
GBA1 Gene Mutations in α-Synucleinopathies—Molecular Mechanisms Underlying Pathology and Their Clinical Significance

 , , ,
, , ,
Abstract
1. Introduction
2. GBA1 Gene Mutations and the Associated Pathologies
2.1. The Structure and Function of the GBA Gene and Its Protein Product, Glucocerebrosidase
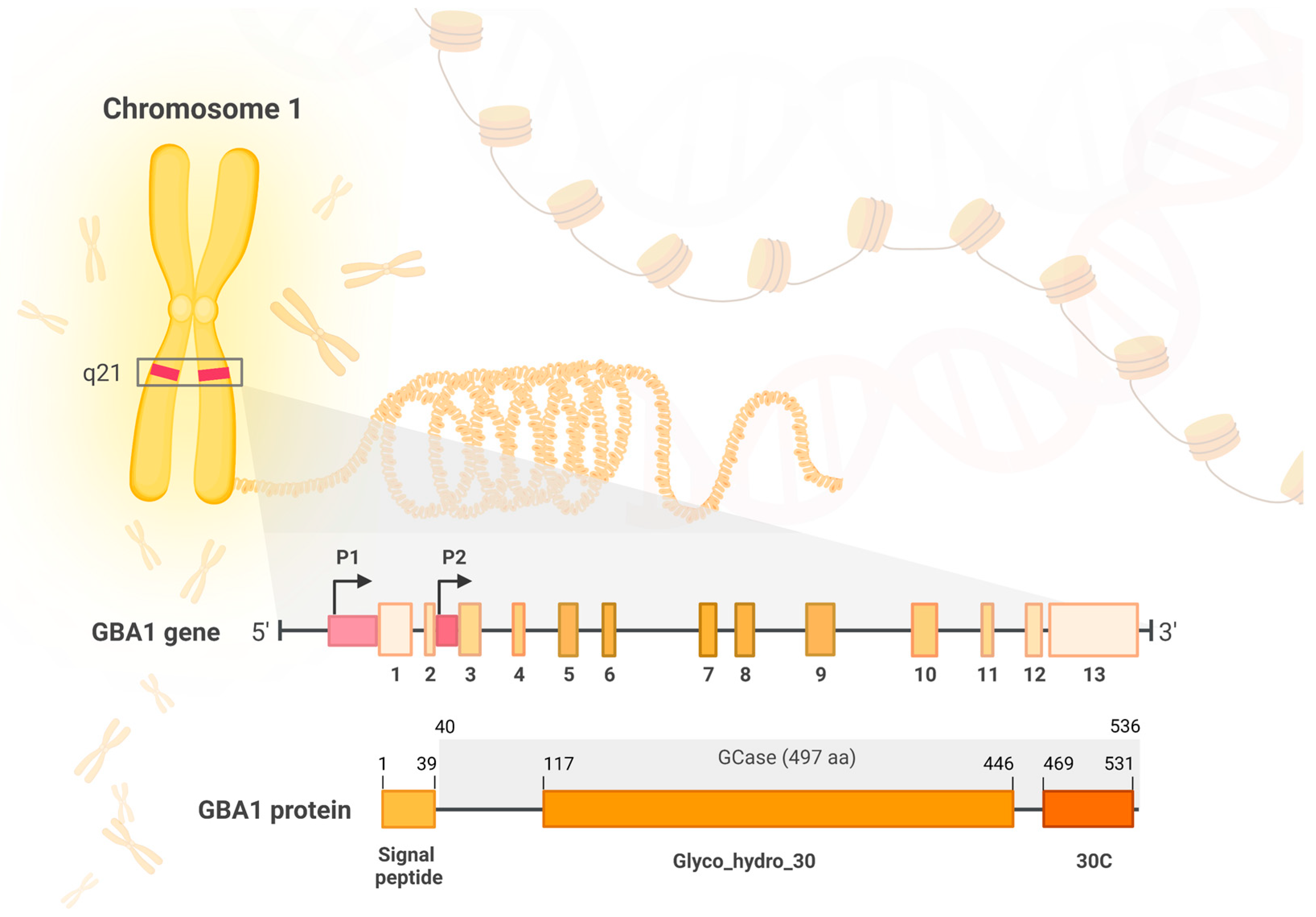
2.1.1. The GBA Gene
2.1.2. GBA1 Product—Glucocerebrosidase
2.2. GBA1 Gene Mutations (Table 1)
2.2.1. GBA1 mutations in Gaucher’s Disease
2.2.2. The Association between Gaucher’s Disease and Parkinson’s Disease
2.2.3. GBA1 Mutations in Parkinson’s Disease
2.2.4. GBA1 Mutations in Dementia with Lewy Bodies

{kind=link}
{kind=link}
{kind=link}
| Common Variant Name (New Nomenclature) | DNA Nucleotide Change | Ethnicity | Disease | Reference |
|---|---|---|---|---|
| N370S (N409S) | c.1226A>G | AJ *, Non-AJ *, Russian, North African | GD type 1; PD; | [21,41,64,67,68,69,70] |
| L444P (L483P) | c.1448T>C | Chinese, Japanese, Caucasian, Canadian, Italian, Brazilian, Greek, Non-AJ, AJ, North African | GD type 2 or 3 or neuropathic; PD; DLB | [41,64,67,70,71,72,73,74] |
| E326K (E365K) | c.1093G>A | Non-AJ, AJ | PD; DLB; RBD | [41,53,64,67] |
| T369M | p.T369M substitution | Non-AJ, AJ | PD; RBD | [41,51,67] |
| RecNciI (4856_4905) | A456P, V460V recombinant | Non-AJ, AJ, Japanese, North African | PD | [41,67,70,75,76] |
| 84GG (L29Afs*18) | c.84dupG | AJ | PD | [41] |
2.3. The Effects of GBA1 Mutations on GCase Activity
2.4. A Correlation between GCase Activity and α-Synuclein Accumulation
3. α-Synucleinopathies Associated with GBA1 Mutations—Clinical Characteristics
3.1. Parkinson’s Disease
3.1.1. Clinical Features
3.1.2. Neuroimaging Features
3.2. Dementia with Lewy Bodies
3.2.1. Clinical Features
3.2.2. Neuroimaging features
3.3. Multiple System Atrophy
4. Mechanisms Underlying the Crosstalk between GCase and α-Synuclein
4.1. Lipid Metabolism
4.2. Autophagic–Lysosomal Pathway, Protein Metabolism and GCase Interaction with Lysosome
4.3. Mitochondrial Dysfunction
4.4. Neuroinflammation
4.5. Glucocerebrosidase and LIMP-2
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mendoza-Velásquez, J.J.; Flores-Vázquez, J.F.; Barrón-Velázquez, E.; Sosa-Ortiz, A.L.; Illigens, B.-M.W.; Siepmann, T. Autonomic Dysfunction in α-Synucleinopathies. Front. Neurol. 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Burré, J. The Synaptic Function of α-Synuclein. J. Parkinsons. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Hagita, H.; Horiguchi, T.; Tanimura, A.; Noma, T. Redefining GBA Gene Structure Unveils the Ability of Cap-Independent, IRES-Dependent Gene Regulation. Commun. Biol. 2022, 5, 639. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 44. [Google Scholar] [CrossRef]
- Woo, E.G.; Tayebi, N.; Sidransky, E. Next-Generation Sequencing Analysis of GBA1: The Challenge of Detecting Complex Recombinant Alleles. Front. Genet. 2021, 12, 684067. [Google Scholar] [CrossRef]
- NCBI. GBA2 Glucosylceramidase Beta 2 [Homo Sapiens (Human)]—Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/57704 (accessed on 27 November 2022).
- NCBI. GBA3 Glucosylceramidase Beta 3 (Gene/Pseudogene) [Homo Sapiens (Human)]—Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/57733 (accessed on 27 November 2022).
- Lerche, S.; Wurster, I.; Roeben, B.; Zimmermann, M.; Riebenbauer, B.; Deuschle, C.; Hauser, A.; Schulte, C.; Berg, D.; Maetzler, W.; et al. Parkinson’s Disease: Glucocerebrosidase Mutation Severity Is Associated with CSF Alpha-Synuclein Profiles. Mov. Disord. 2020, 35, 495–499. [Google Scholar] [CrossRef]
- Surface, M.; Balwani, M.; Waters, C.; Haimovich, A.; Gan-Or, Z.; Marder, K.S.; Hsieh, T.; Song, L.; Padmanabhan, S.; Hsieh, F.; et al. Plasma Glucosylsphingosine in GBA1 Mutation Carriers with and without Parkinson’s Disease. Mov. Disord. 2022, 37, 416–421. [Google Scholar] [CrossRef]
- Gatti, M.; Magri, S.; Di Bella, D.; Sarto, E.; Taroni, F.; Mariotti, C.; Nanetti, L. Spastic Paraplegia Type 46: Novel and Recurrent GBA2 Gene Variants in a Compound Heterozygous Italian Patient with Spastic Ataxia Phenotype. Neurol. Sci. 2021, 42, 4741–4745. [Google Scholar] [CrossRef]
- Sultana, S.; Reichbauer, J.; Schüle, R.; Mochel, F.; Synofzik, M.; van der Spoel, A.C. Lack of Enzyme Activity in GBA2 Mutants Associated with Hereditary Spastic Paraplegia/Cerebellar Ataxia (SPG46). Biochem. Biophys. Res. Commun. 2015, 465, 35–40. [Google Scholar] [CrossRef]
- Dekker, N.; Voorn-Brouwer, T.; Verhoek, M.; Wennekes, T.; Narayan, R.S.; Speijer, D.; Hollak, C.E.M.; Overkleeft, H.S.; Boot, R.G.; Aerts, J.M.F.G. The Cytosolic β-Glucosidase GBA3 Does Not Influence Type 1 Gaucher Disease Manifestation. Blood Cells Mol. Dis. 2011, 46, 19–26. [Google Scholar] [CrossRef]
- Huh, Y.E.; Chiang, M.S.R.; Locascio, J.J.; Liao, Z.; Liu, G.; Choudhury, K.; Kuras, Y.I.; Tuncali, I.; Videnovic, A.; Hunt, A.L.; et al. β-Glucocerebrosidase Activity in GBA -Linked Parkinson Disease. Neurology 2020, 95, e685–e696. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, H.; Maor, G.; Chicco, G.; Filocamo, M.; Zimran, A.; Horowitz, M. UPR Activation and CHOP Mediated Induction of GBA1 Transcription in Gaucher Disease. Blood Cells Mol. Dis. 2018, 68, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.-Y.; Lo, R.C.N.; Ho, P.W.-L.; Liu, H.-F.; Chang, E.E.S.; Leung, C.-T.; Malki, Y.; Choi, Z.Y.-K.; Wong, W.Y.; Kung, M.H.-W.; et al. LRRK2, GBA and Their Interaction in the Regulation of Autophagy: Implications on Therapeutics in Parkinson’s Disease. Transl. Neurodegener. 2022, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Stubblefield, B.K.; McGlinchey, R.P.; McMahon, B.; Ory, D.S.; Sidransky, E. A Characterization of Gaucher IPS-Derived Astrocytes: Potential Implications for Parkinson’s Disease. Neurobiol. Dis. 2020, 134, 104647. [Google Scholar] [CrossRef]
- Menozzi, E.; Schapira, A.H.V. Enhancing the Activity of Glucocerebrosidase as a Treatment for Parkinson Disease. CNS Drugs 2020, 34, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Sedor, S.; McGeary, I.; Cornblath, E.J.; Peng, C.; Riddle, D.M.; Li, H.L.; Zhang, B.; Brown, H.J.; Olufemi, M.F.; et al. Glucocerebrosidase Activity Modulates Neuronal Susceptibility to Pathological α-Synuclein Insult. Neuron 2020, 105, 822–836. [Google Scholar] [CrossRef] [PubMed]
- Amin, J.; Erskine, D.; Donaghy, P.C.; Surendranathan, A.; Swann, P.; Kunicki, A.P.; Boche, D.; Holmes, C.; McKeith, I.G.; O’Brien, J.T.; et al. Inflammation in Dementia with Lewy Bodies. Neurobiol. Dis. 2022, 168, 105698. [Google Scholar] [CrossRef]
- Kuo, S.-H.; Tasset, I.; Cheng, M.M.; Diaz, A.; Pan, M.-K.; Lieberman, O.J.; Hutten, S.J.; Alcalay, R.N.; Kim, S.; Ximénez-Embún, P.; et al. Mutant Glucocerebrosidase Impairs α-Synuclein Degradation by Blockade of Chaperone-Mediated Autophagy. Sci. Adv. 2022, 8, eabm6393. [Google Scholar] [CrossRef]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher Disease: Mutation and Polymorphism Spectrum in the Glucocerebrosidase Gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- Smith, L.; Mullin, S.; Schapira, A.H.V. Insights into the Structural Biology of Gaucher Disease. Exp. Neurol. 2017, 298, 180–190. [Google Scholar] [CrossRef]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray Structure of Human Acid-β-glucosidase, the Defective Enzyme in Gaucher Disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef]
- Amaral, C.E.d.M.; Lopes, P.F.; Ferreira, J.C.C.; Alves, E.A.C.; Montenegro, M.V.B.; da Costa, E.T.; Yamada, E.S.; Cavalcante, F.O.Q.; Santana-da-Silva, L.C. GBA Mutations p.N370S and p.L444P Are Associated with Parkinson’s Disease in Patients from Northern Brazil. Arq. Neuropsiquiatr. 2019, 77, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Marlet, F.R.; Cerri, S.; Schapira, A.H.V.; Blandini, F.; Di Monte, D.A. Sphingolipid Changes in Parkinson L444P GBA Mutation Fibroblasts Promote α-Synuclein Aggregation. Brain 2022, 145, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Liou, B.; Zhang, W.; Fannin, V.; Quinn, B.; Ran, H.; Xu, K.; Setchell, K.D.R.; Witte, D.; Grabowski, G.A.; Sun, Y. Combination of Acid β-Glucosidase Mutation and Saposin C Deficiency in Mice Reveals Gba1 Mutation Dependent and Tissue-Specific Disease Phenotype. Sci. Rep. 2019, 9, 5571. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Wolf, P.; Chiang, M.S.R.; Helesicova, K.; Zhang, X.K.; Merchant, K.; Hutten, S.J.; Scherzer, C.; Caspell-Garcia, C.; Blauwendraat, C.; et al. Longitudinal Measurements of Glucocerebrosidase Activity in Parkinson’s Patients. Ann. Clin. Transl. Neurol. 2020, 7, 1816–1830. [Google Scholar] [CrossRef]
- Dupuis, L.; Chauvet, M.; Bourdelier, E.; Dussiot, M.; Belmatoug, N.; Le Van Kim, C.; Chêne, A.; Franco, M. Phagocytosis of Erythrocytes from Gaucher Patients Induces Phenotypic Modifications in Macrophages, Driving Them toward Gaucher Cells. Int. J. Mol. Sci. 2022, 23, 7640. [Google Scholar] [CrossRef]
- Stone, W.L.; Basit, H.; Master, S.R. Gaucher Disease; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Furderer, M.L.; Hertz, E.; Lopez, G.J.; Sidransky, E. Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism. Int. J. Mol. Sci. 2022, 23, 5842. [Google Scholar] [CrossRef]
- Beutler, E.; Gelbart, T.; Scott, C.R. Hematologically Important Mutations: Gaucher Disease. Blood Cells, Mol. Dis. 2005, 35, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, P. Expression and Functional Characterization of Mutated Glucocerebrosidase Alleles Causing Gaucher Disease in Spanish Patients. Blood Cells Mol. Dis. 2004, 32, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Lopez, G. The Link between the GBA Gene and Parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef]
- Malini, E.; Grossi, S.; Deganuto, M.; Rosano, C.; Parini, R.; Dominisini, S.; Cariati, R.; Zampieri, S.; Bembi, B.; Filocamo, M.; et al. Functional Analysis of 11 Novel GBA Alleles. Eur. J. Hum. Genet. 2014, 22, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shu, L.; Sun, Q.; Zhou, X.; Pan, H.; Guo, J.; Tang, B. Integrated Genetic Analysis of Racial Differences of Common GBA Variants in Parkinson’s Disease: A Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Leonart, L.P.; Fachi, M.M.; Böger, B.; da Silva, M.R.; Szpak, R.; Lombardi, N.F.; Pedroso, M.L.A.; Pontarolo, R. A Systematic Review and Meta-Analyses of Longitudinal Studies on Drug Treatments for Gaucher Disease. Ann. Pharmacother. 2022, 10600280221108443. [Google Scholar] [CrossRef] [PubMed]
- Chavananon, S.; Sripornsawan, P.; Songthawee, N.; Chotsampancharoen, T. Successful Treatment of Gaucher Disease With Matched Sibling Hematopoietic Stem Cell Transplantation: A Case Report and Literature Review. J. Pediatr. Hematol. Oncol. 2021, 43, e1153–e1155. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Hefti, F.; Sevigny, J. Gene Therapy for Parkinson’s Disease Associated with GBA1 Mutations. J. Parkinsons. Dis. 2021, 11, S183–S188. [Google Scholar] [CrossRef] [PubMed]
- Goker-Alpan, O. Parkinsonism among Gaucher Disease Carriers. J. Med. Genet. 2004, 41, 937–940. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and Dementia in GBA-Associated Parkinson’s Disease: The Mutation Matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Senkevich, K.; Gan-Or, Z. Autophagy Lysosomal Pathway Dysfunction in Parkinson’s Disease; Evidence from Human Genetics. Parkinsonism Relat. Disord. 2020, 73, 60–71. [Google Scholar] [CrossRef]
- Balestrino, R.; Tunesi, S.; Tesei, S.; Lopiano, L.; Zecchinelli, A.L.; Goldwurm, S. Penetrance of Glucocerebrosidase (GBA) Mutations in Parkinson’s Disease: A Kin Cohort Study. Mov. Disord. 2020, 35, 2111–2114. [Google Scholar] [CrossRef] [PubMed]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaiti, B.; Bonaiti-Pellie, C.; Brice, A.; et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Thaler, A.; Gurevich, T.; Bar Shira, A.; Gana Weisz, M.; Ash, E.; Shiner, T.; Orr-Urtreger, A.; Giladi, N.; Mirelman, A. A “Dose” Effect of Mutations in the GBA Gene on Parkinson’s Disease Phenotype. Parkinsonism Relat. Disord. 2017, 36, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Avenali, M.; Blandini, F.; Cerri, S. Glucocerebrosidase Defects as a Major Risk Factor for Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 97. [Google Scholar] [CrossRef]
- Velez-Pardo, C.; Lorenzo-Betancor, O.; Jimenez-Del-Rio, M.; Moreno, S.; Lopera, F.; Cornejo-Olivas, M.; Torres, L.; Inca-Martinez, M.; Mazzetti, P.; Cosentino, C.; et al. The Distribution and Risk Effect of GBA Variants in a Large Cohort of PD Patients from Colombia and Peru. Parkinsonism Relat. Disord. 2019, 63, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Gruschus, J.M.; Velayati, A.; Sidransky, E.; Lee, J.C. Saposin C Protects Glucocerebrosidase against α-Synuclein Inhibition. Biochemistry 2013, 52, 7161–7163. [Google Scholar] [CrossRef]
- Petrucci, S.; Ginevrino, M.; Trezzi, I.; Monfrini, E.; Ricciardi, L.; Albanese, A.; Avenali, M.; Barone, P.; Bentivoglio, A.R.; Bonifati, V.; et al. GBA-Related Parkinson’s Disease: Dissection of Genotype-Phenotype Correlates in a Large Italian Cohort. Mov. Disord. 2020, 35, 2106–2111. [Google Scholar] [CrossRef]
- Mallett, V.; Ross, J.P.; Alcalay, R.N.; Ambalavanan, A.; Sidransky, E.; Dion, P.A.; Rouleau, G.A.; Gan-Or, Z. GBA p.T369M Substitution in Parkinson Disease: Polymorphism or Association? A Meta-Analysis. Neurol. Genet. 2016, 2, e104. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jing, Y.; Lun, P.; Liu, X.; Sun, P. Association of Gender and Age at Onset with Glucocerebrosidase Associated Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neurol. Sci. 2021, 42, 2261–2271. [Google Scholar] [CrossRef]
- Berge-Seidl, V.; Pihlstrøm, L.; Maple-Grødem, J.; Forsgren, L.; Linder, J.; Larsen, J.P.; Tysnes, O.-B.; Toft, M. The GBA Variant E326K Is Associated with Parkinson’s Disease and Explains a Genome-Wide Association Signal. Neurosci. Lett. 2017, 658, 48–52. [Google Scholar] [CrossRef]
- Heijer, J.M.; Cullen, V.C.; Quadri, M.; Schmitz, A.; Hilt, D.C.; Lansbury, P.; Berendse, H.W.; Berg, W.D.J.; Bie, R.M.A.; Boertien, J.M.; et al. A Large-Scale Full GBA1 Gene Screening in Parkinson’s Disease in the Netherlands. Mov. Disord. 2020, 35, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Mahungu, A.C.; Anderson, D.G.; Rossouw, A.C.; van Coller, R.; Carr, J.A.; Ross, O.A.; Bardien, S. Screening of the Glucocerebrosidase (GBA) Gene in South Africans of African Ancestry with Parkinson’s Disease. Neurobiol. Aging 2020, 88, 156.e11–156.e14. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.L.; Lohmann, K.; Tan, A.H.; Tay, Y.W.; Ibrahim, K.A.; Abdul Aziz, Z.; Mawardi, A.S.; Puvanarajah, S.D.; Lim, T.T.; Looi, I.; et al. Glucocerebrosidase (GBA) Gene Variants in a Multi-Ethnic Asian Cohort with Parkinson’s Disease: Mutational Spectrum and Clinical Features. J. Neural Transm. 2022, 129, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Barkhuizen, M.; Anderson, D.G.; van der Westhuizen, F.H.; Grobler, A.F. A Molecular Analysis of the GBA Gene in Caucasian South Africans with Parkinson’s Disease. Mol. Genet. Genomic Med. 2017, 5, 147–156. [Google Scholar] [CrossRef]
- Tipton, P.W.; Soto-Beasley, A.I.; Walton, R.L.; Soler-Rangel, S.; Romero-Osorio, Ó.; Díaz, C.; Moreno-López, C.L.; Ross, O.A.; Wszolek, Z.K.; Cerquera-Cleves, C. Prevalence of GBA p.K198E Mutation in Colombian and Hispanic Populations. Parkinsonism Relat. Disord. 2020, 73, 16–18. [Google Scholar] [CrossRef]
- Liu, C.-C.; Murray, M.E.; Li, X.; Zhao, N.; Wang, N.; Heckman, M.G.; Shue, F.; Martens, Y.; Li, Y.; Raulin, A.-C.; et al. APOE3-Jacksonville (V236E) Variant Reduces Self-Aggregation and Risk of Dementia. Sci. Transl. Med. 2021, 13, eabc9375. [Google Scholar] [CrossRef]
- Nalls, M.A.; Duran, R.; Lopez, G.; Kurzawa-Akanbi, M.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M.; Theuns, J.; Crosiers, D.; Cras, P.; et al. A Multicenter Study of Glucocerebrosidase Mutations in Dementia With Lewy Bodies. JAMA Neurol. 2013, 70, 727. [Google Scholar] [CrossRef]
- Guerreiro, R.; Escott-Price, V.; Darwent, L.; Parkkinen, L.; Ansorge, O.; Hernandez, D.G.; Nalls, M.A.; Clark, L.; Honig, L.; Marder, K.; et al. Genome-Wide Analysis of Genetic Correlation in Dementia with Lewy Bodies, Parkinson’s and Alzheimer’s Diseases. Neurobiol. Aging 2016, 38, 214.e7–214.e10. [Google Scholar] [CrossRef]
- Rongve, A.; Witoelar, A.; Ruiz, A.; Athanasiu, L.; Abdelnour, C.; Clarimon, J.; Heilmann-Heimbach, S.; Hernández, I.; Moreno-Grau, S.; de Rojas, I.; et al. GBA and APOE Ε4 Associate with Sporadic Dementia with Lewy Bodies in European Genome Wide Association Study. Sci. Rep. 2019, 9, 7013. [Google Scholar] [CrossRef]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.-P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. Diagnosis and Management of Dementia with Lewy Bodies. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef]
- Liu, L.; Li, J.; Quan, W.; Qin, Y.; Zhang, Q.; Pei, X.; Su, H.; Xu, J.; Chen, J. Effect of GBA Gene Variants on Clinical Characteristics of Dementia with Lewy Bodies: A Review and Meta-Analyses. Neurol. Sci. 2022, 43, 3541–3550. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.F.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Factor, S.A.; Wood-Siverio, C.; et al. GBA Variants Are Associated with a Distinct Pattern of Cognitive Deficits in Parkinson’s Disease. Mov. Disord. 2016, 31, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Perez-Roca, L.; Adame-Castillo, C.; Campdelacreu, J.; Ispierto, L.; Vilas, D.; Rene, R.; Alvarez, R.; Gascon-Bayarri, J.; Serrano-Munoz, M.A.; Ariza, A.; et al. Glucocerebrosidase MRNA Is Diminished in Brain of Lewy Body Diseases and Changes with Disease Progression in Blood. Aging Dis. 2018, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.C.; Pankratz, N.; Marek, D.K.; Pauciulo, M.W.; Elsaesser, V.E.; Halter, C.A.; Rudolph, A.; Wojcieszek, J.; Pfeiffer, R.F.; Foroud, T.; et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 2009, 72, 310–316. [Google Scholar] [CrossRef]
- Emelyanov, A.; Boukina, T.; Yakimovskii, A.; Usenko, T.; Drosdova, A.; Zakharchuk, A.; Andoskin, P.; Dubina, M.; Schwarzman, A.; Pchelina, S. Glucocerebrosidase Gene Mutations Are Associated with Parkinson’s Disease in Russia. Mov. Disord. 2012, 27, 158–159. [Google Scholar] [CrossRef]
- Saunders-Pullman, R.; Hagenah, J.; Dhawan, V.; Stanley, K.; Pastores, G.; Sathe, S.; Tagliati, M.; Condefer, K.; Palmese, C.; Brüggemann, N.; et al. Gaucher Disease Ascertained through a Parkinson’s Center: Imaging and Clinical Characterization. Mov. Disord. 2010, 25, 1364–1372. [Google Scholar] [CrossRef]
- Lesage, S.; Condroyer, C.; Hecham, N.; Anheim, M.; Belarbi, S.; Lohman, E.; Viallet, F.; Pollak, P.; Abada, M.; Durr, A.; et al. Mutations in the Glucocerebrosidase Gene Confer a Risk for Parkinson Disease in North Africa. Neurology 2011, 76, 301–303. [Google Scholar] [CrossRef]
- Tan, E.-K.; Tong, J.; Fook-Chong, S.; Yih, Y.; Wong, M.-C.; Pavanni, R.; Zhao, Y. Glucocerebrosidase Mutations and Risk of Parkinson Disease in Chinese Patients. Arch. Neurol. 2007, 64, 1056. [Google Scholar] [CrossRef]
- De Marco, E.V.; Annesi, G.; Tarantino, P.; Rocca, F.E.; Provenzano, G.; Civitelli, D.; Cirò Candiano, I.C.; Annesi, F.; Carrideo, S.; Condino, F.; et al. Glucocerebrosidase Gene Mutations Are Associated with Parkinson’s Disease in Southern Italy. Mov. Disord. 2008, 23, 460–463. [Google Scholar] [CrossRef]
- Bras, J.M.; Singleton, A. Genetic Susceptibility in Parkinson’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 597–603. [Google Scholar] [CrossRef]
- Kalinderi, K.; Bostantjopoulou, S.; Paisan-Ruiz, C.; Katsarou, Z.; Hardy, J.; Fidani, L. Complete Screening for Glucocerebrosidase Mutations in Parkinson Disease Patients from Greece. Neurosci. Lett. 2009, 452, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Morgan, A.; Lang, A.E.; Salehi-Rad, S.; Kawarai, T.; Meng, Y.; Ray, P.N.; Farrer, L.A.; St George-Hyslop, P.; Rogaeva, E. Analysis of the Glucocerebrosidase Gene in Parkinson’s Disease. Mov. Disord. 2005, 20, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, J.; Mizuta, I.; Toyoda, A.; Ashida, R.; Takahashi, Y.; Goto, J.; Fukuda, Y.; Date, H.; Iwata, A.; Yamamoto, M.; et al. Mutations for Gaucher Disease Confer High Susceptibility to Parkinson Disease. Arch. Neurol. 2009, 66, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, S.S.; Petersen, D.; Marlet, F.R.; Kücükköse, E.; Galvagnion, C. The Interplay between Glucocerebrosidase, α-Synuclein and Lipids in Human Models of Parkinson’s Disease. Biophys. Chem. 2021, 273, 106534. [Google Scholar] [CrossRef] [PubMed]
- Doeppner, R.; Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Levy, O.A.; Waters, C.H.; Fahn, S.; Ford, B.; Kuo, S.-H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase Activity in Parkinson’s Disease with and without GBA Mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef]
- Kedariti, M.; Frattini, E.; Baden, P.; Cogo, S.; Civiero, L.; Ziviani, E.; Zilio, G.; Bertoli, F.; Aureli, M.; Kaganovich, A.; et al. LRRK2 Kinase Activity Regulates GCase Level and Enzymatic Activity Differently Depending on Cell Type in Parkinson’s Disease. Npj Park. Dis. 2022, 8, 92. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Li, J.-D. The Roles of Post-Translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef]
- Beavan, M.; McNeill, A.; Proukakis, C.; Hughes, D.A.; Mehta, A.; Schapira, A.H.V. Evolution of Prodromal Clinical Markers of Parkinson Disease in a GBA Mutation–Positive Cohort. JAMA Neurol. 2015, 72, 201. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced Glucocerebrosidase Is Associated with Increased α-Synuclein in Sporadic Parkinson’s Disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef]
- Elabi, O.; Gaceb, A.; Carlsson, R.; Padel, T.; Soylu-Kucharz, R.; Cortijo, I.; Li, W.; Li, J.-Y.; Paul, G. Human α-Synuclein Overexpression in a Mouse Model of Parkinson’s Disease Leads to Vascular Pathology, Blood Brain Barrier Leakage and Pericyte Activation. Sci. Rep. 2021, 11, 1120. [Google Scholar] [CrossRef]
- Yap, T.L.; Velayati, A.; Sidransky, E.; Lee, J.C. Membrane-Bound α-Synuclein Interacts with Glucocerebrosidase and Inhibits Enzyme Activity. Mol. Genet. Metab. 2013, 108, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gegg, M.; Chau, D.; Schapira, A. Glucocerebrosidase Activity, Cathepsin D and Monomeric α-Synuclein Interactions in a Stem Cell Derived Neuronal Model of a PD Associated GBA1 Mutation. Neurobiol. Dis. 2020, 134, 104620. [Google Scholar] [CrossRef] [PubMed]
- Menozzi, E.; Schapira, A.H.V. Exploring the Genotype-Phenotype Correlation in GBA-Parkinson Disease: Clinical Aspects, Biomarkers, and Potential Modifiers. Front. Neurol. 2021, 12, 694764. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Machado De Oliveira, L.; Reis Barbosa, E.; Aquino, C.C.; Puppi Munhoz, R.; Fasano, A.; Cury, R.G. Deep Brain Stimulation in Patients with Mutations in Parkinson’s Disease-Related Genes: A Systematic Review. Clin. Pract. 2019, 6, 359–368. [Google Scholar] [CrossRef]
- Malek, N.; Weil, R.S.; Bresner, C.; Lawton, M.A.; Grosset, K.A.; Tan, M.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Foltynie, T.; et al. Features of GBA-Associated Parkinson’s Disease at Presentation in the UK Tracking Parkinson’s Study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 702–709. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Caccappolo, E.; Mejia-Santana, H.; Tang, M.-X.; Rosado, L.; Orbe Reilly, M.; Ruiz, D.; Ross, B.; Verbitsky, M.; Kisselev, S.; et al. Cognitive Performance of GBA Mutation Carriers with Early-Onset PD: The CORE-PD Study. Neurology 2012, 78, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Marković, I.; Kresojević, N.; Kostić, V.S. Glucocerebrosidase and Parkinsonism: Lessons to Learn. J. Neurol. 2016, 263, 1033–1044. [Google Scholar] [CrossRef]
- Winder-Rhodes, S.E.; Evans, J.R.; Ban, M.; Mason, S.L.; Williams-Gray, C.H.; Foltynie, T.; Duran, R.; Mencacci, N.E.; Sawcer, S.J.; Barker, R.A. Glucocerebrosidase Mutations Influence the Natural History of Parkinson’s Disease in a Community-Based Incident Cohort. Brain 2013, 136, 392–399. [Google Scholar] [CrossRef]
- Jesús, S.; Huertas, I.; Bernal-Bernal, I.; Bonilla-Toribio, M.; Cáceres-Redondo, M.T.; Vargas-González, L.; Gómez-Llamas, M.; Carrillo, F.; Calderón, E.; Carballo, M.; et al. GBA Variants Influence Motor and Non-Motor Features of Parkinson’s Disease. PLoS ONE 2016, 11, e0167749. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, D.A.; McCarthy, A.; Soto-Beasley, A.I.; Walton, R.L.; Magennis, B.; McLaughlin, R.L.; Hardiman, O.; Ross, O.A.; Lynch, T. Association Between Glucocerebrosidase Mutations and Parkinson’s Disease in Ireland. Front. Neurol. 2020, 11, 527. [Google Scholar] [CrossRef] [PubMed]
- Stoker, T.B.; Camacho, M.; Winder-Rhodes, S.; Liu, G.; Scherzer, C.R.; Foltynie, T.; Evans, J.; Breen, D.P.; Barker, R.A.; Williams-Gray, C.H. Impact of GBA1 Variants on Long-Term Clinical Progression and Mortality in Incident Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Pal, G.; Mangone, G.; Hill, E.J.; Ouyang, B.; Liu, Y.; Lythe, V.; Ehrlich, D.; Saunders-Pullman, R.; Shanker, V.; Bressman, S.; et al. Parkinson Disease and Subthalamic Nucleus Deep Brain Stimulation: Cognitive Effects in GBA Mutation Carriers. Ann. Neurol. 2022, 91, 424–435. [Google Scholar] [CrossRef]
- Leocadi, M.; Canu, E.; Donzuso, G.; Stojkovic, T.; Basaia, S.; Kresojević, N.; Stankovic, I.; Sarasso, E.; Piramide, N.; Tomic, A.; et al. Longitudinal Clinical, Cognitive, and Neuroanatomical Changes over 5 Years in GBA-Positive Parkinson’s Disease Patients. J. Neurol. 2022, 269, 1485–1500. [Google Scholar] [CrossRef]
- Szwedo, A.A.; Dalen, I.; Pedersen, K.F.; Camacho, M.; Bäckström, D.; Forsgren, L.; Tzoulis, C.; Winder-Rhodes, S.; Hudson, G.; Liu, G.; et al. GBA and APOE Impact Cognitive Decline in Parkinson’s Disease: A 10-Year Population-Based Study. Mov. Disord. 2022, 37, 1016–1027. [Google Scholar] [CrossRef]
- Omer, N.; Giladi, N.; Gurevich, T.; Bar-Shira, A.; Gana-Weisz, M.; Glinka, T.; Goldstein, O.; Kestenbaum, M.; Cedarbaum, J.M.; Mabrouk, O.S.; et al. Glucocerebrosidase Activity Is Not Associated with Parkinson’s Disease Risk or Severity. Mov. Disord. 2022, 37, 190–195. [Google Scholar] [CrossRef]
- Moran, E.E.; Bressman, S.B.; Ortega, R.A.; Raymond, D.; Nichols, W.C.; Palmese, C.A.; Elango, S.; Swan, M.; Shanker, V.; Perera, I.; et al. Cognitive Functioning of Glucocerebrosidase (GBA) Non-Manifesting Carriers. Front. Neurol. 2021, 12, 635958. [Google Scholar] [CrossRef]
- Swan, M.; Doan, N.; Ortega, R.A.; Barrett, M.; Nichols, W.; Ozelius, L.; Soto-Valencia, J.; Boschung, S.; Deik, A.; Sarva, H.; et al. Neuropsychiatric Characteristics of GBA-Associated Parkinson Disease. J. Neurol. Sci. 2016, 370, 63–69. [Google Scholar] [CrossRef]
- Wilson, H.; de Natale, E.; Politis, M. Microstructural Changes in GBA Mutation Carriers with Depression. In Movement Disorders; John Wiley & Sons: Hoboken, NJ, USA, 2020. [Google Scholar]
- Mullin, S.; Beavan, M.; Bestwick, J.; McNeill, A.; Proukakis, C.; Cox, T.; Hughes, D.; Mehta, A.; Zetterberg, H.; Schapira, A.H.V. Evolution and Clustering of Prodromal Parkinsonian Features in GBA1 Carriers. Mov. Disord. 2019, 34, 1365–1373. [Google Scholar] [CrossRef]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.-K.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-Associated Parkinson’s Disease: Reduced Survival and More Rapid Progression in a Prospective Longitudinal Study. Mov. Disord. 2015, 30, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Avenali, M.; Toffoli, M.; Mullin, S.; McNeil, A.; Hughes, D.A.; Mehta, A.; Blandini, F.; Schapira, A.H.V. Evolution of Prodromal Parkinsonian Features in a Cohort of GBA Mutation-Positive Individuals: A 6-Year Longitudinal Study. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1091–1097. [Google Scholar] [CrossRef]
- Iwaki, H.; Blauwendraat, C.; Leonard, H.L.; Liu, G.; Maple-Grødem, J.; Corvol, J.C.; Pihlstrøm, L.; Van Nimwegen, M.; Hutten, S.J.; Nguyen, K.D.H.; et al. Genetic Risk of Parkinson Disease and Progression: An Analysis of 13 Longitudinal Cohorts. Neurol. Genet. 2019, 5, e354. [Google Scholar] [CrossRef] [PubMed]
- Schindlbeck, K.A.; Vo, A.; Nguyen, N.; Tang, C.C.; Niethammer, M.; Dhawan, V.; Brandt, V.; Saunders-Pullman, R.; Bressman, S.B.; Eidelberg, D. LRRK2 and GBA Variants Exert Distinct Influences on Parkinson’s Disease-Specific Metabolic Networks. Cereb. Cortex 2020, 30, 2867–2878. [Google Scholar] [CrossRef]
- Greuel, A.; Trezzi, J.; Glaab, E.; Ruppert, M.C.; Maier, F.; Jäger, C.; Hodak, Z.; Lohmann, K.; Ma, Y.; Eidelberg, D.; et al. GBA Variants in Parkinson’s Disease: Clinical, Metabolomic, and Multimodal Neuroimaging Phenotypes. Mov. Disord. 2020, 35, 2201–2210. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Uribe, L.; Cho, H.R.; Caspell-Garcia, C.; Coffey, C.S.; Siderowf, A.; Trojanowski, J.Q.; Shaw, L.M.; Seibyl, J.; Singleton, A.; et al. Clinical and Dopamine Transporter Imaging Characteristics of Non-Manifest LRRK2 and GBA Mutation Carriers in the Parkinson’s Progression Markers Initiative (PPMI): A Cross-Sectional Study. Lancet Neurol. 2020, 19, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Brumm, M.C.; Uribe, L.; Caspell-Garcia, C.; Coffey, C.S.; Siderowf, A.; Alcalay, R.N.; Trojanowski, J.Q.; Shaw, L.M.; Seibyl, J.; et al. Clinical and Dopamine Transporter Imaging Characteristics of Leucine Rich Repeat Kinase 2 (LRRK2) and Glucosylceramidase Beta (GBA) Parkinson’s Disease Participants in the Parkinson’s Progression Markers Initiative: A Cross-Sectional Study. Mov. Disord. 2020, 35, 833–844. [Google Scholar] [CrossRef]
- Arkadir, D.; Dinur, T.; Becker Cohen, M.; Revel-Vilk, S.; Tiomkin, M.; Brüggemann, N.; Cozma, C.; Rolfs, A.; Zimran, A. Prodromal Substantia Nigra Sonography Undermines Suggested Association between Substrate Accumulation and the Risk for GBA -Related Parkinson’s Disease. Eur. J. Neurol. 2019, 26, 1013–1018. [Google Scholar] [CrossRef]
- Gao, Y.; Nie, K.; Huang, B.; Mei, M.; Guo, M.; Xie, S.; Huang, Z.; Wang, L.; Zhao, J.; Zhang, Y.; et al. Changes of Brain Structure in Parkinson’s Disease Patients with Mild Cognitive Impairment Analyzed via VBM Technology. Neurosci. Lett. 2017, 658, 121–132. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, C.; Yao, J.; Chen, X.; Gao, F.; Jiang, S.; Chen, W.; Zhou, J.; Wang, G. Protein-Based Amide Proton Transfer-Weighted MR Imaging of Amnestic Mild Cognitive Impairment. NeuroImage Clin. 2020, 25, 102153. [Google Scholar] [CrossRef]
- Filippi, M.; Balestrino, R.; Basaia, S.; Agosta, F. Neuroimaging in Glucocerebrosidase-Associated Parkinsonism: A Systematic Review. Mov. Disord. 2022, 37, 1375–1393. [Google Scholar] [CrossRef] [PubMed]
- Price, A.; Farooq, R.; Yuan, J.-M.; Menon, V.B.; Cardinal, R.N.; O’Brien, J.T. Mortality in Dementia with Lewy Bodies Compared with Alzheimer’s Dementia: A Retrospective Naturalistic Cohort Study. BMJ Open 2017, 7, e017504. [Google Scholar] [CrossRef]
- Power, J.H.T.; Barnes, O.L.; Chegini, F. Lewy Bodies and the Mechanisms of Neuronal Cell Death in Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathol. 2017, 27, 3–12. [Google Scholar] [CrossRef]
- Amin, J.; Holmes, C.; Dorey, R.B.; Tommasino, E.; Casal, Y.R.; Williams, D.M.; Dupuy, C.; Nicoll, J.A.R.; Boche, D. Neuroinflammation in Dementia with Lewy Bodies: A Human Post-Mortem Study. Transl. Psychiatry 2020, 10, 267. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, I.; Arai, T.; Hasegawa, M. The Basis of Clinicopathological Heterogeneity in TDP-43 Proteinopathy. Acta Neuropathol. 2019, 138, 751–770. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Prokop, S.; Gorion, K.-M.M.; Kim, J.D.; Sorrentino, Z.A.; Bell, B.M.; Manaois, A.N.; Chakrabarty, P.; Davies, P.; Giasson, B.I. Tau Ser208 Phosphorylation Promotes Aggregation and Reveals Neuropathologic Diversity in Alzheimer’s Disease and Other Tauopathies. Acta Neuropathol. Commun. 2020, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Calil, V.; Sudo, F.K.; Santiago-Bravo, G.; Lima, M.A.; Mattos, P. Anosognosia in Dementia with Lewy Bodies: A Systematic Review. Arq. Neuropsiquiatr. 2021, 79, 334–342. [Google Scholar] [CrossRef]
- Shiner, T.; Mirelman, A.; Gana Weisz, M.; Bar-Shira, A.; Ash, E.; Cialic, R.; Nevler, N.; Gurevich, T.; Bregman, N.; Orr-Urtreger, A.; et al. High Frequency of GBA Gene Mutations in Dementia With Lewy Bodies Among Ashkenazi Jews. JAMA Neurol. 2016, 73, 1448. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Prada-Dacasa, P.; Santos, C.; Adame-Castillo, C.; Campdelacreu, J.; Reñé, R.; Gascón-Bayarri, J.; Ispierto, L.; Álvarez, R.; Ariza, A.; et al. GBA Mutations Are Associated With Earlier Onset and Male Sex in Dementia With Lewy Bodies. Mov. Disord. 2016, 31, 1066–1070. [Google Scholar] [CrossRef]
- Asselta, R.; Rimoldi, V.; Siri, C.; Cilia, R.; Guella, I.; Tesei, S.; Soldà, G.; Pezzoli, G.; Duga, S.; Goldwurm, S. Glucocerebrosidase Mutations in Primary Parkinsonism. Parkinsonism Relat. Disord. 2014, 20, 1215–1220. [Google Scholar] [CrossRef]
- Matar, E.; Ehgoetz Martens, K.A.; Halliday, G.M.; Lewis, S.J.G. Clinical Features of Lewy Body Dementia: Insights into Diagnosis and Pathophysiology. J. Neurol. 2020, 267, 380–389. [Google Scholar] [CrossRef]
- Moylett, S.; Price, A.; Cardinal, R.N.; Aarsland, D.; Mueller, C.; Stewart, R.; O’Brien, J.T. Clinical Presentation, Diagnostic Features, and Mortality in Dementia with Lewy Bodies. J. Alzheimer’s Dis. 2019, 67, 995–1005. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Koss, D.J.; Erskine, D.; Walker, L.; Kurzawa-Akanbi, M.; Burn, D.; Donaghy, P.; Morris, C.; Taylor, J.-P.; Thomas, A.; et al. Dementia with Lewy Bodies: An Update and Outlook. Mol. Neurodegener. 2019, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.A.; Hasegawa, M. Prion-like Spreading of Pathological α-Synuclein in Brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Lee, J.; Monteiro, O.; Woerman, A.L.; Lazar, A.A.; Condello, C.; Paras, N.A.; Prusiner, S.B. Different α-Synuclein Prion Strains Cause Dementia with Lewy Bodies and Multiple System Atrophy. Proc. Natl. Acad. Sci. USA 2022, 119, e2113489119. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Tian, M.; Yao, Q.; Li, Q.; Tang, F.; Xiao, C.; Shi, J.; Chen, J. Neuroimaging Alterations in Dementia with Lewy Bodies and Neuroimaging Differences between Dementia with Lewy Bodies and Alzheimer’s Disease: An Activation Likelihood Estimation Meta-analysis. CNS Neurosci. Ther. 2022, 28, 183–205. [Google Scholar] [CrossRef]
- Ferreira, D. Structural Imaging in Dementia with Lewy Bodies: The Potential of Multivariate Data Analysis. Psychiatry Res. Neuroimaging 2020, 306, 111180. [Google Scholar] [CrossRef]
- Komatsu, J.; Samuraki, M.; Nakajima, K.; Arai, H.; Arai, H.; Arai, T.; Asada, T.; Fujishiro, H.; Hanyu, H.; Iizuka, O.; et al. 123-I-MIBG Myocardial Scintigraphy for the Diagnosis of DLB: A Multicentre 3-Year Follow-up Study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1167–1173. [Google Scholar] [CrossRef]
- Kantarci, K.; Boeve, B.F.; Przybelski, S.A.; Lesnick, T.G.; Chen, Q.; Fields, J.; Schwarz, C.G.; Senjem, M.L.; Gunte, J.L.; Jack, C.R.; et al. FDG PET Metabolic Signatures Distinguishing Prodromal DLB and Prodromal AD. NeuroImage Clin. 2021, 31, 102754. [Google Scholar] [CrossRef]
- Caminiti, S.P.; Sala, A.; Iaccarino, L.; Beretta, L.; Pilotto, A.; Gianolli, L.; Iannaccone, S.; Magnani, G.; Padovani, A.; Ferini-Strambi, L.; et al. Brain Glucose Metabolism in Lewy Body Dementia: Implications for Diagnostic Criteria. Alzheimers Res. Ther. 2019, 11, 20. [Google Scholar] [CrossRef]
- Hemminghyth, M.S.; Chwiszczuk, L.J.; Rongve, A.; Breitve, M.H. The Cognitive Profile of Mild Cognitive Impairment Due to Dementia with Lewy Bodies—An Updated Review. Front. Aging Neurosci. 2020, 12, 597579. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.; Bancher, C.; Eckert, A.; Förstl, H.; Frölich, L.; Hort, J.; Korczyn, A.D.; Kressig, R.W.; Levin, O.; Palomo, M.S.M. Management of Mild Cognitive Impairment (MCI): The Need for National and International Guidelines. World J. Biol. Psychiatry 2020, 21, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Hershey, L.A.; Coleman-Jackson, R. Pharmacological Management of Dementia with Lewy Bodies. Drugs Aging 2019, 36, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, J.; Matsukawa, T.; Sasaki, H.; Yabe, I.; Matsushima, M.; Dürr, A.; Brice, A.; Takashima, H.; Kikuchi, A.; Aoki, M.; et al. Variants Associated with Gaucher Disease in Multiple System Atrophy. Ann. Clin. Transl. Neurol. 2015, 2, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Wernick, A.I.; Walton, R.L.; Koga, S.; Soto-Beasley, A.I.; Heckman, M.G.; Gan-Or, Z.; Ren, Y.; Rademakers, R.; Uitti, R.J.; Wszolek, Z.K.; et al. GBA Variation and Susceptibility to Multiple System Atrophy. Parkinsonism Relat. Disord. 2020, 77, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Gruschus, J.M.; Velayati, A.; Westbroek, W.; Goldin, E.; Moaven, N.; Sidransky, E.; Lee, J.C. α-Synuclein Interacts with Glucocerebrosidase Providing a Molecular Link between Parkinson and Gaucher Diseases. J. Biol. Chem. 2011, 286, 28080–28088. [Google Scholar] [CrossRef] [PubMed]
- Gruschus, J.M. An Evolutionary Affair—The Connection between Gaucher Disease and Parkinson’s Disease. In Synuclein and the Coelacanth; Elsevier: Amsterdam, The Netherlands, 2021; pp. 159–179. [Google Scholar]
- Tayebi, N.; Parisiadou, L.; Berhe, B.; Gonzalez, A.N.; Serra-Vinardell, J.; Tamargo, R.J.; Maniwang, E.; Sorrentino, Z.; Fujiwara, H.; Grey, R.J.; et al. Glucocerebrosidase Haploinsufficiency in A53T α-Synuclein Mice Impacts Disease Onset and Course. Mol. Genet. Metab. 2017, 122, 198–208. [Google Scholar] [CrossRef]
- Bae, E.-J.; Yang, N.-Y.; Song, M.; Lee, C.S.; Lee, J.S.; Jung, B.C.; Lee, H.-J.; Kim, S.; Masliah, E.; Sardi, S.P.; et al. Glucocerebrosidase Depletion Enhances Cell-to-Cell Transmission of α-Synuclein. Nat. Commun. 2014, 5, 4755. [Google Scholar] [CrossRef]
- Brekk, O.R.; Moskites, A.; Isacson, O.; Hallett, P.J. Lipid-Dependent Deposition of Alpha-Synuclein and Tau on Neuronal Secretogranin II-Positive Vesicular Membranes with Age. Sci. Rep. 2018, 8, 15207. [Google Scholar] [CrossRef]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef]
- Moratalla, R.; Fan, D.; Zheng, W.; Fan, D. Glucocerebrosidase Mutations Cause Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease: Pathogenesis and Therapeutic Implications. Front. Aging Neurosci. 2022, 14, 851135. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.-H.; Tasset, I.; Cuervo, A.M.; Sulzer, D. Misfolded GBA/β-Glucocerebrosidase Impairs ER-Quality Control by Chaperone-Mediated Autophagy in Parkinson Disease. Autophagy 2022, 18, 3050–3052. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D. A Modulator of Wild-Type Glucocerebrosidase Improves Pathogenic Phenotypes in Dopaminergic Neuronal Models of Parkinson’s Disease. Sci. Transl. Med. 2019, 11, eaau6870. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. IPSC-Derived Neurons from GBA1-Associated Parkinson’s Disease Patients Show Autophagic Defects and Impaired Calcium Homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.V.; Duchen, M.R. Mitochondria and Quality Control Defects in a Mouse Model of Gaucher Disease—Links to Parkinson’s Disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef]
- Stojkovska, I.; Krainc, D.; Mazzulli, J.R. Molecular Mechanisms of α-Synuclein and GBA1 in Parkinson’s Disease. Cell Tissue Res. 2018, 373, 51–60. [Google Scholar] [CrossRef]
- Enquist, I.B.; Bianco, C.L.; Ooka, A.; Nilsson, E.; Månsson, J.E.; Ehinger, M.; Richter, J.; Brady, R.O.; Kirik, D.; Karlsson, S. Murine Models of Acute Neuronopathic Gaucher Disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17483–17488. [Google Scholar] [CrossRef]
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.-H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial Dysfunction and Mitophagy Defect Triggered by Heterozygous GBA Mutations. Autophagy 2019, 15, 113–130. [Google Scholar] [CrossRef]
- Chahine, L.M.; Qiang, J.; Ashbridge, E.; Minger, J.; Yearout, D.; Horn, S.; Colcher, A.; Hurtig, H.I.; Lee, V.M.-Y.; Van Deerlin, V.M.; et al. Clinical and Biochemical Differences in Patients Having Parkinson Disease With vs Without GBA Mutations. JAMA Neurol. 2013, 70, 852. [Google Scholar] [CrossRef]
- Kam, T.-I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and Astrocyte Dysfunction in Parkinson’s Disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal Storage and Impaired Autophagy Lead to Inflammasome Activation in Gaucher Macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and Its Relevance to Parkinson Disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Rothaug, M.; Zunke, F.; Mazzulli, J.R.; Schweizer, M.; Altmeppen, H.; Lüllmann-Rauch, R.; Kallemeijn, W.W.; Gaspar, P.; Aerts, J.M.; Glatzel, M.; et al. LIMP-2 Expression Is Critical for β-Glucocerebrosidase Activity and α-Synuclein Clearance. Proc. Natl. Acad. Sci. USA 2014, 111, 15573–15578. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granek, Z.; Barczuk, J.; Siwecka, N.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. GBA1 Gene Mutations in α-Synucleinopathies—Molecular Mechanisms Underlying Pathology and Their Clinical Significance. Int. J. Mol. Sci. 2023, 24, 2044. https://doi.org/10.3390/ijms24032044
Granek Z, Barczuk J, Siwecka N, Rozpędek-Kamińska W, Kucharska E, Majsterek I. GBA1 Gene Mutations in α-Synucleinopathies—Molecular Mechanisms Underlying Pathology and Their Clinical Significance. International Journal of Molecular Sciences. 2023; 24(3):2044. https://doi.org/10.3390/ijms24032044
Chicago/Turabian StyleGranek, Zuzanna, Julia Barczuk, Natalia Siwecka, Wioletta Rozpędek-Kamińska, Ewa Kucharska, and Ireneusz Majsterek. 2023. "GBA1 Gene Mutations in α-Synucleinopathies—Molecular Mechanisms Underlying Pathology and Their Clinical Significance" International Journal of Molecular Sciences 24, no. 3: 2044. https://doi.org/10.3390/ijms24032044
APA StyleGranek, Z., Barczuk, J., Siwecka, N., Rozpędek-Kamińska, W., Kucharska, E., & Majsterek, I. (2023). GBA1 Gene Mutations in α-Synucleinopathies—Molecular Mechanisms Underlying Pathology and Their Clinical Significance. International Journal of Molecular Sciences, 24(3), 2044. https://doi.org/10.3390/ijms24032044