumov, V.T.; Mamedov, K.S. Synthesis and study of chelate compounds of metals containing spatially hindered phenol. Koord. Khim. 1981, 7, 66–72. [Google Scholar]
- Stoe&Cie. X-AREA, X-RED32; Stoe&Cie: Darmstadt, Germany, 2012. [Google Scholar]
- Farrugia, L. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Shutkov, I.A.; Okulova, Y.N.; Tyurin, V.Y.; Sokolova, E.V.; Babkov, D.A.; Spasov, A.A.; Gracheva, Y.A.; Schmidt, C.; Kirsanov, K.I.; Shtil, A.A.; et al. Ru(III) Complexes with Lonidamine-Modified Ligands. Int. J. Mol. Sci. 2021, 22, 13468. [Google Scholar] [CrossRef]
- Bachurin, S.O.; Makhaeva, G.F.; Shevtsova, E.F.; Aksinenko, A.Y.; Grigoriev, V.V.; Shevtsov, P.N.; Goreva, T.V.; Epishina, T.A.; Kovaleva, N.V.; Pushkareva, E.A.; et al. Conjugation of Aminoadamantane and γ-Carboline Pharmacophores Gives Rise to Unexpected Properties of Multifunctional Ligands. Molecules 2021, 26, 5527. [Google Scholar] [CrossRef]
- Shelanski, M.L.; Gaskin, F.; Cantor, C.R. Microtubule assembly in the absence of added nucleotides. Proc. Natl. Acad. Sci. USA 1973, 70, 765–768. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | L | 2 |
|---|---|---|
| Empirical formula | C21H27NO2 | C25H36ClNO2Sn0.50 |
| Fw T, K | 325.43 293(2) | 477.33 293(2) |
| Space group | P212121 | P21/n |
| Syngony | Orthorhombic | Monoclinic |
| a (Å) | 6.0356(4) | 9.5216(5) |
| b (Å) | 17.5484(14) | 11.7902(7) |
| c (Å) | 17.7780(12) | 22.4464(11) |
| α (°) | 90.00 | 90.00 |
| β (°) | 90.00 | 98.820(3) |
| γ (°) | 90.00 | 90.00 |
| V (Å3) | 1883.0(2) | 2490.1(2) |
| Z λ | 4 MoKα | 4 CuKα |
| Δρmax/Δρmin(e/Å3) | 0.212/−0.210 | 2.028/−1.732 |
| μ (mm−1) | 0.073 | 5.396 |
| GOOF | 0.864 | 1.122 |
| R1/wR2(I ≥ 2σ(I) | 0.0458/0.1172 | 0.0772/0.2112 |
| L | 2 | ||
|---|---|---|---|
| Selected bond distances (Å) | |||
| O(1)-C(4) O(2)-C(13) N(1)-C(7) N(1)-C(1) | 1.365(3) 1.342(3) 1.272(3) 1.419(3) | Sn(1)-C(22) Sn(1)-O(2) Sn(1)-Cl(1) | 2.140(7) 2.258(4) 2.6123(18) |
| Angles (°) | |||
| C(4)-O(1)-H(31) C(13)-O(2)-H(32) C(7)-N(1)-C(1) C(7)-N(1)-H(32) C(1)-N(1)-H(32) C(6)-C(1)-N(1) C(2)-C(1)-N(1) O(1)-C(4)- C(5) O(1)-C(4)-C(3) N(1)-C(7)-C(8) N(1) C(7)-H(7) O(2)-C(13)-C(12) O(2)-C(13)-C(8) | 113(2) 103.9(17) 123.4(2) 101.4(14) 134.8(14) 116.1(2) 124.5(2) 115.2(2) 122.6(2) 121.8(2) 124.3(14) 119.6(2) 120.5(2) | C(22)-Sn(1)-O(2) C(22)i-Sn(1)-O(2) C(22)-Sn(1)-O(2)i C(22)i-Sn(1)-O(2)i O(2)-Sn(1)-O(2)i C(22)-Sn(1)-Cl(1) C(22)i -Sn(1)-Cl(1) O(2)-Sn(1)-Cl(1) O(2)i -Sn(1)-Cl(1) C(22)-Sn(1)-Cl(1)i C(22)i -Sn(1)-Cl(1)i O(2)-Sn(1)-Cl(1)i O(2)i -Sn(1)-Cl(1)i | 88.31(19) 91.69(19) 91.69(19) 88.31(19) 180.00(18) 91.89(15) 88.11(15) 92.11(12) 87.89(12) 88.11(15) 91.89(15) 87.89(12) 92.11(12) |
| Compound | TEAC (CUPRAC) |
|---|---|
| L | 0.81 ± 0.03 [13] |
| 1 | 3.37 ± 0.03 |
| 2 | 2.78 ± 0.04 |
| 3 | 1.19 ± 0.09 |
| 4 | 2.64 ± 0.07 |
| 5 | 2.31 ± 0.06 |
| Compound | 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|---|
| EC50, µM | 16 ± 2 | 20 ± 3 | 30 ± 3 | 19 ± 2 | 23 ± 2 | |
| s | 0.32 | 0.4 | 0.30 | 0.38 | 0.23 | |
| s−1 | 3.13 | 2.5 | 3.36 | 2.60 | 4.32 | |
| C | k (L mol−1 s−1) | |||||
| 0.06 mM | * | 49 ± 5 | * | * | * | |
| 0.04 mM | 31 ± 4 | 29 ± 3 | 5 ± 1 | 49 ± 4 | 10 ± 1 | |
| 0.02 mM | 13 ± 4 | 2 ± 0.5 | 1 ± 0.2 | 3 ± 0.5 | 3 ± 0.4 | |
| 0.01 mM | 1 ± 0.3 | 0.7 ± 0.1 | 0.9 ± 0.1 | 2 ± 0.1 | 0.7 ± 0.1 | |
| C, μM | I, % |
|---|---|
| 0.8 | 22.66 |
| 8.3 | 27.59 |
| 16.6 | 31.21 |
| 33.3 | 39.55 |
| Compound | IC50, µM | |||
|---|---|---|---|---|
| HCT-116 | MCF-7 | A-549 | WI-38 | |
| 1 | 58.2 ± 14 | 80.6 ± 6 | 89 ± 17 | 58.2 ± 8 |
| 2 | 1.6 ± 0.2 | 4.3 ± 0.5 | 5.7 ± 1 | 1.6 ± 0.2 |
| 3 | 3.8 ± 0.5 | 6.3 ± 0.7 | 7.7 ± 1.4 | 2.3 ± 0.3 |
| 4 | 3.1 ± 0.4 | 5.1 ± 0.6 | 6.8 ± 1.8 | 2.1 ± 0.3 |
| 5 | 0.32 ± 0.08 | 0.29 ± 0.03 | 0.30 ± 0.04 | 0.2 ± 0.05 |
| cisplatin | 9.04 ± 0.7 | 11 ± 1 | 16.7 ± 3 | 4.8 ± 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonenko, T.A.; Gracheva, Y.A.; Shpakovsky, D.B.; Vorobyev, M.A.; Mazur, D.M.; Tafeenko, V.A.; Oprunenko, Y.F.; Shevtsova, E.F.; Shevtsov, P.N.; Nazarov, A.A.; et al. Biological Activity of Novel Organotin Compounds with a Schiff Base Containing an Antioxidant Fragment. Int. J. Mol. Sci. 2023, 24, 2024. https://doi.org/10.3390/ijms24032024
Antonenko TA, Gracheva YA, Shpakovsky DB, Vorobyev MA, Mazur DM, Tafeenko VA, Oprunenko YF, Shevtsova EF, Shevtsov PN, Nazarov AA, et al. Biological Activity of Novel Organotin Compounds with a Schiff Base Containing an Antioxidant Fragment. International Journal of Molecular Sciences. 2023; 24(3):2024. https://doi.org/10.3390/ijms24032024
Chicago/Turabian StyleAntonenko, Taisiya A., Yulia A. Gracheva, Dmitry B. Shpakovsky, Mstislav A. Vorobyev, Dmitrii M. Mazur, Victor A. Tafeenko, Yury F. Oprunenko, Elena F. Shevtsova, Pavel N. Shevtsov, Alexey A. Nazarov, and et al. 2023. "Biological Activity of Novel Organotin Compounds with a Schiff Base Containing an Antioxidant Fragment" International Journal of Molecular Sciences 24, no. 3: 2024. https://doi.org/10.3390/ijms24032024
APA StyleAntonenko, T. A., Gracheva, Y. A., Shpakovsky, D. B., Vorobyev, M. A., Mazur, D. M., Tafeenko, V. A., Oprunenko, Y. F., Shevtsova, E. F., Shevtsov, P. N., Nazarov, A. A., & Milaeva, E. R. (2023). Biological Activity of Novel Organotin Compounds with a Schiff Base Containing an Antioxidant Fragment. International Journal of Molecular Sciences, 24(3), 2024. https://doi.org/10.3390/ijms24032024